Co-reporter:Minjun Wang;Feng Liu;Jianbo Hu
Industrial & Engineering Chemistry Research October 14, 2015 Volume 54(Issue 40) pp:9805-9812
Publication Date(Web):Publication Date (Web): September 24, 2015
DOI:10.1021/acs.iecr.5b01944
Oxyfuel combustion using O2–CO2 mixed gas as the oxidant is a promising technology for effectively capturing high concentration CO2. Chemical looping air separation (CLAS) is a novel approach with a low energy footprint for O2–CO2 mixed gas production. In this work, a series of Zr-doped Cu-based oxygen carriers were prepared by the sol–gel combustion method and used to produce the O2–CO2 gas mixture. The effects of doping Zr on phase structure, surface topography, O2 absorption/desorption, and cyclic reaction of Cu-based oxygen carriers were studied by experiments. The influencing mechanism of Zr doping on O2 release of Cu-based oxygen carrier was investigated using density functional theory (DFT) calculations. The results indicate that the addition of Zr enlarges the specific surface area and refines the grains. The Zr-doped Cu-based oxygen carriers have high desorption reaction rates. The oxygen release capacity increases with the increasing Zr doping amount. The cyclic operation indicates that the Zr-doped Cu-based oxygen carriers have high cyclic stability. The DFT calculation results show that, compared with the pure CuO, the Zr-doped CuO has lower oxygen vacancy formation energy, lower energy barriers for oxygen desorption and O anion diffusion, and narrower band gap, which indicates that the Zr-doped CuO releases oxygen more easily than the pure CuO and has a lower energy barrier of electron transfer, thus leading to a higher reactivity.
Co-reporter:Yingju Yang, Jing Liu, Bingkai Zhang, Feng Liu
Chemical Engineering Journal 2017 Volume 308(Volume 308) pp:
Publication Date(Web):15 January 2017
DOI:10.1016/j.cej.2016.09.128
•Hg0 oxidation by HCl on MnFe2O4 sorbent was investigated using DFT method.•HgCl and HgCl2 are adsorbed on MnFe2O4 surface in the chemisorption manner.•Hg0 oxidation by HCl over MnFe2O4 follows the Langmuir-Hinshelwood mechanism.•The first step (Hg0 → HgCl) is the rate-determining step of mercury oxidation.Magnetic manganese ferrite (MnFe2O4) with spinel structure has received considerable attention for its potential application in gaseous pollutants removal because of its surface redox reactivity properties. Density functional theory (DFT) calculations were performed to investigate heterogeneous mercury oxidation by HCl over MnFe2O4 surface. The results indicate that Hg0 is chemically adsorbed on MnFe2O4 (1 0 0) surface with an adsorption energy of −60.82 kJ/mol. HCl can decompose on MnFe2O4 surface to form active surface chlorine species for mercury oxidation. Both intermediate (HgCl) and final (HgCl2) products are adsorbed on MnFe2O4 (1 0 0) surface in a dissociative adsorption manner. Hg0 oxidation by HCl over MnFe2O4 surface follows the Langmuir-Hinshelwood mechanism in which a bimolecular reaction occurs between adsorbed Hg0 and active surface chlorine species. The mercury oxidation process over MnFe2O4 surface was investigated by examining the energy profile of reaction pathway. Heterogeneous Hg0 oxidation by HCl occurs through a two-step reaction pathway (Hg0 → HgCl → HgCl2) in which the first step (Hg0 → HgCl) is the rate-determining step.
