Co-reporter:Ken S. Feldman and Tamara S. Folda
The Journal of Organic Chemistry 2016 Volume 81(Issue 11) pp:4566-4575
Publication Date(Web):May 13, 2016
DOI:10.1021/acs.joc.6b00348
A synthesis route to the pentacyclic alkaloid (−)-gilbertine, which features a cyclization cascade passing through a transient indolidene intermediate, was pursued. A key stereochemical relationship was set via a Nicholas-type enolate alkylation. Ultimately, undesired C–N cyclization thwarted the final projected C–C bond forming ring closure, and gilbertine could not be prepared by this route.
Co-reporter:Ken S. Feldman, Inanllely Y. Gonzalez, and Christopher M. Glinkerman
The Journal of Organic Chemistry 2015 Volume 80(Issue 23) pp:11849-11862
Publication Date(Web):September 8, 2015
DOI:10.1021/acs.joc.5b01777
Tetracyclic products featuring predominantly a trans-hexahydroindane unit annelated onto the C(2)/C(3) positions of indole can be accessed by intramolecular cyclocondensation of tethered alkenyl sulfides with either indolidene or indolidenium cation intermediates. Studies with geometrically pure E- and Z-alkenyl sulfide isomers reveal a likely dichotomy of reaction paths that provide mixtures of both regioisomers and stereoisomers of the hexahydroindane adducts.
Co-reporter:Ken S. Feldman ; Inanllely Y. Gonzalez ;Christopher M. Glinkerman
Journal of the American Chemical Society 2014 Volume 136(Issue 43) pp:15138-15141
Publication Date(Web):October 16, 2014
DOI:10.1021/ja508421e
C(2)–C(3) cyclopentannelated indole constructs are prepared by either (a) a cyclization cascade of an alkenyl sulfide tethered to a 2-azido-1-allenylbenzene core or (b) cationic cyclization of a tethered alkenyl sulfide with a putative 2-indolidenium cation. In both cases, issues of C–C versus C–N bond formation emerge, and the results indicate that the former is favored.
Co-reporter:Ken S. Feldman and Brandon R. Selfridge
The Journal of Organic Chemistry 2013 Volume 78(Issue 9) pp:4499-4511
Publication Date(Web):April 15, 2013
DOI:10.1021/jo4005074
The enantiomer of the bicyclic lomaiviticin aglycone A core was prepared via a two-directional, divergent approach featuring (1) a double Ireland Claisen rearrangement to establish key core bonds with correct relative stereochemistry and (2) a double olefin metathesis reaction to deliver both cyclohexene rings of the target.
Co-reporter:Ken S. Feldman, Joshua F. Antoline
Tetrahedron 2013 69(5) pp: 1434-1445
Publication Date(Web):
DOI:10.1016/j.tet.2012.12.032
Co-reporter:Ken S. Feldman and Joshua F. Antoline
Organic Letters 2012 Volume 14(Issue 3) pp:934-937
Publication Date(Web):January 13, 2012
DOI:10.1021/ol203463n
The pentacyclic alkaloid (±)-meloscine was prepared in 19 steps through a reaction sequence that features a putative azatrimethylenemethane intermediate, generated through cascade cyclization of an allenyl azide substrate, to deliver the core azabicyclo[3.3.0]octadiene substructure. Subsequent manipulation of the peripheral functionality then delivered (±)-meloscine.
Co-reporter:Ken S. Feldman and Brandon R. Selfridge
Organic Letters 2012 Volume 14(Issue 21) pp:5484-5487
Publication Date(Web):October 17, 2012
DOI:10.1021/ol302567f
The bicyclic core of ent-lomaiviticin A was prepared in 11 operations from (S)-1-phenyl-2-propyn-1-ol in a two-directional route that features (1) a double Ireland Claisen rearrangement and (2) a double olefin metathesis reaction to form the key C–C bonds of the target.
Co-reporter:Ken S. Feldman, Brandon R. Selfridge
Tetrahedron Letters 2012 Volume 53(Issue 7) pp:825-828
Publication Date(Web):15 February 2012
DOI:10.1016/j.tetlet.2011.12.011
The diastereoselectivity of Ireland–Claisen rearrangements of allylic glycolates is dependent on the E:Z ratio of the silyl ketene acetals, the alkene geometry in the allyl unit, and the transition state topography. High yields and excellent diastereoselectivities (>95:5) have been achieved for selected substrates, including those with R2 = ethyl that results in a newly formed quaternary center. A discussion of the scope, selectivities, and transition state models will be presented.
