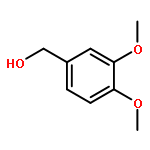
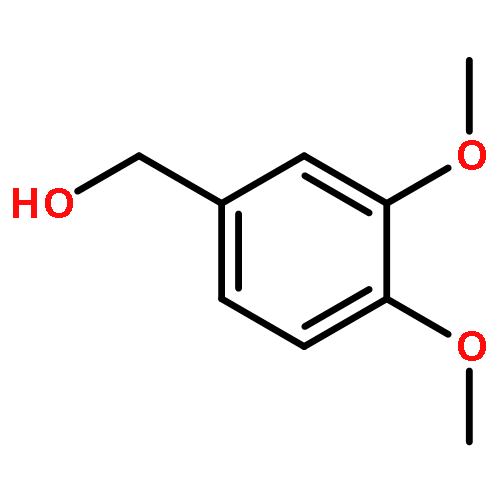
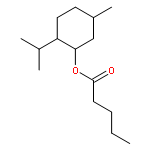
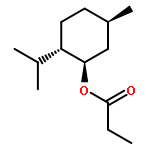
Co-reporter:Duc-Hiep Bach, Jian-Yu Liu, Won Kyung Kim, Ji-Young Hong, So Hyun Park, Donghwa Kim, Si-Ning Qin, Thi-Thu-Trang Luu, Hyen Joo Park, Yong-Nan Xu, Sang Kook Lee
Bioorganic & Medicinal Chemistry 2017 Volume 25, Issue 13(Issue 13) pp:
Publication Date(Web):1 July 2017
DOI:10.1016/j.bmc.2017.04.027
The overproduction of nitric oxide (NO) plays an important role in a variety of pathophysiological processes, including inflammation. Therefore, the suppression of NO production is a promising target in the design of anti-inflammatory agents. In the present study, a series of phthalimide analogs was synthesized, and their anti-inflammatory activities were evaluated using lipopolysaccharide (LPS)-stimulated NO production in cultured murine macrophage RAW264.7 cells. A structure-activity relationship study showed that the free hydroxyl group at C-4 and C-6 and the bulkiness of the N-substituted alkyl chain are associated with biological activity. Among the series of phthalimide derivatives, compound IIh exhibited potent inhibitory activity, with an IC50 value of 8.7 µg/mL. Further study revealed that the inhibitory activity of compound IIh was correlated with the down-regulation of the mRNA and protein expression of LPS-stimulated inducible nitric oxide synthase (iNOS). Compound IIh also suppressed the induction of the pro-inflammatory cytokines tumor necrosis factor-α and interleukin-1β in LPS-stimulated RAW 264.7 cells. The anti-inflammatory activity of compound IIh was also found to be associated with the suppression of the Toll-like receptor (TLR)4 signaling pathway by down-regulating the activation of interferon regulatory factor 3 (IRF-3) and interferon-β and signal transducer expression. These findings demonstrate that novel phthalimides might be potential candidates for the development of anti-inflammatory agents.New series of phthalimides were synthesized including the C7-alkyl chain as a key finding and structure-activity study with the inhibition of nitric oxide production was investigated.Download high-res image (91KB)Download full-size image
Co-reporter:Yanhua Fan, Jianyu Liu, Dan Liu, Zhipeng Zhou, Ying Bao, Jian Wang, Qingchun Zhao, Yongnan Xu
Environmental Toxicology and Pharmacology 2017 Volume 49(Volume 49) pp:
Publication Date(Web):1 January 2017
DOI:10.1016/j.etap.2016.09.012
•We report the synthesis and biological evaluation of NSCA-1.•NSCA-1 exerts synergistic cytotoxicity with Adriacin in HepG2-ADR cells.•NSCA-1 significantly decreased the expression of p-Akt and p-GSK3β.•NSCA-1enhanced the sensitivity of HepG2-ADR cells to Adriacin via P-gp inhibition.•NSCA-1 enhanced Adriacin induced apoptosis via p53 dependent pathway.Coumalamide derivatives are one of 2-pyrones derivatives, exerting multifunctional bioactivity. An array of coumalamide derivatives have been developed and presented good antiproliferative properties on cancer cells. However, the synthesis of 5-substituted coumalamide derivatives has not yet been published. Resistance to chemotherapeutic drugs is a major obstacle in hepatocellular carcinoma therapy. Recent evidence suggests that overexpression of constitutively active Akt confers on cancer cells resistance