Co-reporter:Jolene P. Reid
Organic & Biomolecular Chemistry 2017 vol. 15(Issue 33) pp:6943-6947
Publication Date(Web):2017/08/23
DOI:10.1039/C7OB01345K
Chiral BINOL-derived phosphoric acids catalyse the transfer hydrogenation of ketimines using Hantszch esters. In many cases the nitrogen on the imine binds to the catalyst through the catalyst hydroxyl group and the nucleophile forms a second hydrogen bond to the phosphoryl oxygen. DFT and ONIOM calculations show that the introduction of an ortho-hydroxyaryl group on the carbon atom of the ketimine leads the reaction to proceed through a 14-membered bifunctional mechanism. The transition states of these reactions involve both hydrogen bonding from the hydroxyl group on the imine and the nucleophile's proton to the phosphate catalyst. This mechanistic pathway is lower in energy than the conventional route, consistent with the experimentally observed increased rates of reaction relative to imines that are not derived from ortho-hydroxybenzophenone. To complement the high-level calculations, an accessible qualitative model has been developed that predicts the correct sense of stereoinduction for all examples.
Co-reporter:K. Ermanis;K. E. B. Parkes;T. Agback;J. M. Goodman
Organic & Biomolecular Chemistry 2017 vol. 15(Issue 42) pp:8998-9007
Publication Date(Web):2017/10/31
DOI:10.1039/C7OB01379E
A large-scale optimisation of density functional theory (DFT) conditions for computational NMR structure elucidation has been conducted by systematically screening the DFT functionals and statistical models. The extended PyDP4 workflow was tested on a diverse and challenging set of 42 biologically active and stereochemically rich compounds, including highly flexible molecules. MMFF/mPW1PW91/M06-2X in combination with a 2 Gaussian, 1 region statistical model was capable of identifying the correct diastereomer among up to an upper limit of 32 potential diastereomers. Overall a 2-fold reduction in structural uncertainty and a 7-fold reduction in model overconfidence have been achieved. Tools for rapid set-up and analysis of computational and experimental results, as well as for the statistical model generation, have been developed and are provided. All of this should facilitate rapid and reliable computational NMR structure elucidation, which has become a valuable tool to natural product chemists and synthetic chemists alike.
Co-reporter:Jolene P. Reid, Luis Simón, and Jonathan M. Goodman
Accounts of Chemical Research 2016 Volume 49(Issue 5) pp:1029
Publication Date(Web):April 29, 2016
DOI:10.1021/acs.accounts.6b00052
Chiral phosphoric acids have become powerful catalysts for the stereocontrolled synthesis of a diverse array of organic compounds. Since the initial report, the development of phosphoric acids as catalysts has been rapid, demonstrating the tremendous generality of this catalyst system and advancing the use of phosphoric acids to catalyze a broad range of asymmetric transformations ranging from Mannich reactions to hydrogenations through complementary modes of activation. These powerful applications have been developed without a clear mechanistic understanding of the reasons for the high level of stereocontrol.This Account describes investigations into the mechanism of the phosphoric acid catalyzed addition of nucleophiles to imines, focusing on binaphthol-based systems. In many cases, the hydroxyl phosphoric acid can form a hydrogen bond to the imine while the P═O interacts with the nucleophile. The single catalyst, therefore, activates both the electrophile and the nucleophile, while holding both in the chiral pocket created by the binaphthol and constrained by substituents at the 3 and 3′ positions.Detailed geometric and energetic information about the transition states can be gained from calculations using ONIOM methods that combine the advantages of DFT with some of the speed of force fields. These high-level calculations give a quantitative account of the selectivity in many cases, but require substantial computational resources. A simple qualitative model is a useful complement to this complex quantitative model.We summarize our calculations into a working model that can readily be sketched by hand and used to work out the likely sense of selectivity for each reaction. The steric demands of the different parts of the reactants determine how they fit into the chiral cavity and which of the competing pathways is favored. The preferred pathway can be found by considering the size of the substituents on the nitrogen and carbon atoms of the imine electrophile, and the position of the nucleophilic site on the nucleophile in relation to the hydrogen-bond which holds it in the catalyst active site.We present a guide to defining the pathway in operation allowing the fast and easy prediction of the stereochemical outcome and provide an overview of the breadth of reactions that can be explained by these models including the latest examples.
