Co-reporter:Takashi Matsuo, Akihiro Hayashi, Masato Abe, Takaaki Matsuda, Yoshio Hisaeda and Takashi Hayashi
Journal of the American Chemical Society October 28, 2009 Volume 131(Issue 42) pp:15124-15125
Publication Date(Web):October 7, 2009
DOI:10.1021/ja907428e
Myoglobin (Mb) and horseradish peroxidase (HRP) were both reconstituted with a meso-unsubstituted iron corrole and their electronic configurations and peroxidase activities were investigated. The appearance of the 540 nm band upon incorporation of the iron corrole into apoMb indicates axial coordination by the proximal histidine imidazole in the Mb heme pocket. Based on 1H NMR measurements using the Evans method, the total magnetic susceptibility of the iron corrole reconstituted Mb was evaluated to be S = 3/2. In contrast, although a band does not appear in the vicinity of 540 nm during reconstitution of the iron corrole into the matrix of HRP, a spectrum similar to that of the iron corrole reconstituted Mb is observed upon the addition of dithionite. This observation suggests that the oxidation state of the corrole iron in the reconstituted HRP can be assigned as +4. The catalytic activities of both proteins toward guaiacol oxidation are quite different; the iron corrole reconstituted HRP decelerates H2O2-dependent oxidation of guaiacol, while the same reaction catalyzed by iron corrole reconstituted Mb has the opposite effect and accelerates the reaction. This finding can be attributed to the difference in the oxidation states of the corrole iron when these proteins are in the resting state.
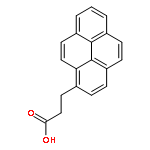
Co-reporter:Akira Fujii, Yutaka Sekiguchi, Hiroyoshi Matsumura, Tsuyoshi Inoue, Wen-Sheng Chung, Shun Hirota, and Takashi Matsuo
Bioconjugate Chemistry 2015 Volume 26(Issue 3) pp:537
Publication Date(Web):February 3, 2015
DOI:10.1021/acs.bioconjchem.5b00026
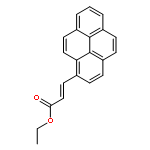
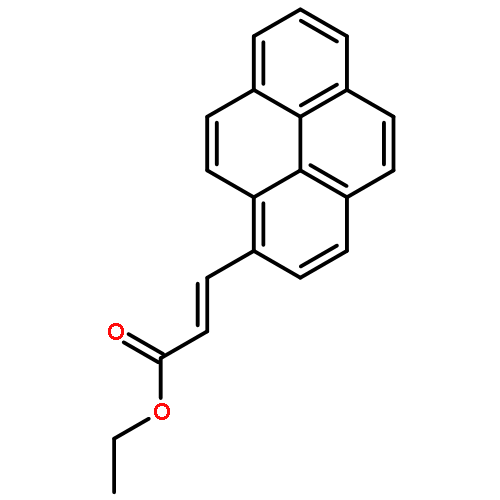
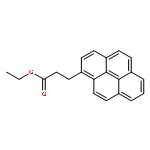
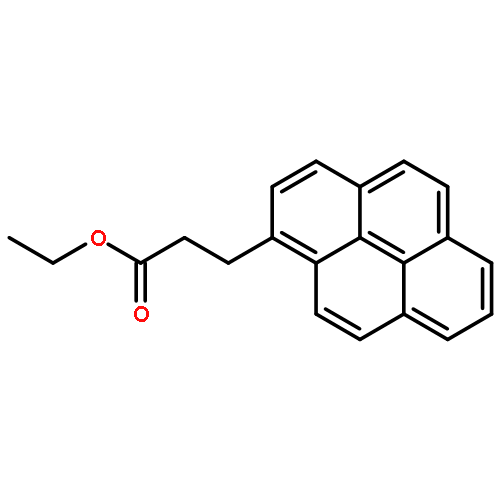
The excimer emission of pyrene is popularly employed for investigating the association between pyrene-labeled biomolecules or between pyrene-labeled places in a biomolecule. The property of pyrene excimer emission is affected by the fluctuation in ring stacking modes, which originates from the structural flexibilities of pyrene probes and/or of labeled places. Investigations of the excimer emission in terms of dynamics of pyrene stacking modes provide the detailed spatial information between pyrene-labeled places. In order to evaluate the effects of probe structures and fluctuation in pyrene–pyrene association modes on their emission properties on protein surface, three types of pyrene probe with different linker lengths were synthesized and conjugated to two cysteine residues in the A55C/C77S/V169C mutant of adenylate kinase (Adk), an enzyme that shows a structural transition between OPEN and CLOSED forms. In the CLOSED form of Adk labeled by a pyrene probe with a short linker, excimer emission was found to be predominated by the ground-state association of pyrenes. The pyrene stacking structure on the protein surface was successfully determined by an X-ray crystallographic analysis. However, the emission decay in the protein suggested the existence of several stacking orientations in solution. With the increase in the linker length, the effect of fluctuation in pyrene association modes on the spectral properties distinctly emerged at both ground and excited states. The combination of steady-state and time-resolved spectroscopic analyses is useful for differentiation in the origin of the excimer emission, which is essential for precisely understanding the interaction fashions between pyrene-labeled biomolecules.
