Co-reporter:Akihito Konishi, Yui Okada, Motohiro Nakano, Kenji Sugisaki, Kazunobu Sato, Takeji Takui, and Makoto Yasuda
Journal of the American Chemical Society November 1, 2017 Volume 139(Issue 43) pp:15284-15284
Publication Date(Web):September 30, 2017
DOI:10.1021/jacs.7b05709
Mesityl derivatives of the unknown dibenzopentalene isomer dibenzo[a,f]pentalene were synthesized. The molecular geometry and physical properties of dibenzo[a,f]pentalene were investigated. Dibenzo[a,f]pentalene combines a large antiaromatic and appreciable singlet open-shell character, properties not shared by well-known isomer dibenzo[a,e]pentalene.
Co-reporter:Yoshihiro Nishimoto, Kyoungmin Kang, and Makoto Yasuda
Organic Letters July 21, 2017 Volume 19(Issue 14) pp:3927-3927
Publication Date(Web):July 12, 2017
DOI:10.1021/acs.orglett.7b01847
(Z)-β-Aryloxyalkenylzincs are synthesized stereoselectively via anti-carbozincation among alkynyl ethers, silyl ketene acetals, and ZnBr2. X-ray analysis revealed the structure of the zinc species is a mononuclear two-coordinate dialkenylzinc that is transformed into functionalized enol ethers as a single isomer in the reaction of various electrophiles.
Co-reporter:Naoto Esumi;Yoshihiro Nishimoto
European Journal of Organic Chemistry 2017 Volume 2017(Issue 19) pp:2831-2835
Publication Date(Web):2017/05/18
DOI:10.1002/ejoc.201700501
A new highly anti-diastereoselective Michael addition of α-alkoxy ketones to α,β-unsaturated ketones was achieved. This method features a dual-catalyst protocol that combines samarium(III) trifluoromethanesulfonate and Bu3SnOMe. The combination of these two catalysts effectively allowed the generation of enolate species from α-alkoxy ketones and produced Michael adducts in high yields with high anti diastereoselectivity. A variety of enones and α-alkoxy ketones were applied to this system to give the anti products. One-pot domino Michael/aldol reactions effectively afforded cyclic enones with a defined configuration.
Co-reporter:Naoto Esumi, Kensuke Suzuki, Yoshihiro Nishimoto, and Makoto Yasuda
Organic Letters 2016 Volume 18(Issue 21) pp:5704-5707
Publication Date(Web):October 25, 2016
DOI:10.1021/acs.orglett.6b02869
We report the visible-light-induced radical coupling reaction of silyl enol ethers with α-bromocarbonyl compounds to give 1,4-dicarbonyls. The reaction was effectively accelerated using an inexpensive organic dye (eosin Y) as a photoredox catalyst. 1,4-Dicarbonyl compounds alone were afforded, without the generation of carbonyl adducts of the α-halocarbonyls, which are usually generated in the presence of fluoride anions or Lewis acids. A variety of silyl enol ethers, α-bromoketones, α-bromoesters, and α-bromoamides were applied to this system to produce the coupling compounds.
Co-reporter:Akihito Konishi, Ryosuke Yasunaga, Kouji Chiba and Makoto Yasuda
Chemical Communications 2016 vol. 52(Issue 16) pp:3348-3351
Publication Date(Web):21 Jan 2016
DOI:10.1039/C6CC00291A
A cage-shaped borate with benzofuran moieties was synthesized. This borate showed a higher degree of catalytic activity for Mukaiyama-aldol type reactions than a simple benzene-based cage-shaped borate induced by self-aggregation. Moreover, the exposure of the complex to black-light irradiation enhanced the catalytic activity.
Co-reporter:Itaru Suzuki;Naoto Esumi ;Dr. Makoto Yasuda
Asian Journal of Organic Chemistry 2016 Volume 5( Issue 2) pp:179-182
Publication Date(Web):
DOI:10.1002/ajoc.201500475
Abstract
Organic dyes are an attractive alternative to the use of transition-metal complexes in photoredox catalysis. Herein, we report an inexpensive organic dye (eosin Y)-catalyzed radical coupling of α-bromocarbonyl compounds with allyltrifluoroborates under visible light irradiation. This reaction was accelerated by either Bu4NF or CsF. Based on mechanistic studies, the cation exchange between potassium allyltrifluoroborate and Bu4NF proceeded to generate KF. The generated KF or CsF acts as a Lewis acid to promote a single electron transfer (SET) from eosin Y to α-halocarbonyls. The protocol was applied to bromoesters, bromoketones, and bromoamides to give the coupling products in high yields.
Co-reporter:Dr. Akihito Konishi;Yohei Minami;Takahisa Hosoi;Kouji Chiba;Dr. Makoto Yasuda
Chemistry - A European Journal 2016 Volume 22( Issue 36) pp:12688-12691
Publication Date(Web):
DOI:10.1002/chem.201603147
Abstract