Co-reporter:Ken S. Feldman and Paiboon Ngernmeesri
Organic Letters 2011 Volume 13(Issue 20) pp:5704-5707
Publication Date(Web):September 29, 2011
DOI:10.1021/ol202535f
The bis indole sponge alkaloid dragmacidin E was synthesized in racemic form over 25 steps starting from 7-benzhydroxyindole. Key steps include (a) a Witkop cyclization to facilitate construction of the indole-spanning seven-membered ring and (b) a cyclodehydrative pyrazinone synthesis that unites the two indole-containing sectors.
Co-reporter:Ken S. Feldman, Ahmed Yimam Nuriye, and Jianfeng Li
The Journal of Organic Chemistry 2011 Volume 76(Issue 12) pp:5042-5060
Publication Date(Web):May 16, 2011
DOI:10.1021/jo200740b
Exploratory oxidative cyclization studies on cyclopentanelated and cyclohexenelated oroidin derivatives utilized Pummerer chemistry to generate pentacyclic structures related to the palau’amine family of sponge metabolites. Stereochemical issues were paramount, and appropriate choice of annelated ring size led to formation of the pentacyclic framework with complete diastereoselectivity for all of the core bonds.
Co-reporter:Ken S. Feldman and Ahmed Yimam Nuriye
Organic Letters 2010 Volume 12(Issue 20) pp:4532-4535
Publication Date(Web):September 14, 2010
DOI:10.1021/ol1018322
A pentacyclic model system featuring the trans azabicyclo[3.3.0]octane unit of dibromopalau’amine was prepared with complete diastereoselectivity in the polycyclic core from a tricyclic precursor. The key transformations of this sequence include (a) a Pummerer reaction-mediated oxidative bicyclization, and (b) a Wolff rearrangement-based ring contraction to deliver the strained azabicyclo[3.3.0]octane core.
Co-reporter:Ken S. Feldman and Paiboon Ngernmeesri
Organic Letters 2010 Volume 12(Issue 20) pp:4502-4505
Publication Date(Web):September 13, 2010
DOI:10.1021/ol1018008
The conversion of a cycloheptannelated indole platform into the heptacyclic core structure of dragmacidin E proceeded over nine steps. Key sequences include a cyclocondensation to form an intermediate dihydropyrazinone ring and the conversion of a cyclic urea into the cyclic guanidine of the target.
Co-reporter:Ken S. Feldman and Matthew D. Fodor
The Journal of Organic Chemistry 2009 Volume 74(Issue 9) pp:3449-3461
Publication Date(Web):March 30, 2009
DOI:10.1021/jo900283g
The sponge-derived alkaloid dibromoagelaspongin was prepared from a dihydrooroidin derivative by exploiting the Pummerer reaction twice in succession. Oxidative cyclization of the substrate’s pyrrole-2-carboxamide function into the imidazole moiety was achieved in a regiospecific manner to establish both C−N bonds to C(6) of the target.
Co-reporter:Ken S. Feldman, Ahmed Yimam Nuriye
Tetrahedron Letters 2009 50(17) pp: 1914-1916
Publication Date(Web):
DOI:10.1016/j.tetlet.2009.02.024
Co-reporter:Ken S. Feldman, D. Keith Hester II, Malliga R. Iyer, Paul J. Munson, Carlos Silva López and Olalla Nieto Faza
The Journal of Organic Chemistry 2009 Volume 74(Issue 14) pp:4958-4974
Publication Date(Web):May 27, 2009
DOI:10.1021/jo900659w
The thermal, photochemical, and photochemical/CuI-mediated cascade cyclizations of a range of substituted 1-(2-azidophenyl)-3-alkenylallenes are described. These reactions provide both 1,2- and 2,3-cyclopentennelated indole products in varying ratios. In most cases, high regioselectivity for the 2,3-annelated isomer can be achieved under the hν/CuI conditions. Computational studies of this multistep reaction support the intermediacy of indolidene intermediates whose electrocyclizations (with or without copper present) define the regioselectivity branch point in the sequence.
Co-reporter:Ken S. Feldman, D. Keith Hester II, John H. Golbeck
Bioorganic & Medicinal Chemistry Letters 2007 Volume 17(Issue 17) pp:4891-4894
Publication Date(Web):1 September 2007
DOI:10.1016/j.bmcl.2007.06.041
A series of 11 simple phylloquinone derivatives, each lacking the extended phytyl side chain but featuring H-bond donor amides at one or both peri positions, were prepared and some salient physical properties were measured. A correlation between both IR frequency and NMR peak position, as indicators of internal H-bond strength, and the quinone half-wave reduction potential, was observed. These data are consistent with the prevailing hypothesis that quinone carbonyl H-bonding in general, and stronger H-bonds in particular, favorably bias the endogenous quinone’s electrochemical potential toward easier reduction.Peri-amide-substituted naphthoquinones were synthesized and their reduction potentials, N–H IR and N–H 1H NMR absorptions were recorded.