to chemotherapy. In this study, we report the synthesis and biological evaluation of a novel N-substituted coumalamide derivative (NSCA-1). The results indicated that NSCA-1 exerts synergistic cytotoxicity with Adriamycin in HepG2/ADR (HepG2/adriamycin) cells. Furthermore, both of the Akt kinase activity and phosphorylated Akt (Ser473) were found to be inhibited by NSCA-1 and subsequently resulting in decreased phosphorylation of GSK-3β. The intracellular accumulation of Adriamycin was also boosted by NSCA-1 via reducing the expression of p-gp. In addition, we found that combined treatment with NSCA-1 enhance cell apoptosis induced by Adriamycin via p53-dependant apoptotic pathway.
Co-reporter:Yanhua Fan, Hongyuan Lu, Hongda Ma, Fan Feng, Xiaolong Hu, Qiao Zhang, Jian Wang, Yongnan Xu and Qingchun Zhao
Food & Function 2015 vol. 6(Issue 12) pp:3746-3759
Publication Date(Web):24 Aug 2015
DOI:10.1039/C5FO00371G
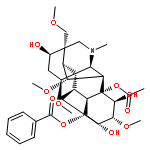
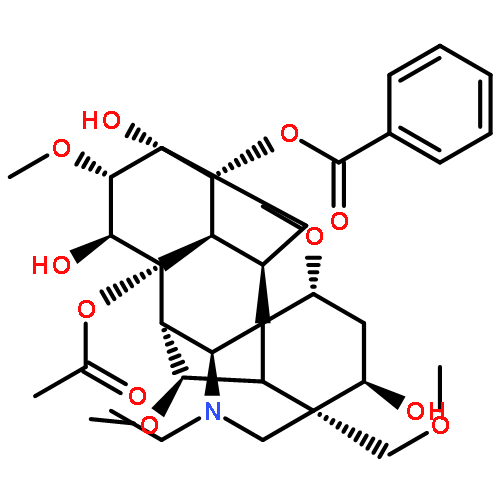
Eriocaulon sieboldianum (Sieb. & Zucc. ex Steud.) is an edible and medicinal plant used in traditional Chinese medicine. Often in combination with other herbs, it is processed into healthcare beverages for expelling wind-heat, protecting eyes, and reducing blood lipids. Besides, its water decoction together with other herbs has been utilized to treat cancer in China. However, the active ingredients and the precise cellular mechanisms of E. sieboldianum remain to be elucidated. The Aurora kinase family plays critical roles in the regulation of cell division and has attracted great attention to the identification of small-molecule Aurora kinase inhibitors for potential treatment of cancer. A molecular docking study was employed for docking of the most bioactive compounds. Hispidulin (HPDL) and quercetin-3-O-(6′′-O-galloyl)-β-D-galactopyranoside (QGGP) were singled out as potent inhibitors of Aurora kinase. Their inhibitory activity towards Aurora kinase was further confirmed by the obvious decrease in autophosphorylation of Aurora-A (Thr288) and Aurora-B (Thr232). Moreover, the induction of cell cycle arrest in HepG2 cells and the suppressed phosphorylation of histone H3 were also consistent with the inhibition of Aurora kinase. The data indicate that the E. sieboldianum extract and its two active compounds, HPDL and QGGP, could effectively induce apoptosis via p53, MAPKs and the mitochondrial apoptotic pathways. These findings could improve the understanding and enhance the development of drugs based on E. sieboldianum and raise its application value in anticancer therapy or prevention. In addition, our results indicated that Aurora kinase might be a novel target of HPDL and QGGP.
Co-reporter:Jinbao Xiang;Hanghang Li;Kai Yang;Lang Yi;Qun Dang
Molecular Diversity 2012 Volume 16( Issue 1) pp:173-181
Publication Date(Web):2012 February
DOI:10.1007/s11030-011-9345-y
Highly substituted novel 4H-pyrimido[1,6-a] pyrimidines were prepared by a trifluoromethanesulfonic acid catalyzed one-pot three-component condensation of 4-aminopyrimidines, aldehydes, and β-ketoesters. A preliminary feasibility study was undertaken on these compounds, to assess the potential production of a library of further diversified compounds by nucleophilic replacement of Cl (R1) or by reaction of electrophiles with the NH2 (R2) group.