The Group 14 enolates play an important part in many organic reactions. Herein, the reduction of an α-bromo ketone with germanium(II) salts cleanly afforded the corresponding germyl enolate as an isolatable species. This experimental reductive generation of a germyl enolate enabled us to characterize both C- and O-bound tautomers derived from an identical precursor and to unveil the tautomeric mechanisms, including the kinetic parameters and the relative stability of these tautomers, along with confirmation from DFT calculations. Moreover, the highly coordinated germyl enolates were isolated by a stabilization process induced by adding ligands. All products were characterized by NMR spectroscopy and X-ray crystallography.
Co-reporter:Makoto Yasuda, Yoshitaka Nagano, Hiroshi Yunoki, Kensuke Tsuruwa, and Akio Baba
Organometallics 2014 Volume 33(Issue 15) pp:3924-3927
Publication Date(Web):August 1, 2014
DOI:10.1021/om500768e
The reaction of homochiral aminoallylic stannanes with aldehydes gave carbonyl adducts of amino alcohols in the presence of either SnCl2 or InBr3. Both additives afforded the products in opposite absolute stereochemical configurations. The controlled chirality was ascribed to the different transmetalation pathways of SnCl2 and InBr3.
Co-reporter:Dr. Yoshihiro Inamoto;Yuta Kaga;Dr. Yoshihiro Nishimoto;Dr. Makoto Yasuda;Dr. Akio Baba
Chemistry - A European Journal 2014 Volume 20( Issue 37) pp:11664-11668
Publication Date(Web):
DOI:10.1002/chem.201403734
Abstract
A sequential addition of silyl cyanide and ketene silyl acetals to esters was achieved by a gallium trihalide catalyst to produce β-cyano-β-siloxy esters. This is the first example of the sequential addition of two different carbon nucleophiles to esters. The employment of lactones provided α,α-disubstituted cyclic ethers with a cyano group and an ester moiety. A variety of esters and lactones are applicable to this reaction system.
Co-reporter:Itaru Suzuki, Makoto Yasuda and Akio Baba
Chemical Communications 2013 vol. 49(Issue 99) pp:11620-11622
Publication Date(Web):27 Sep 2013
DOI:10.1039/C3CC46570E
The coupling of acetals with various alkynes was achieved using only 1 mol% of inexpensive and mild Lewis acid ZnCl2, which furnished propargyl ethers. The coupling was catalyzed by Zn(OMe)Cl, which was generated in situ to form an alkynylzinc species. This protocol was allowed to expand to a one-pot subsequent reaction with allylchlorosilane to obtain a 1,4-enyne product.
Co-reporter:Hideto Nakajima, Makoto Yasuda and Akio Baba
Dalton Transactions 2012 vol. 41(Issue 22) pp:6602-6606
Publication Date(Web):03 Apr 2012
DOI:10.1039/C2DT30266G
Stable hexanuclear lithium phenolate bearing a cage-shaped tripodal ligand was isolated, which had a hexagonal-prismatic Li6O6 core at room temperature, because of the hard mobility of the ligand and its reduction of the problematic steric repulsion. The properties of the lithium phenolates were analyzed by X-ray crystallography and NMR spectroscopy.
Co-reporter:Hideto Nakajima;Dr. Makoto Yasuda;Ryosuke Takeda ;Dr. Akio Baba
Angewandte Chemie International Edition 2012 Volume 51( Issue 16) pp:3867-3870
Publication Date(Web):
DOI:10.1002/anie.201200346
Co-reporter:Hideto Nakajima;Dr. Makoto Yasuda;Ryosuke Takeda ;Dr. Akio Baba
Angewandte Chemie 2012 Volume 124( Issue 16) pp:3933-3936
Publication Date(Web):
DOI:10.1002/ange.201200346
Co-reporter:Dr. Makoto Yasuda;Hideto Nakajima;Ryosuke Takeda;Sachiko Yoshioka;Satoshi Yamasaki;Kouji Chiba;Dr. Akio Baba
Chemistry - A European Journal 2011 Volume 17( Issue 14) pp:3856-3867
Publication Date(Web):
DOI:10.1002/chem.201002789
Abstract
Boron complexes that contain new tridentate ligands, tris(o-oxyaryl)methanes and -silanes, were prepared. These complexes had a cage-shaped structure around a boron center and showed higher Lewis acidity and catalytic activity than open-shaped boron compounds. The cage-shaped ligands determined the properties of the borates by altering the geometry and were consistently bound to the metal center by chelation. The synthesized compounds were L⋅B(OC6H4)3CH, L⋅B(OC6H4)3SiMe, and its derivatives (L=THF or pyridine as an external ligand). Theoretical calculations suggested that the cage-shaped borates had a large dihedral angle (Cipso-O-B-O) compared with open-shaped borates. The geometric effect due to the dihedral angle means that compared with open-shaped, the cage-shaped borates have