Co-reporter:Jolene P. Reid
Journal of the American Chemical Society 2016 Volume 138(Issue 25) pp:7910-7917
Publication Date(Web):May 26, 2016
DOI:10.1021/jacs.6b02825
BINOL-derived phosphoric acids provide effective asymmetric catalysis for many organic reactions. Catalysts based on this scaffold show a large structural diversity, especially in the 3,3′ substituents, and little is known about the molecular requirements for high selectivity. As a result, selection of the best catalyst for a particular transformation requires a trial and error screening process, as the size of the 3,3′ substituents is not simply related to their efficacy: the right choice is neither too large nor too small. We have developed an approach to identify and quantify structural features on the catalyst that determine selectivity. We show that the application of quantitative steric parameters (a new measure, AREA(θ), and rotation barrier) to an imine hydrogenation reaction allows the identification of catalyst features necessary for efficient stereoinduction, validated by QM/MM hybrid calculations.
Co-reporter:Kristaps Ermanis, Kevin E. B. Parkes, Tatiana Agback and Jonathan M. Goodman
Organic & Biomolecular Chemistry 2016 vol. 14(Issue 16) pp:3943-3949
Publication Date(Web):23 Mar 2016
DOI:10.1039/C6OB00015K
The DP4 parameter, which provides a confidence level for NMR assignment, has been widely used to help assign the structures of many stereochemically-rich molecules. We present an improved version of the procedure, which can be downloaded as Python script instead of running within a web-browser, and which analyses output from open-source molecular modelling programs (TINKER and NWChem) in addition to being able to use output from commercial packages (Schrodinger's Macromodel and Jaguar; Gaussian). The new open-source workflow incorporates a method for the automatic generation of diastereomers using InChI strings and has been tested on a range of new structures. This improved workflow permits the rapid and convenient computational elucidation of structure and relative stereochemistry.
Co-reporter:Timothy E. H. Allen, Sonia Liggi, Jonathan M. Goodman, Steve Gutsell, and Paul J. Russell
Chemical Research in Toxicology 2016 Volume 29(Issue 10) pp:1611
Publication Date(Web):September 7, 2016
DOI:10.1021/acs.chemrestox.6b00101
Molecular initiating events (MIEs) can be boiled down to chemical interactions. Chemicals that interact must have intrinsic properties that allow them to exhibit this behavior, be these properties stereochemical, electronic, or otherwise. In an attempt to discover some of these chemical characteristics, we have constructed structural alert-style structure–activity relationships (SARs) to computationally predict MIEs. This work utilizes chemical informatics approaches, searching the ChEMBL database for molecules that bind to a number of pharmacologically important human toxicology targets, including G-protein coupled receptors, enzymes, ion channels, nuclear receptors, and transporters. By screening these compounds to find common 2D fragments and combining this approach with a good understanding of the literature, bespoke 2D structural alerts have been written. These SARs form the beginning of a tool for screening novel chemicals to establish the kind of interactions that they may be able to make in humans. These SARs have been run through an internal validation to test their quality, and the results of this are also discussed. MIEs have proven to be difficult to find and characterize, but we believe we have taken a key first step with this work.
Co-reporter:Timothy E. H. AllenJonathan M. Goodman, Steve GutsellPaul J. Russell
Chemical Research in Toxicology 2016 Volume 29(Issue 12) pp:
Publication Date(Web):November 16, 2016
DOI:10.1021/acs.chemrestox.6b00341
The adverse outcome pathway (AOP) framework provides an alternative to traditional in vivo experiments for the risk assessment of chemicals. AOPs consist of a number of key events (KEs) linked by key event relationships across a range of biological organization backed by scientific evidence. The first KE in the pathway is the molecular initiating event (MIE)—the initial chemical trigger that starts an AOP. Over the past 3 years the AOP conceptual framework has gained a large amount of momentum in toxicology as an alternative to animal methods, and so the MIE has come into the spotlight. What is an MIE? How can MIEs be measured or predicted? What research is currently contributing to our understanding of MIEs? In this Perspective we outline answers to these key questions.
Co-reporter:Matthew N. Grayson and Jonathan M. Goodman
The Journal of Organic Chemistry 2015 Volume 80(Issue 4) pp:2056-2061
Publication Date(Web):January 28, 2015
DOI:10.1021/jo502616a
Density functional theory calculations suggest that asymmetric boronate addition to o-quinone methides proceeds via a Lewis acid catalyzed process through a closed six-membered transition structure. The BINOL-derived catalyst undergoes an exchange process with the original ethoxide boronate ligands. This activation mode successfully accounts for the sense and level of enantioselectivity observed experimentally. A qualitative model which accurately predicts the observed enantioselectivity has been developed and is consistent with results from our study of ketone propargylation demonstrating the reaction model’s generality. The effects of replacing the BINOL framework with H8–BINOL have been rationalized.