![Thiocyanic acid, tricyclo[3.3.1.13,7]dec-2-yl ester](http://img.cochemist.com/ccimg/153800/153757-17-6.png)
![Thiocyanic acid, tricyclo[3.3.1.13,7]dec-2-yl ester](http://img.cochemist.com/ccimg/153800/153757-17-6_b.png)







![[6-(4-methoxy-phenyl)-imidazo[2,1-b ]thiazol-3-yl]-acetic Acid](http://img.cochemist.com/ccimg/68400/68347-95-5.png)
![[6-(4-methoxy-phenyl)-imidazo[2,1-b ]thiazol-3-yl]-acetic Acid](http://img.cochemist.com/ccimg/68400/68347-95-5_b.png)


![2-(6-phenylimidazo[2,1-b][1,3]thiazol-3-yl)acetic Acid](http://img.cochemist.com/ccimg/68400/68347-91-1.png)
![2-(6-phenylimidazo[2,1-b][1,3]thiazol-3-yl)acetic Acid](http://img.cochemist.com/ccimg/68400/68347-91-1_b.png)
![Olean-12-en-28-oic acid,3-[[O-6-deoxy-a-L-mannopyranosyl-(1?2)-O-[b-D-glucopyranosyl-(1?4)]-a-L-arabinopyranosyl]oxy]-23-hydroxy-, (3b,4a)-](http://img.cochemist.com/ccimg/68100/68027-15-6.png)
![Olean-12-en-28-oic acid,3-[[O-6-deoxy-a-L-mannopyranosyl-(1?2)-O-[b-D-glucopyranosyl-(1?4)]-a-L-arabinopyranosyl]oxy]-23-hydroxy-, (3b,4a)-](http://img.cochemist.com/ccimg/68100/68027-15-6_b.png)




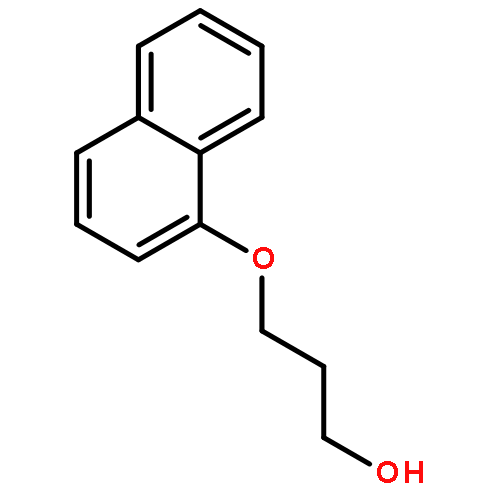






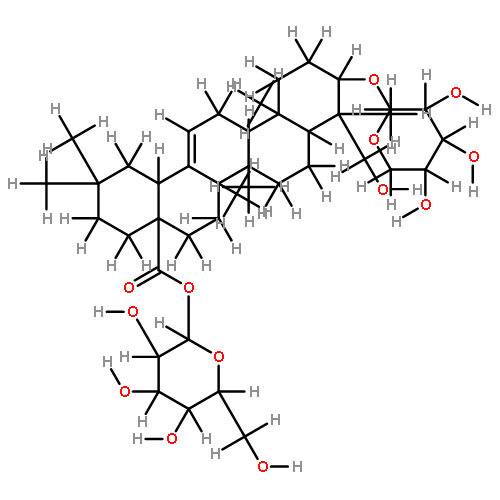


![Tetracosanamide,N-[(1S,2S,3R)-2,3-dihydroxy-1-(hydroxymethyl)heptadecyl]-](http://img.cochemist.com/ccimg/34500/34437-74-6.png)
![Tetracosanamide,N-[(1S,2S,3R)-2,3-dihydroxy-1-(hydroxymethyl)heptadecyl]-](http://img.cochemist.com/ccimg/34500/34437-74-6_b.png)