Co-reporter:Yingju Yang, Jing Liu, Zhen Wang, Feng Liu
Fuel Processing Technology 2017 Volume 159(Volume 159) pp:
Publication Date(Web):1 May 2017
DOI:10.1016/j.fuproc.2017.01.035
Fe2O3 is one of the catalytically active compositions present in fly ash for mercury oxidation. Fe2O3 sample was prepared by a precipitation method. The textural property and surface chemical state of Fe2O3 sample were characterized by Brunauer-Emmett-Teller (BET) surface area and X-ray photoelectron spectroscopy (XPS), respectively. A series of experiments were conducted in a fixed-bed reactor to investigate the heterogeneous mercury oxidation by HCl on Fe2O3 surface. Fe2O3 can obtain a maximum mercury oxidation efficiency of 89.5% at 100 °C. Heterogeneous Hg0 oxidation by HCl over Fe2O3 surface occurs through the gas-solid reaction between gas-phase Hg0 and active surface chlorine species generated from the dissociation of HCl. Based on the experimental results, a detailed eight-step heterogeneous reaction kinetic model of mercury oxidation over Fe2O3 was proposed to predict mercury oxidation in the presence of Fe2O3 and HCl. This heterogeneous model was validated by comparison to different experimental data. The results of kinetic calculations show that model prediction is in good agreement with experimental results obtained by two research groups. Mercury oxidation process over Fe2O3 surface can be described using this heterogeneous kinetic model.
Co-reporter:Yingju Yang, Jing Liu, Bingkai Zhang, Yongchun Zhao, Xiaoyi Chen, Fenghua Shen
Chemical Engineering Journal 2017 Volume 317(Volume 317) pp:
Publication Date(Web):1 June 2017
DOI:10.1016/j.cej.2017.02.060
•Ce0.5W0.5/Ti catalyst showed more than 95% Hg oxidation efficiency at 200–350 °C.•The effects of flue gas compositions on Hg oxidation were studied.•Hg oxidation by HCl over CeW/Ti catalysts follows the Eley-Rideal mechanism.•Reaction pathway and energy profile of Hg oxidation on surface were studied by DFT.Ceria-based catalyst has been regarded as a very promising material for mercury oxidation due to its high catalytic oxidation activity. A series of CeO2 − WO3/TiO2 (CeW/Ti) catalysts were synthesized by an ultrasound-assisted impregnation method and tested for their catalytic activity of mercury oxidation in coal-fired flue gas. CeW/Ti catalysts with a CeO2:WO3:TiO2 mass ratio of 0.5:0.5:1 exhibited the highest catalytic activity for mercury oxidation in the wide temperature window of 200–350 °C. O2 promoted mercury oxidation by regenerating the chemisorbed oxygen and lattice oxygen of the catalysts. NO and SO2 could enhance mercury oxidation in the absence of HCl. Mercury oxidation over CeW/Ti catalysts was significantly enhanced when HCl was added to the simulated flue gas, over 95% mercury oxidation efficiency was obtained. H2O weakly inhibits mercury oxidation in flue gas. Mercury oxidation by HCl over CeW/Ti catalysts follows the Eley-Rideal mechanism, in which gas-phase Hg0 reacts with active surface chlorine species generated from HCl dissociation. First principles calculations based on the density functional theory (DFT) were used to elucidate the process of mercury oxidation over CeW/Ti catalysts. Compared to the one-step mercury oxidation process (Hg0→HgCl2), the dominant reaction pathway of mercury oxidation is a two-step process (Hg0→HgCl→HgCl2) in which the second step (HgCl→HgCl2) with an energy barrier of 50.11 kJ/mol is the rate-limiting step.
Co-reporter:Jianbo Hu, Yang Liu, Jing Liu, Chenkai Gu
Fuel 2017 Volume 200(Volume 200) pp:
Publication Date(Web):15 July 2017
DOI:10.1016/j.fuel.2017.03.079
The water vapor and trace gas impurities play important roles on CO2 capture performance using porous materials. We investigated the effects of H2O, SO2, and O2 on CO2 capture in a series of ZIFs with same SOD topology but different functional groups, such as ZIF-8, ZIF-90, ZIF-Cl, ZIF-NO2, and SALEM-2 by grand canonical Monte Carlo simulations. The results show that H2O and SO2 show a cooperative effect on CO2 adsorption in ZIF-NO2 and ZIF-90 containing polar functional groups, which results in enhanced CO2/N2 selectivity in presence of H2O or SO2. Especially, the presence of H2O or SO2 can enhance the CO2 uptake amounts and the CO2/N2 selectivity in ZIF-NO2 at all H2O or SO2 concentration. On the other hand, the presence of H2O, SO2 has negligible effect on CO2 adsorption and CO2/N2 separation in ZIF-8, ZIF-Cl, and SALEM-2. The presence of O2 has no effect on CO2 capture in all of the materials investigated. Our results indicate that, by changing the functionalities of ZIF materials, H2O and SO2 may play a positive role during CO2 adsorption and separation process.