Co-reporter:Lois M. Overvoorde, Matthew N. Grayson, Yi Luo, and Jonathan M. Goodman
The Journal of Organic Chemistry 2015 Volume 80(Issue 5) pp:2634-2640
Publication Date(Web):February 5, 2015
DOI:10.1021/jo5028134
The reaction of tryptamine and (2-oxocyclohexyl)acetic acid can be catalyzed by 3,3′-bis(triphenylsilyl)-1,1′-bi-2-naphthol phosphoric acid to give an asymmetric β-carboline. This reaction was first studied by Holloway et al. ( Org. Lett. 2010, 12, 4720−4723 ), but their mechanistic work did not explain the high stereoselectivity achieved. This study uses density functional theory and hybrid quantum mechanics/molecular mechanics calculations to investigate this reaction and provide a model to explain its outcome. The step leading to diastereo- and enantioselectivity is an asymmetric Pictet–Spengler reaction involving an N-acyliminium ion bound to the catalyst in a bidentate fashion. This interaction occurs via hydrogen bonds between the two terminal oxygen atoms of the catalyst phosphate group and the hydrogen atoms at N and C2 of the substrate indole group. These bonds hold the transition structure rigidly and thus allow the catalyst triphenylsilyl groups to influence the enantioselectivity.
Co-reporter:Timothy E. H. Allen, Jonathan M. Goodman, Steve Gutsell, and Paul J. Russell
Chemical Research in Toxicology 2014 Volume 27(Issue 12) pp:2100
Publication Date(Web):October 29, 2014
DOI:10.1021/tx500345j
Consumer and environmental safety decisions are based on exposure and hazard data, interpreted using risk assessment approaches. The adverse outcome pathway (AOP) conceptual framework has been presented as a logical sequence of events or processes within biological systems which can be used to understand adverse effects and refine current risk assessment practices in ecotoxicology. This framework can also be applied to human toxicology and is explored on the basis of investigating the molecular initiating events (MIEs) of compounds. The precise definition of the MIE has yet to reach general acceptance. In this work we present a unified MIE definition: an MIE is the initial interaction between a molecule and a biomolecule or biosystem that can be causally linked to an outcome via a pathway. Case studies are presented, and issues with current definitions are addressed. With the development of a unified MIE definition, the field can look toward defining, classifying, and characterizing more MIEs and using knowledge of the chemistry of these processes to aid AOP research and toxicity risk assessment. We also present the role of MIE research in the development of in vitro and in silico toxicology and suggest how, by using a combination of biological and chemical approaches, MIEs can be identified and characterized despite a lack of detailed reports, even for some of the most studied molecules in toxicology.
Co-reporter:Matthew N. Grayson
Journal of the American Chemical Society 2013 Volume 135(Issue 16) pp:6142-6148
Publication Date(Web):March 22, 2013
DOI:10.1021/ja3122137
1,1'-Bi-2-naphthol (BINOL)-derived phosphoric acids catalyze the asymmetric propargylation of aldehydes. Density functional theory (DFT) calculations showed that the reaction proceeds via a six-membered transition structure (TS) in which the catalyst Brønsted acidic site interacts with the pseudoaxial cyclic boronate oxygen and the phosphoryl oxygen interacts with the formyl proton. This model accurately predicts the stereochemical outcome observed experimentally. Replacement of the phosphoric acid hydroxyl group with an N-triflyl moiety has been included in the model by calculation and a broader understanding achieved by qualitative assessment of similar reactions. We present a qualitative guide to rationalizing the experimental outcome and use this to make a prediction which was confirmed experimentally.
Co-reporter:Jack N. Gibb and Jonathan M. Goodman
Organic & Biomolecular Chemistry 2013 vol. 11(Issue 1) pp:90-97
Publication Date(Web):08 Nov 2012
DOI:10.1039/C2OB26547H
Polyurethane foams are widely used materials and control of their physical properties is a significant challenge. Management of cyclo-trimerisation during the polymerisation process is vital when tailoring the mechanical properties of the foam. Proazaphosphatranes are known to efficiently catalyse the cyclo-trimerisation of organic isocyanates, giving high purity isocyanurate with little uretdione by-product. The mechanism of this catalysis was previously unknown, although some zwitterionic intermediates have been identified spectroscopically. We have investigated a nucleophilic-catalysis reaction pathway involving sequential addition of methyl isocyanate to activated zwitterionic intermediates using density functional theory calculations. Evidence for significant transannulation by the proazaphosphatrane nitrogen was found for all intermediates, offering stabilisation of the phosphonium cation. Steric crowding at the proazaphosphatrane nucleophilic phosphorus gives rise to a preference for direct isocyanurate formation rather than via the uretdione, in sharp contrast to the uncatalysed system which has been found to preferentially proceed via the kinetic uretdione product. The investigations suggest the mechanism of proazaphosphatrane catalysed cyclo-oligomerisation does not proceed via the uretdione product, and hence why little of this impurity is observed experimentally.