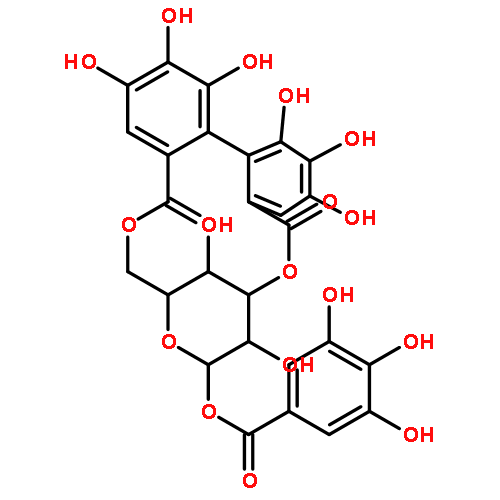



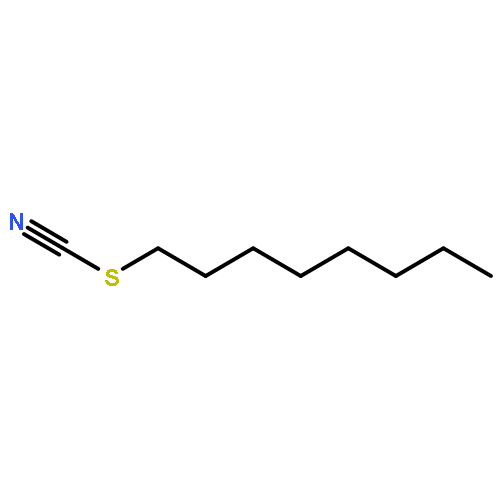
![Tricyclo[3.3.1.13,7]decane, 2-iodo-](http://img.cochemist.com/ccimg/19000/18971-91-0.png)
![Tricyclo[3.3.1.13,7]decane, 2-iodo-](http://img.cochemist.com/ccimg/19000/18971-91-0_b.png)
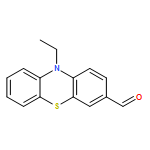



![(3β)-3-{[4-O-(β-D-Glucopyranosyl)-α-L-arabinopyranosyl]oxy}-23-hy droxyolean-12-en-28-oic acid](http://img.cochemist.com/ccimg/17300/17233-22-6.png)
![(3β)-3-{[4-O-(β-D-Glucopyranosyl)-α-L-arabinopyranosyl]oxy}-23-hy droxyolean-12-en-28-oic acid](http://img.cochemist.com/ccimg/17300/17233-22-6_b.png)
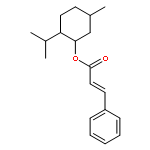

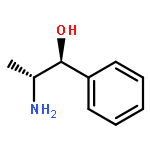






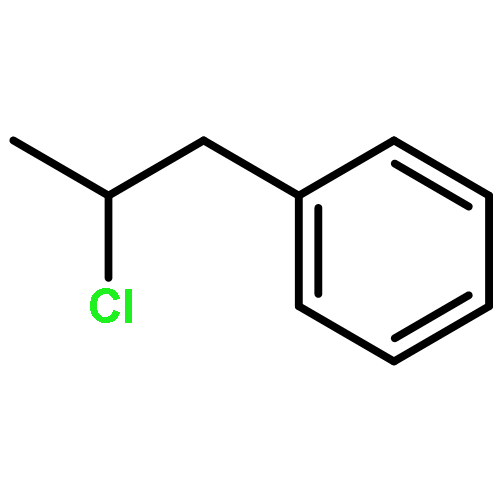




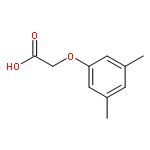
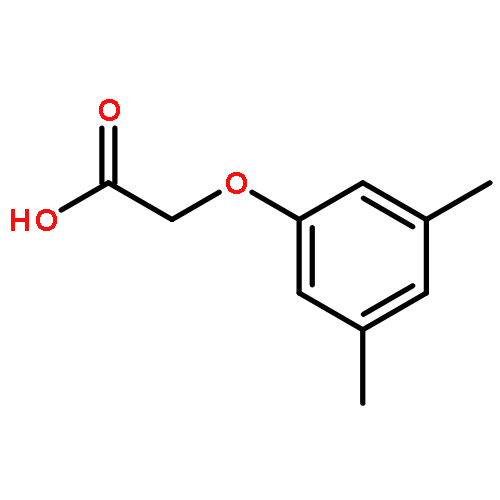

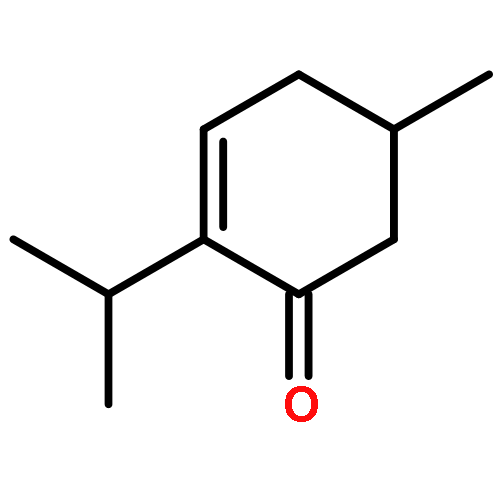








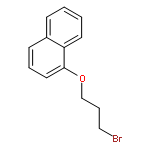
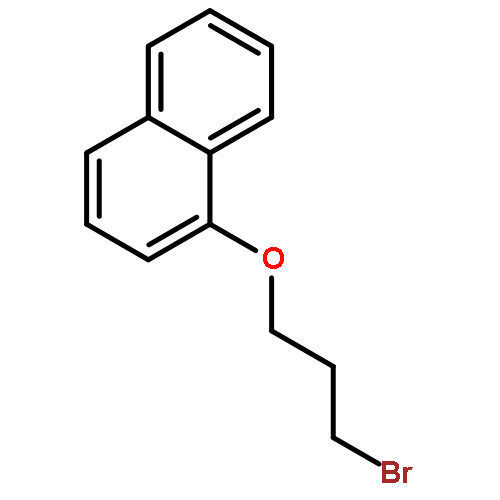