Co-reporter:Zhen Wang, Jing Liu, Bingkai Zhang, Yingju Yang, Zhen Zhang, and Sen Miao
Environmental Science & Technology 2016 Volume 50(Issue 10) pp:5398-5404
Publication Date(Web):May 2, 2016
DOI:10.1021/acs.est.6b00549
Catalytic oxidation of elemental mercury (Hg0) through a selective catalytic reduction (SCR) system is a promising method to reduce mercury emissions from coal-burning power plants. The density functional theory (DFT) and periodic slab models were used to study the reaction mechanism of Hg0 oxidation by HBr on V2O5/TiO2 SCR catalyst surface. The interaction mechanisms of Hg0, HBr, HgBr, and HgBr2 on V2O5/TiO2(001) were investigated. The oxidation reaction energy profiles and the corresponding geometries of the intermediates, final states, and transition states were researched. The results indicate that Hg0 and HgBr2 are weakly adsorbed on the oxygen sites of the V2O5/TiO2(001) surface with physisorption. HgBr is chemically adsorbed on the surface. HBr is dissociatively adsorbed on the surface with an energy barrier of 85.59 kJ/mol. The reaction of Hg0 oxidation by HBr follows the Eley–Rideal mechanism: Hg0 interacts with a surface Br from HBr dissociation to form HgBr, and surface HgBr further interacts with HBr to form HgBr2, last HgBr2 desorbs from the surface. Comparing the energy pathway of Hg0 oxidation over V2O5/TiO2(001) surface by HBr to that of HCl, it is found that the dissociation energy barrier of HBr is lower than that of HCl, the formation and desorption energy barriers of HgBr2 are also lower than that of HgCl2, which explains why HBr is much more effective than HCl in promoting Hg0 oxidation.
Co-reporter:Minjun Wang, Jing Liu, Fenghua Shen, Hao Cheng, Jinxin Dai, Yan Long
Applied Surface Science 2016 Volume 367() pp:485-492
Publication Date(Web):30 March 2016
DOI:10.1016/j.apsusc.2016.01.240
Highlights
- •
The stability and reaction mechanism of CuO supported on ZrO2 were studied by DFT.
- •
ZrO2 provides a high resistance to CuO sintering.
- •
ZrO2 promotes the activity of CuO for CO oxidation in fuel reactor.
- •
The energy barriers are low enough for CuO/ZrO2 oxidation reaction in air reactor.
Co-reporter:Jianbo Hu
The Journal of Physical Chemistry C 2016 Volume 120(Issue 19) pp:10311-10319
Publication Date(Web):May 3, 2016
DOI:10.1021/acs.jpcc.6b01119
Metal organic frameworks (MOFs) represent a new kind of porous solid and have shown promising potential for carbon dioxide (CO2) capture and separation. In this work, grand canonical Monte Carlo simulations were performed to explore lithium alkoxide functionalization for improving CO2 adsorption capacity in HKUST-1, MOF-143, and MOF-399. The results show that lithium alkoxide functionalization remarkably improves the CO2 uptake ability in all three kinds of MOFs at 298 K, especially at the low-pressure range. The CO2 uptake amount in lithium-functionalized HKUST-1 increased more than 1700% compared with its unfunctionalized form at 1 kPa. Furthermore, the extension of organic linkers leads to lower CO2 adsorption capacity at the low-pressure range due to the lower isosteric heat, but higher CO2 adsorption capacity at the high-pressure range resulting from the increase of total free volume. Specifically, the incorporation of Li atoms onto the frameworks induced a shift of preferential adsorption sites for CO2. The CO2 molecules were first adsorbed around the Li atoms in the three lithium-functionalized MOFs.