Co-reporter:Takashi Matsuo, Shun Hirota
Bioorganic & Medicinal Chemistry 2014 Volume 22(Issue 20) pp:5638-5656
Publication Date(Web):15 October 2014
DOI:10.1016/j.bmc.2014.06.021
Recent development in biochemical experiment techniques and bioinformatics has enabled us to create a variety of artificial biocatalysts with protein scaffolds (namely ‘artificial enzymes’). The construction methods of these catalysts include genetic mutation, chemical modification using synthetic molecules and/or a combination of these methods. Designed evolution strategy based on the structural information of host proteins has become more and more popular as an effective approach to construct artificial protein-based biocatalysts with desired reactivities. From the viewpoint of application of artificial enzymes for organic synthesis, recently constructed artificial enzymes mediating oxidation, reduction and C–C bond formation/cleavage are introduced in this review article.
Co-reporter:Akira Fujii, Shun Hirota, and Takashi Matsuo
Bioconjugate Chemistry 2013 Volume 24(Issue 7) pp:1218
Publication Date(Web):May 29, 2013
DOI:10.1021/bc400160m
Adenylate kinase shows a conformational transition (OPEN and CLOSED forms) during substrate binding and product release to mediate the phosphoryl transfer between ADP and ATP/AMP. The protein motional characteristics will be useful to construct switching systems of fluorophore properties caused by the catalytic cycle of the enzyme. This paper demonstrates in situ reversible switching of a fluorophore property driven by the conformational transition of the enzyme. The pyrene-conjugated mutant adenylate kinase is able to switch the monomer/excimer emission property of pyrene on addition of ADP or P1P5-di(adenosine-5′)pentaphosphate (Ap5A, a transition state analog). The observation under the dilute condition (∼0.1 μM) indicates that the emission spectral change was caused by the motion of a protein molecule and not led by protein–protein interactions through π–π stacking of pyrene rings. The switching can be reversibly conducted by using hexokinase-coupling reaction. The fashion of the changes in emission intensities at various ligand concentrations is different between ADP, Mg2+-bound ADP, and Mg2+-bound Ap5A. The emission property switching is repeatable by a sequential addition of a substrate in a one-pot process. It is proposed that the property of a synthetic molecule on the enzyme surface is switchable in response to the catalytic cycle of adenylate kinase.
Co-reporter:Takashi Matsuo ; Takefumi Yoshida ; Akira Fujii ; Keiya Kawahara ;Shun Hirota
Organometallics 2013 Volume 32(Issue 19) pp:5313-5319
Publication Date(Web):September 19, 2013
DOI:10.1021/om4005302
The efficiency of ring-closing metathesis catalyzed by a Hoveyda–Grubbs type catalyst in water can be enhanced by addition of a chloride salt under neutral conditions. UV–vis spectroscopic study showed that a characteristic band of the catalyst around 380 nm remained over 16 h in the presence of KCl, whereas the band distinctly decreased in the absence of KCl. The disappearance of the band is ascribed to a displacement of a chloride ligand by a water molecule or a hydroxide anion. The spectral changes can be related to the metathesis activity. The experimental results indicate that avoidance of the chloride ligand loss is important to maintain the metathesis activity in water.