Co-reporter:Matthew N. Grayson and Jonathan M. Goodman
The Journal of Organic Chemistry 2013 Volume 78(Issue 17) pp:8796-8801
Publication Date(Web):August 15, 2013
DOI:10.1021/jo401611q
1,1′-Bi-2-naphthol (BINOL)-derived catalysts catalyze the asymmetric propargylation of ketones. Density functional theory (DFT) calculations show that the reaction proceeds via a closed six-membered transition structure (TS) in which the chiral catalyst undergoes an exchange process with the original cyclic boronate ligand. This leads to a Lewis acid type activation mode, not a Brønsted acid process, which accurately predicts the stereochemical outcome observed experimentally.
Co-reporter:Matthew N. Grayson ; Silvina C. Pellegrinet
Journal of the American Chemical Society 2012 Volume 134(Issue 5) pp:2716-2722
Publication Date(Web):January 2, 2012
DOI:10.1021/ja210200d
BINOL-derived phosphoric acids catalyze the asymmetric allylboration of aldehydes. DFT and QM/MM hybrid calculations showed that the reaction proceeds via a transition state involving both a hydrogen-bonding interaction from the catalyst hydroxyl group to the pseudoaxial oxygen of the cyclic boronate and a stabilizing interaction from the phosphoryl oxygen of the catalyst to the formyl hydrogen of the aldehyde. These interactions lower the energy of the transition structure and provide extra rigidity to the system. This mechanistic pathway is consistent with the experimentally observed enantioselectivity except in one case. We have used our model’s predictions to guide our own experimental work. The conflict is resolved in favor of our calculations.
Co-reporter:Luis Simón
Journal of the American Chemical Society 2012 Volume 134(Issue 40) pp:16869-16876
Publication Date(Web):September 11, 2012
DOI:10.1021/ja307712y
The mechanism of the reaction between di-tert-butyl azadicarboxylate and 1,3-dicarbonyl compounds catalyzed by an axially chiral guanidine is investigated by density functional theory methods. The results show that the catalyst acts simultaneously as a Brønsted base and an acid catalyst, and the mechanism is similar to that of the related BINOP organocatalysts. Surprisingly, cyclic and acyclic β-keto esters yield opposite enantiomers; the calculations demonstrate that this is a consequence of the preferred enolate geometry in the transition structures. Literature evidence suggests that other organocatalytic reactions show similar behavior.
Co-reporter:Luis Simón and Jonathan M. Goodman
Organic & Biomolecular Chemistry 2012 vol. 10(Issue 9) pp:1905-1913
Publication Date(Web):23 Dec 2011
DOI:10.1039/C2OB06717J
We recently reported crystallographic evidence that the hydrogen bonds which can stabilize oxygen-centered negative charge within enzyme oxyanion holes are rarely found in the place they should be expected on the basis of the analysis of small-molecule crystal structures. We investigated this phenomenon using calculations on simplified active site models. A recent paper suggested that several aspects of the analysis required further exploration. In this paper we: (i) review the results of our crystallographic study; (ii) report molecular dynamics studies which investigate the effect of protein movement; (iii) report ONIOM calculations which trace the reaction coordinate for an oxyanion hole reaction in the presence of a complete enzyme active site. These results show that the limitations of gas phase calculations on simplified models do not invalidate our comparison of competing active site geometries. These new results reaffirm the conclusion that oxyanion holes are not usually stabilized by planar arrangements of H-bonds, and that this sub-optimal transition state stabilization leads to better overall catalysis.
Co-reporter:Russell H. Currie ;Dr. Jonathan M. Goodman
Angewandte Chemie International Edition 2012 Volume 51( Issue 19) pp:4695-4697
Publication Date(Web):
DOI:10.1002/anie.201109080
Co-reporter:Russell H. Currie ;Dr. Jonathan M. Goodman
Angewandte Chemie 2012 Volume 124( Issue 19) pp:4773-4775
Publication Date(Web):
DOI:10.1002/ange.201109080
Co-reporter:Luis Simón and Jonathan M. Goodman
Organic & Biomolecular Chemistry 2011 vol. 9(Issue 3) pp:689-700
Publication Date(Web):25 Oct 2010
DOI:10.1039/C0OB00477D
There have been many comparisons of computational methods applied to ground states, but studies of organic reactions usually require calculations on transition states, and these provide a different test of the methods. We present calculations of the geometries of nineteen covalent-bond forming transition states using HF and twelve different functionals, including GGA, hybrid-GGA and hybrid meta-GGA approaches. For the calculation of the TS geometries, the results suggest that B3LYP is only slightly less accurate than newer, computationally more expensive methods, and is less sensitive to choice of integration grid. We conclude that the use of B3LYP and related functionals is still appropriate for many studies of organic reaction mechanisms.