Co-reporter:Fenghua Shen, Jing Liu, Zhen Zhang, and Jinxin Dai
Environmental Science & Technology 2015 Volume 49(Issue 22) pp:13716-13723
Publication Date(Web):October 21, 2015
DOI:10.1021/acs.est.5b03626
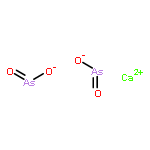
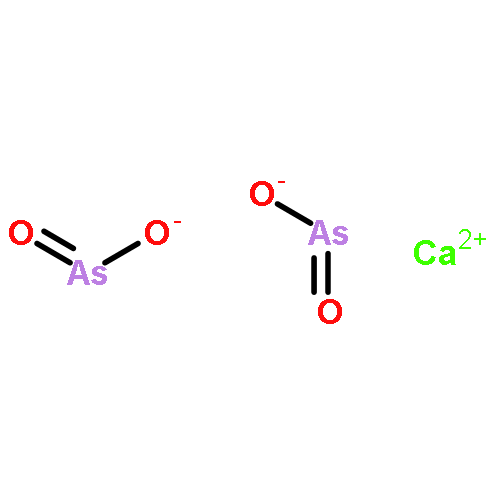
The kinetic behavior of arsenic (As) release during coal combustion and pyrolysis in a fluidized bed was investigated by applying an on-line analysis system of trace elements in flue gas. This system, based on inductively coupled plasma optical emission spectroscopy (ICP-OES), was developed to measure trace elements concentrations in flue gas quantitatively and continuously. Obvious variations of arsenic concentration in flue gas were observed during coal combustion and pyrolysis, indicating strong influences of atmosphere and temperature on arsenic release behavior. Kinetic laws governing the arsenic release during coal combustion and pyrolysis were determined based on the results of instantaneous arsenic concentration in flue gas. A second-order kinetic law was determined for arsenic release during coal combustion, and the arsenic release during coal pyrolysis followed a fourth-order kinetic law. The results showed that the arsenic release rate during coal pyrolysis was faster than that during coal combustion. Thermodynamic calculations were carried out to identify the forms of arsenic in vapor and solid phases during coal combustion and pyrolysis, respectively. Ca3(AsO4)2 and Ca(AsO2)2 are the possible species resulting from As–Ca interaction during coal combustion. Ca(AsO2)2 is the most probable species during coal pyrolysis.
Co-reporter:Bingkai Zhang
The Journal of Physical Chemistry C 2015 Volume 119(Issue 27) pp:15047-15055
Publication Date(Web):June 10, 2015
DOI:10.1021/acs.jpcc.5b00645
CeO2-based catalysts have been regarded as potential materials for Hg removal due to high catalytic performance, nontoxicity, and low cost. Density functional theory calculations were performed to investigate the mercury oxidation mechanism by HCl over a CeO2 catalyst. The thermodynamic stability analysis suggests that the stoichiometric CeO2(111) is the most stable surface. The protonated CeO2 surfaces takes place at low oxygen partial pressures, and the chlorinated CeO2 surfaces can stably exist under low HCl concentrations. The adsorption energies and geometries show that Hg0 is physically adsorbed on oxygen sites of the CeO2(111) surface and HCl is chemically adsorbed on the CeO2(111) surface. HCl can dissociate on the CeO2(111) surface with a low barrier. The Hg oxidation is most likely to proceed with the Eley–Rideal mechanism at the first step (Hg → HgCl), followed by the Langmuir–Hinshelwood mechanism at the second step (HgCl → HgCl2). In the whole Hg oxidation reaction, the formation of HgCl2 is the rate-determining step. The low energy barriers for the oxidation reaction of Hg on CeO2 make it an attractive alternative catalyst for Hg oxidation.