a greater Lewis acidity. The introduction of electron-withdrawing groups on the aryl moieties in the cage-shaped framework increased the Lewis acidity. Substitution of a bridgehead Si for a bridgehead C decreased the Lewis acidity of the boron complexes because the large silicon atom reduces the dihedral angle of Cipso-O-B-O. The ligand-exchange rates of the para-fluoro-substituted compound B(OC6H3F)3CH and the ortho-phenyl-substituted compound B(OC6H3Ph)3CH were less than that of the unsubstituted borate B(OC6H4)3CH. The ligand-exchange rate of B(OC6H4)3SiMe was much faster than that of B(OC6H4)3CH. A hetero Diels–Alder reaction and Mukaiyama-type aldol reactions were more effectively catalyzed by cage-shaped borates than by the open-shaped borate B(OPh)3 or by the strong Lewis acid BF3⋅OEt2. The cage-shaped borates with the bulky substituents at the ortho-positions selectively catalyzed the reaction with less sterically hindered substrates, while the unsubstituted borate showed no selectivity.
Co-reporter:Hideto Nakajima, Makoto Yasuda, Kouji Chiba and Akio Baba
Chemical Communications 2010 vol. 46(Issue 26) pp:4794-4796
Publication Date(Web):25 May 2010
DOI:10.1039/C0CC00253D
Cage-shaped gallium complexes with a back-shielding framework of tris(m-oxybenzyl)arene were synthesized, their bottom arene rings tuned their characteristic Lewis acidity, which was supported by theoretical calculation as well as catalytic application in a hetero Diels–Alder reaction.
Co-reporter:Makoto Yasuda;Masahiko Haga;Yasunori Nagaoka;Akio Baba
European Journal of Organic Chemistry 2010 Volume 2010( Issue 28) pp:5359-5363
Publication Date(Web):
DOI:10.1002/ejoc.201000956
Abstract
The reductive system of allyl bromide and indium(0) in water generated monoallylindium(III) dibromide and diallylindium(III) bromide. These compounds were characterized by X-ray analysis after complexation with 3,5-dibromopyridine or 4-(dimethylamino)pyridine, respectively. Both isolated complexes showed high nucleophilicity and reacted with benzaldehyde to give the allylation product. The diallylindium(III) bromide was less stable in water than the monoallylindium(III) dibromide. The reaction of the diallylindium(III) with benzhydrol gave a (μ-alkoxido)indium species that showed nucleophilicity. This result suggests that allyl(μ-hydroxido)indium also acted as a nucleophile in an aqueous Barbie-type reaction system.
Co-reporter:Makoto Yasuda;Masahiko Haga;Yasunori Nagaoka;Akio Baba
European Journal of Organic Chemistry 2010 Volume 2010( Issue 28) pp:
Publication Date(Web):
DOI:10.1002/ejoc.201090076
Abstract
The cover picture shows the species generated in the reductive system of allyl bromide with indium(0) in water. They include monoallylindium(III) dibromide, diallylindium(III) bromide, and allyl(μ-oxido)indium(III). These compounds were characterized on the basis of X-ray analysis. The allylindium compounds showed nucleophilicity towards carbonyl compounds with different reactivities. Details are discussed in the Short Communication by M. Yasuda, A. Baba et al. on p. 5359 ff.
Co-reporter:Makoto Yasuda, Kensuke Tsuruwa, Tatsuya Azuma, Srinivasarao Arulananda Babu, Akio Baba
Tetrahedron 2009 65(46) pp: 9569-9574
Publication Date(Web):
DOI:10.1016/j.tet.2009.09.063
Co-reporter:Makoto Yasuda, Tatsuya Azuma, Kensuke Tsuruwa, Srinivasarao Arulananda Babu, Akio Baba
Tetrahedron Letters 2009 50(26) pp: 3209-3212
Publication Date(Web):
DOI:10.1016/j.tetlet.2009.01.137
Co-reporter:Makoto Yasuda, Kenji Shimizu, Satoshi Yamasaki and Akio Baba
Organic & Biomolecular Chemistry 2008 vol. 6(Issue 15) pp:2790-2795
Publication Date(Web):28 May 2008
DOI:10.1039/B804589E
The reaction of secondary alcohols 1 with chlorodimethylsilane (HSiMe2Cl) proceeded in the presence of a catalytic amount of GaCl3/diethyl tartrate to give the corresponding organic chlorides 3. In the catalytic cycle, the reaction of diethyl tartrate 4a with HSiMe2Cl 2 gives the chlorosilyl ether 5 with generation of H2. Alcohol-exchange between the formed chlorosilyl ether 5 and the substrate alcohol 1 affords alkoxychlorosilane 6, which reacts with catalytic GaCl3 to give the chlorinated product 3. The moderate Lewis acidity of GaCl3 facilitates chlorination. Strong Lewis acids did not give product due to excessive affinity for the oxy-functionalities. Although tertiary alcohols were chlorinated by this system even in the absence of diethyl tartrate, certain alcohols that are less likely to give carbocationic species were effectively chlorinated using the GaCl3/diethyl tartrate system.