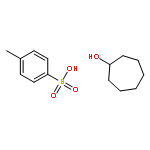
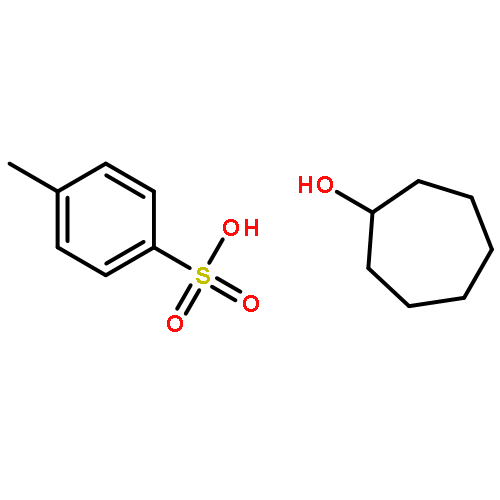
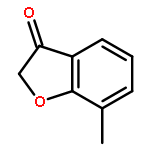
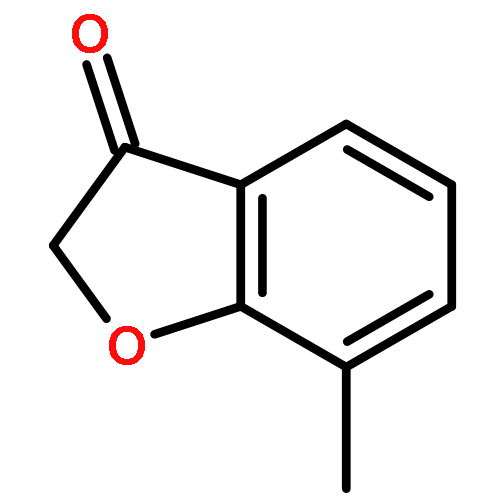
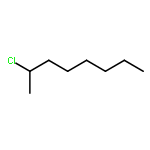



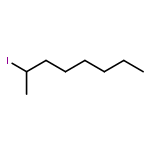



![5-hydroxy-2-(4-methoxyphenyl)-7-[(2s,3r,4s,5s,6r)-3,4,5-trihydroxy-6-[[(2r,3r,4r,5r,6s)-3,4,5-trihydroxy-6-methyloxan-2-yl]oxymethyl]oxan-2-yl]oxychromen-4-one](http://img.cochemist.com/ccimg/500/480-36-4.png)
![5-hydroxy-2-(4-methoxyphenyl)-7-[(2s,3r,4s,5s,6r)-3,4,5-trihydroxy-6-[[(2r,3r,4r,5r,6s)-3,4,5-trihydroxy-6-methyloxan-2-yl]oxymethyl]oxan-2-yl]oxychromen-4-one](http://img.cochemist.com/ccimg/500/480-36-4_b.png)


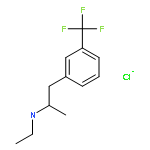
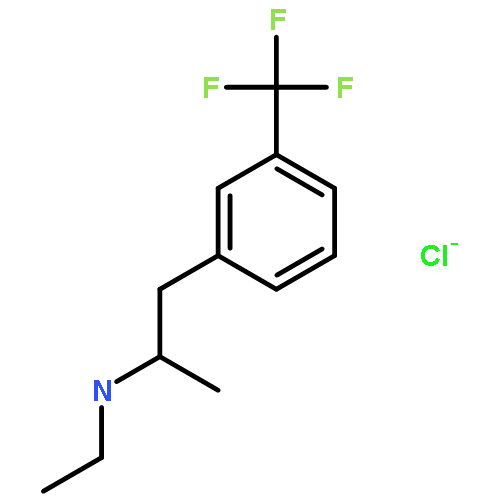
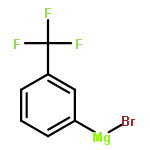

![1-[(E)-2-phenylethenyl]piperidine](http://img.cochemist.com/ccimg/400/332-15-0.png)
![1-[(E)-2-phenylethenyl]piperidine](http://img.cochemist.com/ccimg/400/332-15-0_b.png)