Co-reporter:Yang Liu ; Jing Liu ; Y.S. Lin ;Ming Chang
The Journal of Physical Chemistry C 2014 Volume 118(Issue 13) pp:6744-6751
Publication Date(Web):February 19, 2014
DOI:10.1021/jp4113969
Understanding the effects of the presence of water vapor and other gas impurities (such as SO2 and O2) in flue gas on sorbent performance is critical to properly evaluate the use of metal organic frameworks for realistic postcombustion CO2 capture. Grand canonical Monte Carlo calculations were performed to investigate the effects of H2O, O2, and SO2 on CO2 adsorption and CO2/N2 separation using ZIF-68. The mechanisms of these effects were also investigated by density functional theory calculations. The presence of H2O affects the CO2 adsorption on ZIF-68 in two opposite ways: the negative effect of competitive adsorption of the impurity gases on the adsorption sites over CO2, and the positive effects of formation of new adsorption sites for CO2. The presence of H2O reduces the CO2 adsorption ability but increases the CO2/N2 separation factor. SO2 is found to be strongly adsorbed in the pores of ZIF-68 and considered as an adverse factor for CO2 adsorption and CO2/N2 separation. The presence of O2 has a negligible effect on CO2 adsorption on ZIF-68.
Co-reporter:Yang Liu, Jing Liu, Ming Chang, and Chuguang Zheng
The Journal of Physical Chemistry C 2012 Volume 116(Issue 32) pp:16985-16991
Publication Date(Web):July 23, 2012
DOI:10.1021/jp302619m
Density functional theory (DFT) calculations are performed to investigate the effect of functionalized linkers on CO2 binding in zeolitic imidazolate frameworks (ZIFs). Three typical ZIFs with different functionalized linkers are considered: ZIF-68 with bIM linker (C6H6 group), ZIF-69 with cbIM linker (chlorine group), and ZIF-78 with nbIM linker (nitro group). Compared to bIM linker, the charge distributions of cbIM linker and nbIM linker have been significantly changed by the substitution of the functional groups and have been proved to have positive effects to enhance the binding capacity of CO2. The withdrawal of electronic charge from the linker leads to enhanced acidity for the hydrogen atoms on linker, which can form weak hydrogen bond interactions with the oxygen atoms of CO2. The binding capacity of nbIM linker for CO2 is much higher than those of bIM and cbIM linkers for CO2, which is due to the strong effect of nitro group on polarizing the CO2 molecule and thus enhances the interaction force between them. The binding energies of CO2 in the IM linkers are within the range of −1.64 to −18.54 kJ/mol, which suggests that the binding belongs to physisorption.
Co-reporter:Yang Liu, Jing Liu, Ming Chang, Chuguang Zheng
Fuel 2012 Volume 95() pp:521-527
Publication Date(Web):May 2012
DOI:10.1016/j.fuel.2011.09.057
Capturing CO2 from the flue gases for sequestration is currently a key issue in environmental protection. Metal–organic frameworks (MOFs) are a new class of porous materials and have shown great promise for CO2 adsorption and separation applications. While the linkers of MOFs influence the CO2 adsorption performance greatly, the mechanism about it is still not clear. In this work, density functional theory calculations were performed to study CO2 adsorption mechanism on linker of isoreticular metal–organic framework-1 (IRMOF-1). The effect of model sizes was investigated by comparing the adsorption energies of different models with CO2 located on the same adsorption sites. The results indicate that model (HCOO)5Zn4O(BDC)Zn4O(HCOO)5 is sufficient to calculate CO2 adsorption on linker of IRMOF-1. Eight different positions on linker of IRMOF-1 with three orientations of CO2 were studied in detail to understand the mechanism of CO2 adsorption. The side position with CO2 parallel attack at hydrogen side of linker edge is the most favorite adsorption site. The effects of chemical modifications on CO2 adsorption were studied, and the adsorption energies were found to be significantly increased. This work will be helpful to design and synthesis new materials that have higher CO2 adsorption abilities.Highlights► The adsorption mechanism of CO2 on linker of MOFs is investigated. ► The models are evaluated by comparing the adsorption energies. ► Eight different positions on linker with three orientations of CO2 are studied. ► Chemical modifications are conducted to enhance the adsorption strength of CO2. ► The highest adsorption energy is obtained by doping MOFs with Li atom.