Co-reporter:Akihito Konishi, Hideto Nakajima, Hikaru Maruyama, Sachiko Yoshioka, Akio Baba, Makoto Yasuda
Polyhedron (29 March 2017) Volume 125() pp:
Publication Date(Web):29 March 2017
DOI:10.1016/j.poly.2016.10.019
A new series of tetrameric aluminum molecules with a Mitsubishi structure, composed of two cage-shaped triphenolic ligands with a tripod-chelated structure, have been synthesized and characterized. Transmetalation from the cage-shaped borates was a proper method for the synthesis of these aluminum complexes. All complexes were determined via X-ray crystallography and NMR measurements. These Lewis acidic aluminum complexes catalyze the hetero Diels Alder reaction of the Danishefsky diene with benzaldehyde.Tetrameric aluminum molecules with a Mitsubishi structure, composed of two cage-shaped triphenolic ligands, have been synthesized and characterized. Transmetalation from the cage-shaped borates was a proper method for the synthesis of these aluminum complexes. These aluminum complexes behaved as the Lewis acid catalyst for a hetero Diels–Alder reaction.
Co-reporter:Makoto Yasuda, Kenji Shimizu, Satoshi Yamasaki and Akio Baba
Organic & Biomolecular Chemistry 2008 - vol. 6(Issue 15) pp:NaN2795-2795
Publication Date(Web):2008/05/28
DOI:10.1039/B804589E
The reaction of secondary alcohols 1 with chlorodimethylsilane (HSiMe2Cl) proceeded in the presence of a catalytic amount of GaCl3/diethyl tartrate to give the corresponding organic chlorides 3. In the catalytic cycle, the reaction of diethyl tartrate 4a with HSiMe2Cl 2 gives the chlorosilyl ether 5 with generation of H2. Alcohol-exchange between the formed chlorosilyl ether 5 and the substrate alcohol 1 affords alkoxychlorosilane 6, which reacts with catalytic GaCl3 to give the chlorinated product 3. The moderate Lewis acidity of GaCl3 facilitates chlorination. Strong Lewis acids did not give product due to excessive affinity for the oxy-functionalities. Although tertiary alcohols were chlorinated by this system even in the absence of diethyl tartrate, certain alcohols that are less likely to give carbocationic species were effectively chlorinated using the GaCl3/diethyl tartrate system.
Co-reporter:Akihito Konishi, Ryosuke Yasunaga, Kouji Chiba and Makoto Yasuda
Chemical Communications 2016 - vol. 52(Issue 16) pp:NaN3351-3351
Publication Date(Web):2016/01/21
DOI:10.1039/C6CC00291A
A cage-shaped borate with benzofuran moieties was synthesized. This borate showed a higher degree of catalytic activity for Mukaiyama-aldol type reactions than a simple benzene-based cage-shaped borate induced by self-aggregation. Moreover, the exposure of the complex to black-light irradiation enhanced the catalytic activity.
Co-reporter:Hideto Nakajima, Makoto Yasuda, Kouji Chiba and Akio Baba
Chemical Communications 2010 - vol. 46(Issue 26) pp:NaN4796-4796
Publication Date(Web):2010/05/25
DOI:10.1039/C0CC00253D
Cage-shaped gallium complexes with a back-shielding framework of tris(m-oxybenzyl)arene were synthesized, their bottom arene rings tuned their characteristic Lewis acidity, which was supported by theoretical calculation as well as catalytic application in a hetero Diels–Alder reaction.
Co-reporter:Itaru Suzuki, Makoto Yasuda and Akio Baba
Chemical Communications 2013 - vol. 49(Issue 99) pp:NaN11622-11622
Publication Date(Web):2013/09/27
DOI:10.1039/C3CC46570E
The coupling of acetals with various alkynes was achieved using only 1 mol% of inexpensive and mild Lewis acid ZnCl2, which furnished propargyl ethers. The coupling was catalyzed by Zn(OMe)Cl, which was generated in situ to form an alkynylzinc species. This protocol was allowed to expand to a one-pot subsequent reaction with allylchlorosilane to obtain a 1,4-enyne product.
Co-reporter:Hideto Nakajima, Makoto Yasuda and Akio Baba
Dalton Transactions 2012 - vol. 41(Issue 22) pp:NaN6606-6606
Publication Date(Web):2012/04/03
DOI:10.1039/C2DT30266G
Stable hexanuclear lithium phenolate bearing a cage-shaped tripodal ligand was isolated, which had a hexagonal-prismatic Li6O6 core at room temperature, because of the hard mobility of the ligand and its reduction of the problematic steric repulsion. The properties of the lithium phenolates were analyzed by X-ray crystallography and NMR spectroscopy.