Co-reporter:Takashi Matsuo, Chie Imai, Takefumi Yoshida, Takashi Saito, Takashi Hayashi and Shun Hirota
Chemical Communications 2012 vol. 48(Issue 11) pp:1662-1664
Publication Date(Web):07 Dec 2011
DOI:10.1039/C2CC16898G
An L-phenylalanyl chloromethylketone-based inhibitor equipped with a Hoveyda–Grubbs catalyst moiety was regioselectively incorporated into the cleft of α-chymotrypsin through the intrinsic inhibition mechanism of the protein to construct an artificial organometallic protein.
Co-reporter:Takashi Matsuo, Yuji Tohi, and Takashi Hayashi
The Journal of Organic Chemistry 2012 Volume 77(Issue 20) pp:8946-8955
Publication Date(Web):October 1, 2012
DOI:10.1021/jo3013379
Porphycene is a structural isomer of porphyrin with 18π-conjugated aromatic character. Porphycene modified with trifluoromethyl (CF3) groups in the periphery of the framework readily affords the isolable 20π-conjugated antiaromatic form through a reaction with a proton-donating reductant. The 20π-conjugated form can be characterized by not only a variety of spectroscopies in solutions but also X-ray crystallography. This paper focuses on the free energy profile in the conversion of the 18π-conjugated porphycene into the 20π-conjugated form. From the results of kinetics, electrochemical measurements, and acid/base titrations, the 20π-conjugated CF3 porphycene is formed by a concerted proton–electron transfer (CPET) from a hydroquinone reagent to the 18π-conjugated form. The hydrogen-atom affinity of the 18π-conjugated CF3 porphycene (for two hydrogen atoms) was calculated to be −490 kJ mol–1, indicating that the N–H bonds in the 20π-conjugated form are rather easily cleaved. This reflects the antiaromatic characteristics of the 20π-conjugated porphycene. We propose that the kinetic and thermochemical analysis using redox potentials and pKa data is applicable for determining the reaction pathway in conversion of aromatic/antiaromatic mode of π-conjugated macrocycles as well as popular investigations for oxidations of organic molecules.
Co-reporter:Dr. Takashi Matsuo;Kazuki Fukumoto;Takuro Watanabe;Dr. Takashi Hayashi
Chemistry – An Asian Journal 2011 Volume 6( Issue 9) pp:2491-2499
Publication Date(Web):
DOI:10.1002/asia.201100107
Abstract
H64D myoglobin mutant was reconstituted with two different types of synthetic hemes that have aromatic rings and a carboxylate-based cluster attached to the terminus of one or both of the heme-propionate moieties, thereby forming a “single-winged cofactor” and “double-winged cofactor,” respectively. The reconstituted mutant myoglobins have smaller Km values with respect to 2-methoxyphenol oxidation activity relative to the parent mutant with native heme. This suggests that the attached moiety functions as a substrate-binding domain. However, the kcat value of the mutant myoglobin with the double-winged cofactor is much lower than that of the mutant with the native heme. In contrast, the mutant reconstituted with the single-winged cofactor has a larger kcat value, thereby resulting in overall catalytic activity that is essentially equivalent to that of the native horseradish peroxidase. Enhanced peroxygenase activity was also observed for the mutant myoglobin with the single-winged cofactor, thus indicating that introduction of an artificial substrate-binding domain at only one of the heme propionates in the H64D mutant is the optimal engineering strategy for improving the peroxidase activity of myoglobin.
Co-reporter:Prosenjit Chattopadhyay, Takashi Matsuo, Takanori Tsuji, Jun Ohbayashi, and Takashi Hayashi
Organometallics 2011 Volume 30(Issue 7) pp:1869-1873
Publication Date(Web):March 9, 2011
DOI:10.1021/om101113x
The reaction of cobalt(III) octaethylcorrole (CoOEC) with ethyl diazoacetate (EDA) under a nitrogen atmosphere led to the formation of bridged complexes with the carbene moiety inserted into one of the cobalt−nitrogen bonds. The bridged complexes underwent unique thermal isomerization, presumably via the transiently formed axial carbene complex. One of the thermodynamic isomers was characterized by 1H NMR and mass spectroscopic methods.