Co-reporter:Bingkai Zhang ; Ye Ai ; Jing Liu ; Sang W. Joo ;Shizhi Qian
The Journal of Physical Chemistry C 2011 Volume 115(Issue 50) pp:24951-24959
Publication Date(Web):November 9, 2011
DOI:10.1021/jp2089388
Ionic current rectification (ICR) refers to a phenomenon that an ionic current flowing through a nanopore exhibits a preferential direction. In this paper, we investigate the ICR phenomenon in a conical nanopore embedded within a dielectric membrane using a continuum-based model, which is composed of the Nernst–Planck (NP) equations for the ionic mass transport, the Poisson equation for the electrostatics, and the Navier–Stokes (NS) equations for the flow field. Different from the existing studies, the emphasis of this investigation is placed on the polarization effect of the dielectric membrane on the ionic current and ICR in a conical nanopore. The results show that the polarization effect can influence the ion current and ICR, especially under the conditions of a relatively low κRt, which is the ratio of the nanopore tip radius and the Debye length, and a relatively low surface charge density of the nanopore.
Co-reporter:Ye Ai, Jing Liu, Bingkai Zhang, Shizhi Qian
Sensors and Actuators B: Chemical 2011 Volume 157(Issue 2) pp:742-751
Publication Date(Web):20 October 2011
DOI:10.1016/j.snb.2011.05.036
A conical nanofluidic field effect transistor (FET) refers to a conical nanopore embedded with an electrically controllable gate electrode. The surface potential of the nanopore can be effectively regulated by manipulating the gate potential applied to the gate electrode, which in turn controls the ionic current through the nanopore. The field effect on the ionic current rectification (ICR) in the conical nanofluidic FET is comprehensively investigated using a continuum model, composed of Nernst–Planck equations for the ionic concentrations, Poisson equation for the electric potential, and Navier–Stokes equations for the flow field. Under the conditions of a low ionic concentration, a low surface charge density of the nanopore, and a high permittivity of the dielectric nanopore, regulation of ICR by FET is significant. The field effect on the ICR with the gate electrode located in the middle region is opposite to that with the gate electrode located near the tip of the nanopore.
Co-reporter:Ye Ai, Jing Liu, Bingkai Zhang, and Shizhi Qian
Analytical Chemistry 2010 Volume 82(Issue 19) pp:8217
Publication Date(Web):August 30, 2010
DOI:10.1021/ac101628e
Field effect regulation of DNA nanoparticle translocation through a nanopore using a gate electrode is investigated using a continuum model, composed of the coupled Poisson−Nernst−Planck equations for the ionic mass transport and the Navier−Stokes equations for the hydrodynamic field. The field effect regulation of the DNA translocation relies on the induced electroosmotic flow (EOF) and the particle−nanopore electrostatic interaction. When the electrical double layers (EDLs) formed adjacent to the DNA nanoparticle and the nanopore wall are overlapped, the particle−nanopore electrostatic interaction could dominate over the EOF effect, which enables the DNA trapping inside the nanopore when the applied electric field is relatively low. However, the particle−nanopore electrostatic interaction becomes negligible if the EDLs are not overlapped. When the applied electric field is relatively high, a negative gate potential can slow down the DNA translocation by an order of magnitude, compared to a floating gate electrode. The field effect control offers a more flexible and electrically compatible approach to regulate the DNA translocation through a nanopore for DNA sequencing.