![Benzenepropanoic acid, α,α,4-trimethyl-β-[(trimethylsilyl)oxy]-, methyl ester](/data/chemimg/3745000/851392-90-0.png)
![Benzenepropanoic acid, α,α,4-trimethyl-β-[(trimethylsilyl)oxy]-, methyl ester](/data/chemimg/3745000/851392-90-0_b.png)
![Benzenepropanoic acid, 4-chloro-α,α-dimethyl-β-[(trimethylsilyl)oxy]-, methyl ester](/data/chemimg/3745000/851392-75-1.png)
![Benzenepropanoic acid, 4-chloro-α,α-dimethyl-β-[(trimethylsilyl)oxy]-, methyl ester](/data/chemimg/3745000/851392-75-1_b.png)


![STANNANE, [(4-BROMOPHENYL)ETHYNYL]TRIBUTYL-](http://img.cochemist.com/ccimg/750600/750594-92-4.png)
![STANNANE, [(4-BROMOPHENYL)ETHYNYL]TRIBUTYL-](http://img.cochemist.com/ccimg/750600/750594-92-4_b.png)
















![Benzenepropanoic acid, 4-methoxy-α,α-dimethyl-β-[(trimethylsilyl)oxy]-, methyl ester](/data/chemimg/3745000/151721-69-6.png)
![Benzenepropanoic acid, 4-methoxy-α,α-dimethyl-β-[(trimethylsilyl)oxy]-, methyl ester](/data/chemimg/3745000/151721-69-6_b.png)
![Silane, [(1-methoxy-1-hexenyl)oxy]trimethyl-, (Z)-](/data/chemimg/1087000/133464-85-4.png)
![Silane, [(1-methoxy-1-hexenyl)oxy]trimethyl-, (Z)-](/data/chemimg/1087000/133464-85-4_b.png)
![Silane, [[(1E)-1-methoxy-1-hexenyl]oxy]trimethyl-](http://img.cochemist.com/ccimg/133500/133464-84-3.png)
![Silane, [[(1E)-1-methoxy-1-hexenyl]oxy]trimethyl-](http://img.cochemist.com/ccimg/133500/133464-84-3_b.png)