Co-reporter:Luis Simón and Jonathan M. Goodman
The Journal of Organic Chemistry 2011 Volume 76(Issue 6) pp:1775-1788
Publication Date(Web):February 10, 2011
DOI:10.1021/jo102410r
BINOL−phosphoric acid catalysts have been used successfully in many reactions involving imines. In this paper, we present a model, based on DFT calculations, for describing the degree and sense of the enantioselectivity of these reactions that is able to predict the correct enantioselectivity for the reactions in more than 40 recent publications. We rationalize the different factors on which the enantioselectivity depends, focusing on the E- or Z-preference of the transition structures and the orientation of the catalyst with respect to the electrophile.
Co-reporter:Luis Simón and Jonathan M. Goodman
The Journal of Organic Chemistry 2010 Volume 75(Issue 3) pp:589-597
Publication Date(Web):December 29, 2009
DOI:10.1021/jo902120s
DFT methods have been used to study the mechanism and the enantioselectivity of the Friedel−Crafts reaction of indoles with acyl and tosyl imides catalyzed by BINOL−phosphoric acid catalysts. The results are in excellent agreement with the experimental enantioselectivities. The energies of the competing transition structures and, thus, the enantioselectivity are rationalized from calculations on a model system. We propose a simple model to predict the absolute configuration of the products.
Co-reporter:Maxim V. Fedorov ; Jonathan M. Goodman ;Stephan Schumm
Journal of the American Chemical Society 2009 Volume 131(Issue 31) pp:10854-10856
Publication Date(Web):July 20, 2009
DOI:10.1021/ja9030374
Molecular dynamics simulations demonstrate that differences in the interaction of sodium and potassium with the carboxylate side chains of α-poly-l-glutamate (α-PGA) have a dramatic effect on the conformational properties of the polypeptide. Potassium ions cluster mainly in the second and third solvation shells of α-PGA because their low charge density makes the electrostatic interactions between them and α-PGA too weak for K+ to compete with water for the first solvation shell of the α-PGA glutamic acid residuals. Unlike sodium ions, they do not switch the conformation of α-PGA from extended to α-helical. Potentials of mean force for pure water, sodium ion solutions, and potassium ion solutions show marked differences in ion association behavior. This supports the idea that Hofmeister effects depend upon direct ion−macromolecule interactions as well as interactions with water molecules in the first solvation shell rather than bulk water structuring.
Co-reporter:Maxim V. Fedorov, Jonathan M. Goodman and Stephan Schumm
Chemical Communications 2009 (Issue 8) pp:896-898
Publication Date(Web):01 Dec 2008
DOI:10.1039/B816055D
A large-scale fully-atomistic molecular dynamics simulation of poly-L-glutamate demonstrates that a small amount of sodium chloride switches the preferred conformation from an extended conformation to a compact α-helix.
Co-reporter:Robert S. Paton and Jonathan M. Goodman
Journal of Chemical Information and Modeling 2009 Volume 49(Issue 4) pp:944-955
Publication Date(Web):March 23, 2009
DOI:10.1021/ci900009f
We have evaluated the performance of a set of widely used force fields by calculating the geometries and stabilization energies for a large collection of intermolecular complexes. These complexes are representative of a range of chemical and biological systems for which hydrogen bonding, electrostatic, and van der Waals interactions play important roles. Benchmark energies are taken from the high-level ab initio values in the JSCH-2005 and S22 data sets. All of the force fields underestimate stabilization resulting from hydrogen bonding, but the energetics of electrostatic and van der Waals interactions are described more accurately. OPLSAA gave a mean unsigned error of 2 kcal mol−1 for all 165 complexes studied, and outperforms DFT calculations employing very large basis sets for the S22 complexes. The magnitude of hydrogen bonding interactions are severely underestimated by all of the force fields tested, which contributes significantly to the overall mean error; if complexes which are predominantly bound by hydrogen bonding interactions are discounted, the mean unsigned error of OPLSAA is reduced to 1 kcal mol−1. For added clarity, web-based interactive displays of the results have been developed which allow comparisons of force field and ab initio geometries to be performed and the structures viewed and rotated in three dimensions.