Co-reporter:Ken S. Feldman
Phytochemistry 2005 Volume 66(Issue 17) pp:1984-2000
Publication Date(Web):September 2005
DOI:10.1016/j.phytochem.2004.11.015
Continuing studies on the total synthesis of ellagitannin plant metabolites have led to the preparation of the dimeric antitumor compound, coriariin A, as well as designed structural analogues. In related investigations, the synthesis of a 2,4-hexahydroxydiphenoyl (HHDP)-bearing glucopyranose structure has been achieved. This species is related to the geraniin family of ellagitannins, and its subsequent chemistry is suggestive of a mechanistic rationale for the observation that the HHDP units within (3,6-bridged)2,4-HHDP-containing ellagitannins invariably are oxidized further in vivo. Companion studies designed to assay the immunomodulatory properties of coriariin A and analogues have led to the thesis that tumor necrosis factor alpha (TNFα) serves as a mediator of this ellagitannin’s tumor remissive activity. Furthermore, certain tannins and tannin analogues appear to act in an immunosuppressive capacity with peripheral blood monocytes that were exposed to the bacterially derived septic shock inducing agent lipid A.The synthesis of the tumoricidal dimeric ellagitannin, coriariin A, is described, along with related studies on the synthesis of a C(2)/C(4)-HHDP-containing model related to a putative geraniin biosynthesis precursor. The results of immunomodulatory assays with tannins and designed analogues attest to the promise that these species have in the distinct areas of tumor remission and anti-sepsis therapy.
Co-reporter:Ken S Feldman, Sarah L Wilson, Michael D Lawlor, Charles H Lang, William J Scheuchenzuber
Bioorganic & Medicinal Chemistry 2002 Volume 10(Issue 1) pp:47-55
Publication Date(Web):January 2002
DOI:10.1016/S0968-0896(01)00251-6
Designed dimeric gallotannin analogues featuring two tetragalloylglucopyranose cores connected by various hydrocarbon linkers inhibit tumor necrosis factor-α secretion from lipopolysaccharide-stimulated human peripheral blood mononuclear cells by up to 53% (5–24 μM concentration range) compared to control. Comparable suppression of tumor necrosis factor-α levels (∼50% vs control) was observed in the plasma of rats co-treated with lipopolysaccharide and specific tannin analogues selected for their lack of interleukin 1-β stimulating activity.Designed dimeric gallotannin analogues as indicated inhibit TNF-α secretion from LPS-stimulated h-PBMCs by up to 53% (5–24 μM) compared to control. Comparable suppression of TNF-α levels (∼50% vs control) was observed in the plasma of rats co-treated with LPS and specific tannin analogues.
Co-reporter:Ken S. Feldman, Kiran Sahasrabudhe, Michael D. Lawlor, Sarah L. Wilson, Charles H. Lang, William J. Scheuchenzuber
Bioorganic & Medicinal Chemistry Letters 2001 Volume 11(Issue 14) pp:1813-1815
Publication Date(Web):23 July 2001
DOI:10.1016/S0960-894X(01)00332-8
The naturally occurring gallotannin β-d-pentagalloylglucose (β-PGG) decreases tumor necrosis factor-alpha (TNF-α) output from human peripheral blood mononucleocytes exposed to lipopolysaccharide (LPS) by as much as 90% (vs control) at ∼5 μM concentration. A qualitatively similar but less pronounced effect (∼50% decrease) was observed in the serum of rats dosed with both LPS and β-PGG. These results may have relevance to therapies that target disease states characterized by an overproduction of TNF-α.The ability of β-PGG to suppress lipopolysaccharide-stimulated TNF-α secretion from human peripheral blood mononucleocytes and in a live rat system was investigated.



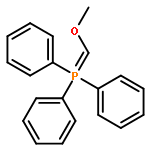
![Phosphorane, [(methylthio)methylene]triphenyl-](http://img.cochemist.com/ccimg/23500/23462-73-9.png)
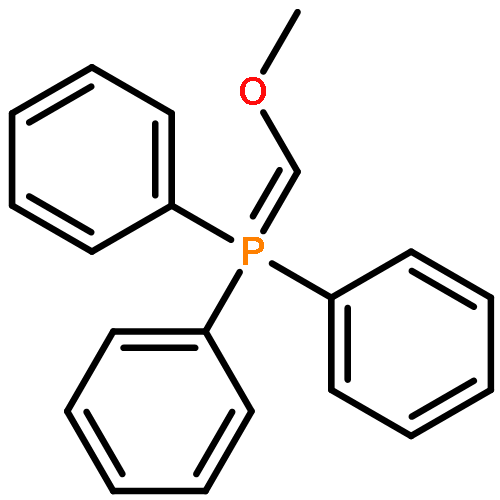
![Phosphorane, [(methylthio)methylene]triphenyl-](http://img.cochemist.com/ccimg/23500/23462-73-9_b.png)