Co-reporter:Jing Liu, Wenqi Qu, Jinzhou Yuan, Shouchun Wang, Jianrong Qiu and Chuguang Zheng
Energy & Fuels 2010 Volume 24(Issue 1) pp:117-122
Publication Date(Web):July 16, 2009
DOI:10.1021/ef9005143
The thermochemical properties of mercury species present in combustion flue gases were studied using quantum mechanical methods combined with effective core potentials (ECP) basis sets. At various levels of theory, the calculated geometries, vibrational frequencies of the species, and the reaction enthalpies were compared with experimental data in order to validate the quantum mechanical method and basis set combination. The results show that the QCISD/RCEP28DVZ combination provides the most accurate results and the B3PW91/RCEP28DVZ and B3LYP/ECP28MWB combinations also perform well. On the basis of the evaluation of theoretical methods and basis sets of quantum chemistry, theoretical exploration on the mercury reaction mechanism in flue gas was conducted on the level of atoms and molecules. The properties of stable minimums were validated by vibration frequencies analysis. The activation energies were calculated by thermal energy calibration (including zero point energy calibration). The reaction rate constants in the temperature range of 298−1500 K were calculated from the transition state theory.
Co-reporter: Sang W. Joo;Sang Yoon Lee; Jing Liu; Shizhi Qian
ChemPhysChem 2010 Volume 11( Issue 15) pp:3281-3290
Publication Date(Web):
DOI:10.1002/cphc.201000433
Abstract
The translation of a charged, elongated cylindrical nanoparticle along the axis of a nanopore driven by an imposed axial salt concentration gradient is investigated using a continuum theory, which consists of the ionic mass conservation equations for the ionic concentrations, the Poisson equation for the electric potential in the solution, and the modified Stokes equations for the hydrodynamic field. The diffusiophoretic motion is driven by the induced electrophoresis and chemiphoresis. The former is driven by the generated overall electric field arising from the difference in the ionic diffusivities and the double layer polarization, while the latter is generated by the induced osmotic pressure gradient around the charged particle. The induced diffusiophoretic motion is investigated as functions of the imposed salt concentration gradient, the ratio of the particle’s radius to the double layer thickness, the cylinder’s aspect ratio (length/radius), the ratio of the nanopore size to the particle size, the surface charge densities of the nanoparticle and the nanopore, and the type of the salt used. The induced diffusiophoretic motion of a nanorod in an uncharged nanopore is mainly governed by the induced electrophoresis, driven by the induced electric field arising from the double layer polarization. The induced particle motion is driven by the induced electroosmotic flow, if the charges of the nanorod and nanopore wall have the same sign.
Co-reporter:J. Liu, Q. Falcoz, D. Gauthier, G. Flamant, C.Z. Zheng
Chemosphere (June 2010) Volume 80(Issue 3) pp:241-247
Publication Date(Web):1 June 2010
DOI:10.1016/j.chemosphere.2010.04.028
The accumulation of toxic metals generated by coal-fired power stations presents a serious threat to the environment. The volatilization behavior of two representative metals (Cd and Zn), and the influence of temperature were investigated during coal combustion. An inductively coupled plasma atomic emission spectrometric (ICP-AES) method was developed to continuously measure the heavy metal concentrations quantitatively in flue gas under combustion conditions in order to track the metal release process. This continuous heavy metal analysis system was implemented by coupling it to two types of high temperature reactors: a bubbling fluidized bed reactor and a fixed bed reactor with diameter of 0.1 m and 0.08 m respectively. For the two metals considered in this study (Cd and Zn), the experimental setup was successfully used to continuously monitor the metal vaporization process during coal combustion independent of reactor design, and at different temperatures. Cd is more easily vaporized than Zn during coal combustion. Temperature significantly influences the metal vaporization process. In general, the higher the temperature, the higher the metal vaporization, although the vaporization is not proportional to temperature. In addition to the experimental study, a thermodynamic calculation was carried out to simulate the heavy metal speciation during coal combustion process. The theoretical volatilization tendency is consistent with the experiment. The thermodynamic calculation identified the formation of binary oxides retarding heavy metal vaporization.