Co-reporter:Jonathan Goodman
Journal of Chemical Information and Modeling 2009 Volume 49(Issue 12) pp:2897-2898
Publication Date(Web):December 4, 2009
DOI:10.1021/ci900437n
Co-reporter:Luis Simón and Jonathan M. Goodman
Organic & Biomolecular Chemistry 2009 vol. 7(Issue 3) pp:483-487
Publication Date(Web):2008/12/03
DOI:10.1039/B817283H
Aminoindanol-derived thioureas catalyze proton transfer and do not stabilise anions in an asymmetric conjugate addition to pyrazole crotonate. Calculations show that the urea H-bonds play different roles in the preferred transition state and in the one leading to the minor enantiomer in the mechanism of hydroxy-thiourea catalyzed conjugate additions to pyrazole crotonates.
Co-reporter:Maxim V. Fedorov, Jonathan M. Goodman, Vladimir V. Kolombet, Stephan Schumm, Ingrid M. Socorro
Journal of Molecular Liquids 2009 Volume 147(1–2) pp:117-123
Publication Date(Web):20 July 2009
DOI:10.1016/j.molliq.2008.10.004
In this study we are trying to understand the effects of aqueous sodium halide solutions on conformational properties of trialanine. We performed long-scale MD simulations of a trialanine peptide in different sodium halide solutions with varying concentrations of the salt (0.20, 0.50, 1.0 and 2.0 M). By clustering the conformational space of the tripeptide, we show that the molecular dynamics trajectories of this molecule form only a few major conformation clusters for each of the sixteen salt solutions under study. This gives us a way to compress the information contained in a terabyte of the simulation data to a small number of representative molecular geometries. These representative structures give insights into molecular mechanism of salt effects on peptide conformations. The results show that salts are able to make significant changes to the conformational landscape of trialanine, and that sodium chloride is particularly effective.
Co-reporter:S. E. Adams, J. M. Goodman, R. J. Kidd, A. D. McNaught, P. Murray-Rust, F. R. Norton, J. A. Townsend and C. A. Waudby
Organic & Biomolecular Chemistry 2004 vol. 2(Issue 21) pp:3067-3070
Publication Date(Web):30 Sep 2004
DOI:10.1039/B411699M
An experimental data checker has been developed that reads, analyses, and cross-correlates experimental information copied and pasted from authors' manuscripts, which will be useful for authors, referees, editors and readers of papers reporting new molecular information, and which makes possible a quantification of the accuracy of journals' data.
Co-reporter:Maxim V. Fedorov, Jonathan M. Goodman and Stephan Schumm
Chemical Communications 2009(Issue 8) pp:NaN898-898
Publication Date(Web):2008/12/01
DOI:10.1039/B816055D
A large-scale fully-atomistic molecular dynamics simulation of poly-L-glutamate demonstrates that a small amount of sodium chloride switches the preferred conformation from an extended conformation to a compact α-helix.
Co-reporter:Luis Simón and Jonathan M. Goodman
Organic & Biomolecular Chemistry 2012 - vol. 10(Issue 9) pp:NaN1913-1913
Publication Date(Web):2011/12/23
DOI:10.1039/C2OB06717J
We recently reported crystallographic evidence that the hydrogen bonds which can stabilize oxygen-centered negative charge within enzyme oxyanion holes are rarely found in the place they should be expected on the basis of the analysis of small-molecule crystal structures. We investigated this phenomenon using calculations on simplified active site models. A recent paper suggested that several aspects of the analysis required further exploration. In this paper we: (i) review the results of our crystallographic study; (ii) report molecular dynamics studies which investigate the effect of protein movement; (iii) report ONIOM calculations which trace the reaction coordinate for an oxyanion hole reaction in the presence of a complete enzyme active site. These results show that the limitations of gas phase calculations on simplified models do not invalidate our comparison of competing active site geometries. These new results reaffirm the conclusion that oxyanion holes are not usually stabilized by planar arrangements of H-bonds, and that this sub-optimal transition state stabilization leads to better overall catalysis.
Co-reporter:Luis Simón and Jonathan M. Goodman
Organic & Biomolecular Chemistry 2011 - vol. 9(Issue 3) pp:NaN700-700
Publication Date(Web):2010/10/25
DOI:10.1039/C0OB00477D
There have been many comparisons of computational methods applied to ground states, but studies of organic reactions usually require calculations on transition states, and these provide a different test of the methods. We present calculations of the geometries of nineteen covalent-bond forming transition states using HF and twelve different functionals, including GGA, hybrid-GGA and hybrid meta-GGA approaches. For the calculation of the TS geometries, the results suggest that B3LYP is only slightly less accurate than newer, computationally more expensive methods, and is less sensitive to choice of integration grid. We conclude that the use of B3LYP and related functionals is still appropriate for many studies of organic reaction mechanisms.
