Co-reporter:Chia-Hua Wu ; Boris Galabov ; Judy I-Chia Wu ; Sonia Ilieva ; Paul von R. Schleyer ;Wesley D. Allen
Journal of the American Chemical Society 2014 Volume 136(Issue 8) pp:3118-3126
Publication Date(Web):January 22, 2014
DOI:10.1021/ja4111946
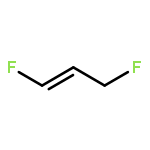
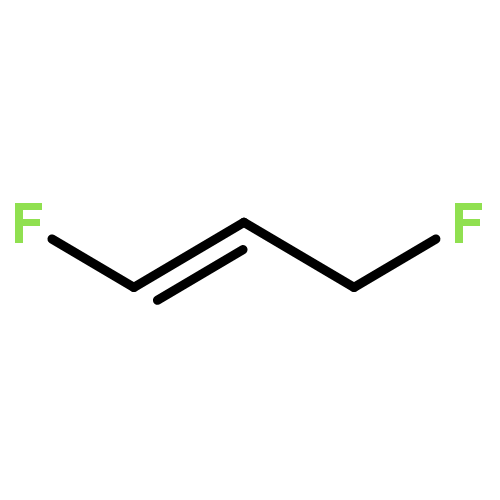
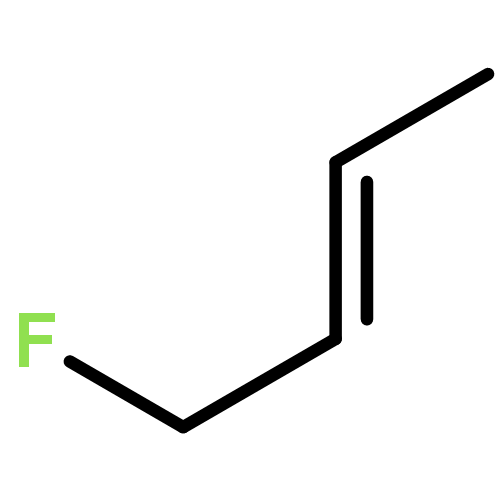
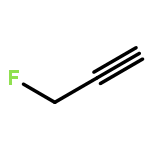
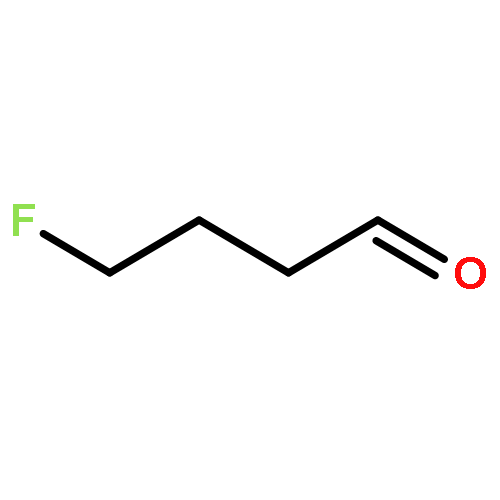
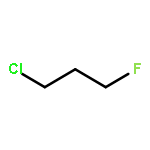
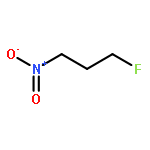
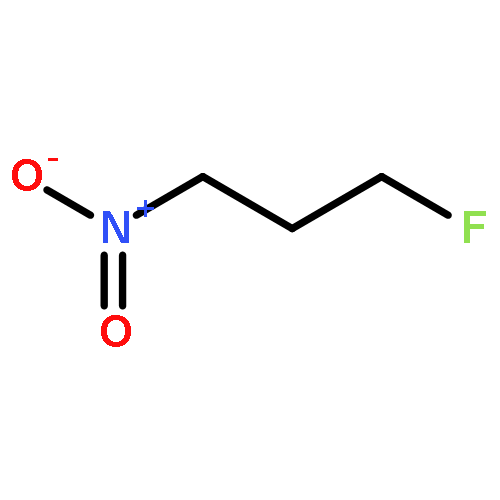
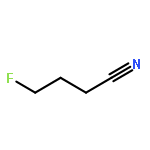
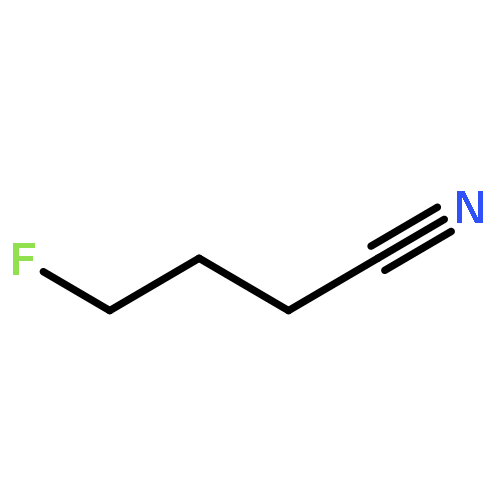
Rigorous quantum chemical investigations of the SN2 identity exchange reactions of methyl, ethyl, propyl, allyl, benzyl, propargyl, and acetonitrile halides (X = F–, Cl–) refute the traditional view that the acceleration of SN2 reactions for substrates with a multiple bond at Cβ (carbon adjacent to the reacting Cα center) is primarily due to π-conjugation in the SN2 transition state (TS). Instead, substrate–nucleophile electrostatic interactions dictate SN2 reaction rate trends. Regardless of the presence or absence of a Cβ multiple bond in the SN2 reactant in a series of analogues, attractive Cβ(δ+)···X(δ–) interactions in the SN2 TS lower net activation barriers (Eb) and enhance reaction rates, whereas repulsive Cβ(δ–)···X(δ–) interactions increase Eb barriers and retard SN2 rates. Block-localized wave function (BLW) computations confirm that π-conjugation lowers the net activation barriers of SN2 allyl (1t, coplanar), benzyl, propargyl, and acetonitrile halide identity exchange reactions, but does so to nearly the same extent. Therefore, such orbital interactions cannot account for the large range of Eb values in these systems.