Co-reporter:Dr. Takashi Matsuo;Kazuki Fukumoto;Takuro Watanabe;Dr. Takashi Hayashi
Chemistry – An Asian Journal 2011 Volume 6( Issue 9) pp:
Publication Date(Web):
DOI:10.1002/asia.201190036
Co-reporter:Takashi Matsuo ; Akihiro Hayashi ; Masato Abe ; Takaaki Matsuda ; Yoshio Hisaeda ;Takashi Hayashi
Journal of the American Chemical Society () pp:
Publication Date(Web):October 7, 2009
DOI:10.1021/ja907428e
Myoglobin (Mb) and horseradish peroxidase (HRP) were both reconstituted with a meso-unsubstituted iron corrole and their electronic configurations and peroxidase activities were investigated. The appearance of the 540 nm band upon incorporation of the iron corrole into apoMb indicates axial coordination by the proximal histidine imidazole in the Mb heme pocket. Based on 1H NMR measurements using the Evans method, the total magnetic susceptibility of the iron corrole reconstituted Mb was evaluated to be S = 3/2. In contrast, although a band does not appear in the vicinity of 540 nm during reconstitution of the iron corrole into the matrix of HRP, a spectrum similar to that of the iron corrole reconstituted Mb is observed upon the addition of dithionite. This observation suggests that the oxidation state of the corrole iron in the reconstituted HRP can be assigned as +4. The catalytic activities of both proteins toward guaiacol oxidation are quite different; the iron corrole reconstituted HRP decelerates H2O2-dependent oxidation of guaiacol, while the same reaction catalyzed by iron corrole reconstituted Mb has the opposite effect and accelerates the reaction. This finding can be attributed to the difference in the oxidation states of the corrole iron when these proteins are in the resting state.
Co-reporter:Takashi Matsuo, Chie Imai, Takefumi Yoshida, Takashi Saito, Takashi Hayashi and Shun Hirota
Chemical Communications 2012 - vol. 48(Issue 11) pp:NaN1664-1664
Publication Date(Web):2011/12/07
DOI:10.1039/C2CC16898G
An L-phenylalanyl chloromethylketone-based inhibitor equipped with a Hoveyda–Grubbs catalyst moiety was regioselectively incorporated into the cleft of α-chymotrypsin through the intrinsic inhibition mechanism of the protein to construct an artificial organometallic protein.








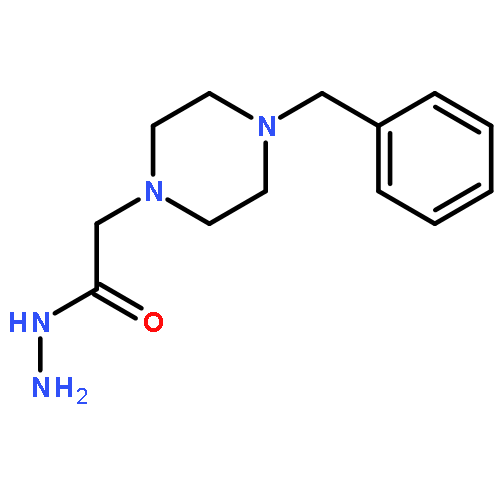
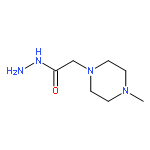
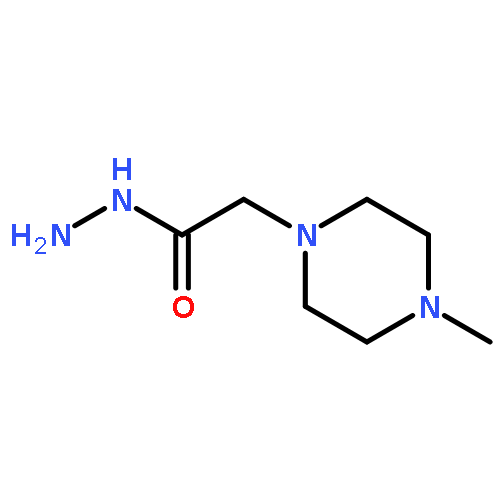
{kind=link}