Co-reporter:Jack N. Gibb and Jonathan M. Goodman
Organic & Biomolecular Chemistry 2013 - vol. 11(Issue 1) pp:NaN97-97
Publication Date(Web):2012/11/08
DOI:10.1039/C2OB26547H
Polyurethane foams are widely used materials and control of their physical properties is a significant challenge. Management of cyclo-trimerisation during the polymerisation process is vital when tailoring the mechanical properties of the foam. Proazaphosphatranes are known to efficiently catalyse the cyclo-trimerisation of organic isocyanates, giving high purity isocyanurate with little uretdione by-product. The mechanism of this catalysis was previously unknown, although some zwitterionic intermediates have been identified spectroscopically. We have investigated a nucleophilic-catalysis reaction pathway involving sequential addition of methyl isocyanate to activated zwitterionic intermediates using density functional theory calculations. Evidence for significant transannulation by the proazaphosphatrane nitrogen was found for all intermediates, offering stabilisation of the phosphonium cation. Steric crowding at the proazaphosphatrane nucleophilic phosphorus gives rise to a preference for direct isocyanurate formation rather than via the uretdione, in sharp contrast to the uncatalysed system which has been found to preferentially proceed via the kinetic uretdione product. The investigations suggest the mechanism of proazaphosphatrane catalysed cyclo-oligomerisation does not proceed via the uretdione product, and hence why little of this impurity is observed experimentally.
Co-reporter:Kristaps Ermanis, Kevin E. B. Parkes, Tatiana Agback and Jonathan M. Goodman
Organic & Biomolecular Chemistry 2016 - vol. 14(Issue 16) pp:NaN3949-3949
Publication Date(Web):2016/03/23
DOI:10.1039/C6OB00015K
The DP4 parameter, which provides a confidence level for NMR assignment, has been widely used to help assign the structures of many stereochemically-rich molecules. We present an improved version of the procedure, which can be downloaded as Python script instead of running within a web-browser, and which analyses output from open-source molecular modelling programs (TINKER and NWChem) in addition to being able to use output from commercial packages (Schrodinger's Macromodel and Jaguar; Gaussian). The new open-source workflow incorporates a method for the automatic generation of diastereomers using InChI strings and has been tested on a range of new structures. This improved workflow permits the rapid and convenient computational elucidation of structure and relative stereochemistry.
Co-reporter:Luis Simón and Jonathan M. Goodman
Organic & Biomolecular Chemistry 2009 - vol. 7(Issue 3) pp:NaN487-487
Publication Date(Web):2008/12/03
DOI:10.1039/B817283H
Aminoindanol-derived thioureas catalyze proton transfer and do not stabilise anions in an asymmetric conjugate addition to pyrazole crotonate. Calculations show that the urea H-bonds play different roles in the preferred transition state and in the one leading to the minor enantiomer in the mechanism of hydroxy-thiourea catalyzed conjugate additions to pyrazole crotonates.