Co-reporter:Judy I. Wu ; James E. Jackson ;Paul von Ragué Schleyer
Journal of the American Chemical Society 2014 Volume 136(Issue 39) pp:13526-13529
Publication Date(Web):September 12, 2014
DOI:10.1021/ja507202f
Computed association energies and dissected nucleus-independent chemical shifts (NICS) document the mutual enhancement (or reduction) of intermolecular interactions and the aromaticity of H-bonded substrates. H-bonding interactions that increase cyclic 4n + 2 π-electron delocalization boost aromaticity. Conversely, such interactions are weakened when aromaticity is decreased as a result of more localized quinoidal π character. Representative examples of the tautomeric equilibria of π-conjugated heterocyclic compounds in protic solvents and other H-bonding environments also illustrate such H-bonding/aromaticity interplay.
Co-reporter:Guillaume Berionni, Judy I-Chia Wu, and Paul v. R. Schleyer
Organic Letters 2014 Volume 16(Issue 23) pp:6116-6119
Publication Date(Web):November 19, 2014
DOI:10.1021/ol5029699
The aromatic character of fused polycyclic systems varies with the nature of their annulated rings. Computed extra cyclic resonance energies (ECREs) reveal that the central six membered rings (6MRs) of the heterocyclic fused congeners 1–5 are “[6]radialene-like”, but that the central 6MRs of triphenylene 9, coronene 10, and isocoronene 11 are “benzene-like.” Comparisons with geometric (harmonic oscillator model of aromaticity, HOMA) and magnetic (nucleus independent chemical shifts, NICS) criteria illustrate the multifaceted nature of aromaticity in 1–11.
Co-reporter:Israel Fernández, Judy I. Wu, and Paul von Ragué Schleyer
Organic Letters 2013 Volume 15(Issue 12) pp:2990-2993
Publication Date(Web):May 31, 2013
DOI:10.1021/ol401154r
Computed aromatic stabilization energies (ASEs) and dissected nucleus independent chemical shifts (NICSπzz) quantify the effect of hyperconjugation on the (anti)aromaticities of the planar conformations of three, five, seven, and nine membered (CnHn)CR2 (R = H, SiH3, F) rings. CH2 and especially C(SiH3)2 groups supply two “pseudo” π electrons hyperconjugatively along with the olefinic π electrons in the ring, whereas a CF2 group acts like a partially vacant p orbital. Following the Hückel rule, compounds with 4n+2 (or 4n) pseudo π electrons are “hyperconjugatively” aromatic (or antiaromatic).
Co-reporter:Judy I. Wu ; Israel Fernández ;Paul v. R. Schleyer
Journal of the American Chemical Society 2012 Volume 135(Issue 1) pp:315-321
Publication Date(Web):December 3, 2012
DOI:10.1021/ja309434t
Like the larger nonplanar Möbius rings, porphyrinoid aromaticity is not due primarily to the macrocyclic π conjugation of the corresponding annulene perimeters. The block-localized wave function (BLW)-derived aromatic stabilization energies (ASE) of several porphyrinoids reveal that, on a per atom basis, the appended 6π electron heterocycles of porphyrinoids confer aromaticity much more effectively than the macrocyclic 4n+2 π electron conjugations. There is no direct relationship between thermochemical stability of porphyrinoids and their macrocyclic 4n or 4n+2 π electron counts. Porphyrinoids having an “antiaromatic” macrocyclic 4n+2 π electron conjugation pathway (e.g., 4) as well as those having no macrocyclic conjugation (e.g., 9) can be stabilized by aromaticity. Computed nucleus independent chemical shifts (NICS) and the anisotropy of the induced current density (ACID) disclose the intricate local versus macrocyclic circulation interplay for several porphyrinoids.