![Silane, trimethyl[(2-methyl-1-phenoxy-1-propenyl)oxy]-](http://img.cochemist.com/ccimg/120700/120615-54-5.png)
![Silane, trimethyl[(2-methyl-1-phenoxy-1-propenyl)oxy]-](http://img.cochemist.com/ccimg/120700/120615-54-5_b.png)












![Silane, [[1-(hexyloxy)ethenyl]oxy]trimethyl-](http://img.cochemist.com/ccimg/93000/92984-28-6.png)
![Silane, [[1-(hexyloxy)ethenyl]oxy]trimethyl-](http://img.cochemist.com/ccimg/93000/92984-28-6_b.png)
![Benzenepropanoic acid, α,α-dimethyl-β-[(trimethylsilyl)oxy]-, methyl ester](/data/chemimg/3745000/92233-94-8.png)
![Benzenepropanoic acid, α,α-dimethyl-β-[(trimethylsilyl)oxy]-, methyl ester](/data/chemimg/3745000/92233-94-8_b.png)


![Silane, [[(1E)-1-ethoxy-3,3-dimethyl-1-butenyl]oxy]trimethyl-](http://img.cochemist.com/ccimg/88300/88255-52-1.png)
![Silane, [[(1E)-1-ethoxy-3,3-dimethyl-1-butenyl]oxy]trimethyl-](http://img.cochemist.com/ccimg/88300/88255-52-1_b.png)
![SILANE, [(1-METHOXY-2-METHYL-1-BUTENYL)OXY]TRIMETHYL-](http://img.cochemist.com/ccimg/84400/84393-12-4.png)
![SILANE, [(1-METHOXY-2-METHYL-1-BUTENYL)OXY]TRIMETHYL-](http://img.cochemist.com/ccimg/84400/84393-12-4_b.png)
![Silane, [(1-methoxy-1-hexenyl)oxy]trimethyl-](http://img.cochemist.com/ccimg/84400/84393-11-3.png)
![Silane, [(1-methoxy-1-hexenyl)oxy]trimethyl-](http://img.cochemist.com/ccimg/84400/84393-11-3_b.png)
![SILANE, [[1-ETHOXY-2-(PHENYLTHIO)ETHENYL]OXY]TRIMETHYL-](http://img.cochemist.com/ccimg/83500/83467-57-6.png)
![SILANE, [[1-ETHOXY-2-(PHENYLTHIO)ETHENYL]OXY]TRIMETHYL-](http://img.cochemist.com/ccimg/83500/83467-57-6_b.png)








![Silane, [(2-ethyl-1-methoxy-1-butenyl)oxy]trimethyl-](http://img.cochemist.com/ccimg/73400/73303-67-0.png)
![Silane, [(2-ethyl-1-methoxy-1-butenyl)oxy]trimethyl-](http://img.cochemist.com/ccimg/73400/73303-67-0_b.png)