![2,4-Cyclohexadien-1-one, 3-methoxy-6-[(4-methoxyphenyl)methylene]-](/data/chemimg/3750300/1646388-71-7.png)
![2,4-Cyclohexadien-1-one, 3-methoxy-6-[(4-methoxyphenyl)methylene]-](/data/chemimg/3750300/1646388-71-7_b.png)
![Phenol, 5-methoxy-2-[(2E)-1-(4-methoxyphenyl)-3-phenyl-2-propen-1-yl]-](/data/chemimg/3750300/1415560-21-2.png)
![Phenol, 5-methoxy-2-[(2E)-1-(4-methoxyphenyl)-3-phenyl-2-propen-1-yl]-](/data/chemimg/3750300/1415560-21-2_b.png)
![Phenol, 5-methoxy-2-[(1S,2E)-1-(4-methoxyphenyl)-3-phenyl-2-propen-1-yl]-](/data/chemimg/3750300/1415560-19-8.png)
![Phenol, 5-methoxy-2-[(1S,2E)-1-(4-methoxyphenyl)-3-phenyl-2-propen-1-yl]-](/data/chemimg/3750300/1415560-19-8_b.png)
![1,3-Benzodioxol-5-ol, 6-[(2E)-1-(4-methoxyphenyl)-3-phenyl-2-propen-1-yl]-](/data/chemimg/3750300/1415559-88-4.png)
![1,3-Benzodioxol-5-ol, 6-[(2E)-1-(4-methoxyphenyl)-3-phenyl-2-propen-1-yl]-](/data/chemimg/3750300/1415559-88-4_b.png)
![1,3-Benzodioxol-5-ol, 6-[(1R,2E)-1-(4-methoxyphenyl)-3-phenyl-2-propen-1-yl]-](/data/chemimg/3750300/1415559-84-0.png)
![1,3-Benzodioxol-5-ol, 6-[(1R,2E)-1-(4-methoxyphenyl)-3-phenyl-2-propen-1-yl]-](/data/chemimg/3750300/1415559-84-0_b.png)

![Boronic acid, B-[(1E)-2-phenylethenyl]-, diethyl ester](/data/chemimg/3750200/1029603-94-8.png)
![Boronic acid, B-[(1E)-2-phenylethenyl]-, diethyl ester](/data/chemimg/3750200/1029603-94-8_b.png)


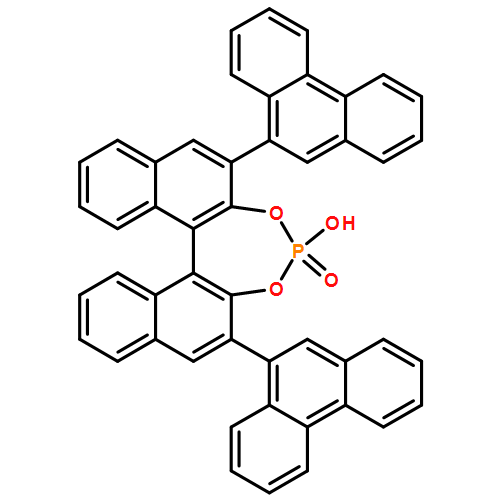


![Dinaphtho[2,1-d:1',2'-f][1,3,2]dioxaphosphepin,4-hydroxy-2,6-bis[2,4,6-tris(1-methylethyl)phenyl]-, 4-oxide, (11bR)-](/data/chemimg/115800/791616-63-2.png)
![Dinaphtho[2,1-d:1',2'-f][1,3,2]dioxaphosphepin,4-hydroxy-2,6-bis[2,4,6-tris(1-methylethyl)phenyl]-, 4-oxide, (11bR)-](/data/chemimg/115800/791616-63-2_b.png)
![Dinaphtho[2,1-d:1',2'-f][1,3,2]dioxaphosphepin,2,6-bis[3,5-bis(trifluoromethyl)phenyl]-4-hydroxy-, 4-oxide, (11bR)-](/data/chemimg/114800/791616-62-1.png)
![Dinaphtho[2,1-d:1',2'-f][1,3,2]dioxaphosphepin,2,6-bis[3,5-bis(trifluoromethyl)phenyl]-4-hydroxy-, 4-oxide, (11bR)-](/data/chemimg/114800/791616-62-1_b.png)










![3-Oxa-9-azoniatricyclo[3.3.1.02,4]nonane,7-[(2-hydroxy-2,2-di-2-thienylacetyl)oxy]-9,9-dimethyl-, (1a,2b,4b,5a,7b)-](http://img.cochemist.com/ccimg/186700/186691-13-4.png)
![3-Oxa-9-azoniatricyclo[3.3.1.02,4]nonane,7-[(2-hydroxy-2,2-di-2-thienylacetyl)oxy]-9,9-dimethyl-, (1a,2b,4b,5a,7b)-](http://img.cochemist.com/ccimg/186700/186691-13-4_b.png)



![[1,1'-Binaphthalene]-2,2'-diol,3,3'-dibromo-, (1R)-](http://img.cochemist.com/ccimg/111800/111795-43-8.png)
![[1,1'-Binaphthalene]-2,2'-diol,3,3'-dibromo-, (1R)-](http://img.cochemist.com/ccimg/111800/111795-43-8_b.png)




![[Binaphthalene]diol](http://img.cochemist.com/ccimg/51800/51746-06-6.png)
![[Binaphthalene]diol](http://img.cochemist.com/ccimg/51800/51746-06-6_b.png)




![Dinaphtho[2,1-d:1',2'-f][1,3,2]dioxaphosphepin,4-hydroxy-2,6-bis(triphenylsilyl)-, 4-oxide, (11bR)-](/data/chemimg/116500/791616-55-2.png)
![Dinaphtho[2,1-d:1',2'-f][1,3,2]dioxaphosphepin,4-hydroxy-2,6-bis(triphenylsilyl)-, 4-oxide, (11bR)-](/data/chemimg/116500/791616-55-2_b.png)