Co-reporter:Yingju Yang, Jing Liu, Bingkai Zhang, Feng Liu
Journal of Hazardous Materials (5 January 2017) Volume 321() pp:154-161
Publication Date(Web):5 January 2017
DOI:10.1016/j.jhazmat.2016.09.007
•Hg adsorption and oxidation mechanisms on MnFe2O4 were studied using DFT method.•Hg0 adsorption on Mn-terminated MnFe2O4 (100) surface is a chemisorption process.•HgO shows high chemical reactivity for its adsorption on MnFe2O4 surface.•The reaction between adsorbed Hg and surface oxygen is the rate-determining step.MnFe2O4 has been regarded as a very promising sorbent for mercury emission control in coal-fired power plants because of its high adsorption capacity, magnetic, recyclable and regenerable properties. First-principle calculations based on density functional theory (DFT) were used to elucidate the mercury adsorption and oxidation mechanisms on MnFe2O4 surface. DFT calculations show that Mn-terminated MnFe2O4 (1 0 0) surface is much more stable than Fe-terminated surface. Hg0 is physically adsorbed on Fe-terminated MnFe2O4 (1 0 0) surface. Hg0 adsorption on Mn-terminated MnFe2O4 (1 0 0) surface is a chemisorption process. The partial density of states (PDOS) analysis indicates that Hg atom interacts strongly with surface Mn atoms through the orbital hybridization. HgO is adsorbed on the MnFe2O4 surface in a chemical adsorption manner. The small HOMO–LUMO energy gap implies that HgO molecular shows high chemical reactivity for HgO adsorption on MnFe2O4 surface. The energy barriers of Hg0 oxidation by oxygen on Fe- and Mn-terminated MnFe2O4 surfaces are 206.37 and 76.07 kJ/mol, respectively. Mn-terminated surface is much more favorable for Hg0 oxidation than Fe-terminated surface. In the whole Hg0 oxidation process, the reaction between adsorbed mercury and surface oxygen is the rate-determining step.Download high-res image (193KB)Download full-size image
Co-reporter:Bingkai Zhang, Jing Liu, Guoliang Dai, Ming Chang, Chuguang Zheng
Proceedings of the Combustion Institute (2015) Volume 35(Issue 3) pp:2855-2865
Publication Date(Web):1 January 2015
DOI:10.1016/j.proci.2014.06.051
The selective catalytic reduction (SCR) for the reduction of NOx can enhance the oxidation of elemental mercury, which is regarded as a low-cost option for mercury control in coal-fired power plants. First-principles calculations based on the density functional theory and the periodic slab models were used to gain a fundamental understanding of mercury oxidation mechanism across V2O5/TiO2 SCR catalyst. The adsorption of Hg0, HCl, HgCl, and HgCl2 on V2O5/TiO2(0 0 1) surface were studied. The energy profile of the oxidation reaction and the structures of related transition states and intermediates were examined. The results show that Hg0 is mainly physically absorbed on vanadyl–oxygen sites of the V2O5/TiO2(0 0 1) surface with an adsorption energy of −27.93 kJ/mol. HCl is chemisorbed on vanadyl–oxygen sites of V2O5/TiO2(0 0 1) surface, and can undergoes dissociation process with an energy barrier of 101.53 kJ/mol to form the vanadium oxy-chloride complex which is essential in Hg0 oxidation reaction on V2O5/TiO2(0 0 1) surface. The mercury oxidation reaction occurs through an Eley–Rideal mechanism in which Hg reacts with HCl that has previously been adsorbed and dissociated on V2O5/TiO2(0 0 1) surface to form surface HgCl, and then surface HgCl reacts with HCl to form HgCl2, finally HgCl2 desorbs from the V2O5/TiO2(0 0 1) surface. In the whole Hg oxidation reaction, the formation of HgCl2 is the rate-determining step based on its high energy barrier.




















































![Methyl (3s,4r)-3-benzoyloxy-8-methyl-8-azabicyclo[3.2.1]octane-4-carboxylate](http://img.cochemist.com/ccimg/100/50-36-2.png)
![Methyl (3s,4r)-3-benzoyloxy-8-methyl-8-azabicyclo[3.2.1]octane-4-carboxylate](http://img.cochemist.com/ccimg/100/50-36-2_b.png)