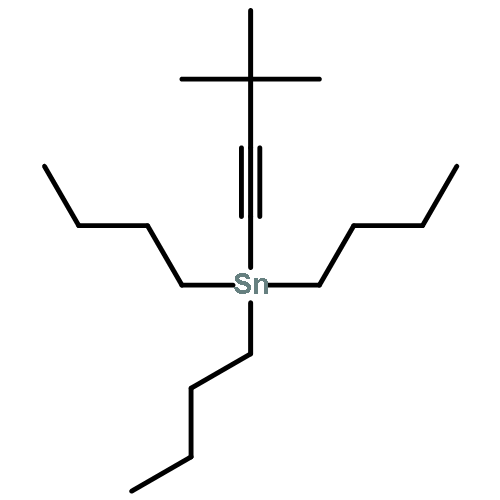











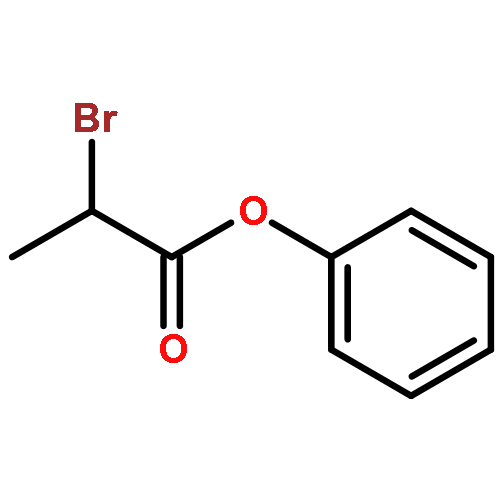



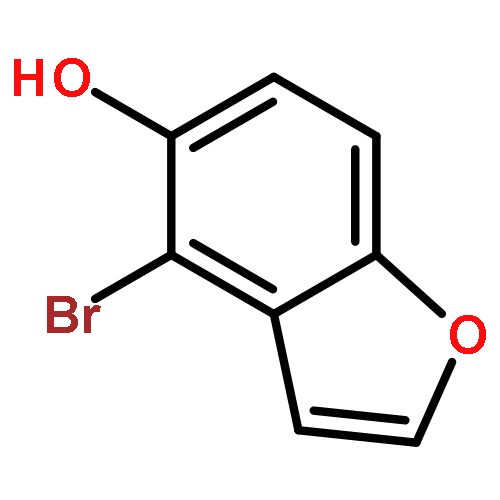


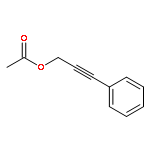





















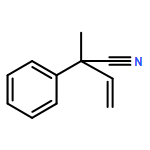
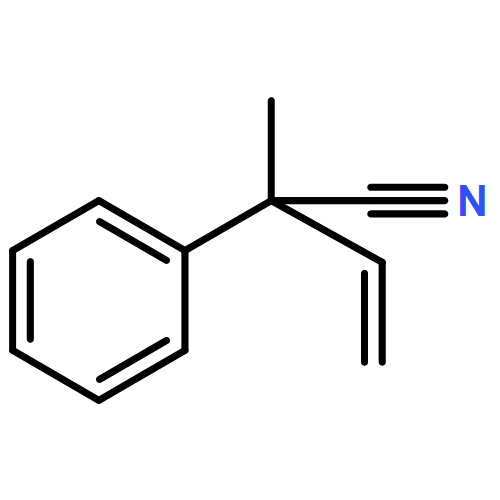




![(5Z)-5-(2-chlorobenzylidene)-3-[2-(morpholin-4-yl)-2-oxoethyl]-2-thioxo-1,3-thiazolidin-4-one](http://img.cochemist.com/ccimg/6400/6335-34-8.png)
![(5Z)-5-(2-chlorobenzylidene)-3-[2-(morpholin-4-yl)-2-oxoethyl]-2-thioxo-1,3-thiazolidin-4-one](http://img.cochemist.com/ccimg/6400/6335-34-8_b.png)




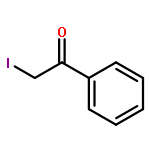
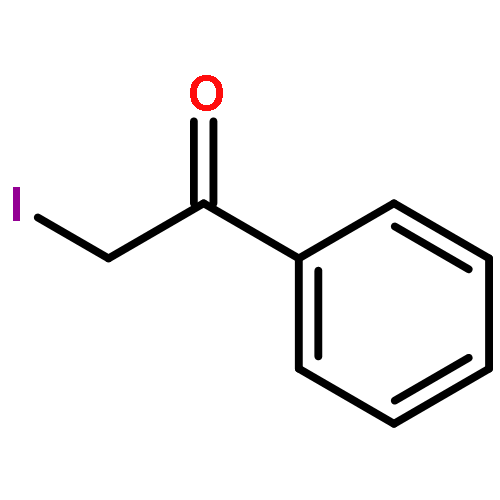




![6H-Dibenzo[b,d]pyran-6-one](http://img.cochemist.com/ccimg/2100/2005-10-9.png)
![6H-Dibenzo[b,d]pyran-6-one](http://img.cochemist.com/ccimg/2100/2005-10-9_b.png)