Co-reporter:Seth B. Herzon and Christopher D. Vanderwal
Chemical Reviews September 27, 2017 Volume 117(Issue 18) pp:11649-11649
Publication Date(Web):September 27, 2017
DOI:10.1021/acs.chemrev.7b00520
Co-reporter:Stephen K. Murphy, Mingshuo Zeng, and Seth B. Herzon
Organic Letters September 15, 2017 Volume 19(Issue 18) pp:
Publication Date(Web):August 31, 2017
DOI:10.1021/acs.orglett.7b02476
An improved synthesis of an eneimide, which is a useful precursor to pleuromutilin-based antibiotics, is reported. This synthesis proceeds in six steps and 17% overall yield (27% based on recovery of a key hydrindenone intermediate) and requires two fewer chromatography steps and five fewer days of reaction time than the previously reported route. The use of expensive, acutely toxic, and precious metal reagents or catalysts has been minimized.
Co-reporter:Alan R. Healy and Seth B. Herzon
Journal of the American Chemical Society October 25, 2017 Volume 139(Issue 42) pp:14817-14817
Publication Date(Web):September 26, 2017
DOI:10.1021/jacs.7b07807
A significant challenge toward studies of the human microbiota involves establishing causal links between bacterial metabolites and human health and disease states. Certain strains of commensal Escherichia coli harbor the 54-kb clb gene cluster which codes for small molecules named precolibactins and colibactins. Several studies suggest colibactins are genotoxins and support a role for clb metabolites in colorectal cancer formation. Significant advances toward elucidating the structures and biosynthesis of the precolibactins and colibactins have been made using genetic approaches, but their full structures remain unknown. In this Perspective we describe recent synthetic efforts that have leveraged biosynthetic advances and shed light on the mechanism of action of clb metabolites. These studies indicate that deletion of the colibactin peptidase ClbP, a modification introduced to promote accumulation of precolibactins, leads to the production of non-genotoxic pyridone-based isolates derived from the diversion of linear biosynthetic intermediates toward alternative cyclization pathways. Furthermore, these studies suggest the active genotoxins (colibactins) are unsaturated imines that are potent DNA damaging agents, thereby confirming an earlier mechanism of action hypothesis. Although these imines have very recently been detected in bacterial extracts, they have to date confounded isolation. As the power of “meta-omics” approaches to natural products discovery further advance, we anticipate that chemical synthetic and biosynthetic studies will become increasingly interdependent.
Co-reporter:Herman Nikolayevskiy;Maung Kyaw Moe Tun;Paul R. Rablen;Choukri Ben Mamoun
Chemical Science (2010-Present) 2017 vol. 8(Issue 7) pp:4867-4871
Publication Date(Web):2017/06/26
DOI:10.1039/C7SC01127J
Ocimicide A1 (1) and the semisynthetic derivative ocimicide A2 (2) are highly potent antimalarial agents efficacious against chloroquine-sensitive and -resistant Plasmodium falciparum strains with IC50 values in the nanomolar and picomolar range, respectively. Members of this family have demonstrated radical cure in rhesus monkeys, without detectable toxicity, but their structure–function relationships and mechanism of action are unknown. Herein we describe a twelve-step synthesis of an advanced N-acylated pentacyclic precursor to the proposed structure of 1 (11% overall yield). Instability and poor P. falciparum growth inhibition of the corresponding free donor–acceptor cyclopropylamine, and large discrepancies between reported and both experimental and DFT-calculated 13C chemical shifts and coupling constants, suggest that substantial revision of the proposed structures may be necessary.
Co-reporter:Mingshuo Zeng;Stephen K. Murphy
Science 2017 Volume 356(Issue 6341) pp:
Publication Date(Web):
DOI:10.1126/science.aan0003
A versatile synthesis of pleuromutilin
Synthetic flexibility is crucial for antibiotic development, because numerous subtle
structural variations can contribute to combating resistant strains. A derivative of the
fungal natural product pleuromutilin was approved a decade ago for treatment of
Gram-positive bacterial skin infections; recent efforts to tune the structure for
activity against Gram-negative bacteria have focused on the stereochemistry at a
particular carbon center. Murphy et al. present a synthetic route to
pleuromutilin that allows the configurations in that segment of the molecule to be
varied, offering a distinct path for structural optimization.
Science, this issue p. 956
Co-reporter:Mengzhao Xue and Seth B. Herzon
Journal of the American Chemical Society 2016 Volume 138(Issue 48) pp:15559-15562
Publication Date(Web):November 14, 2016
DOI:10.1021/jacs.6b09657
(−)-Lomaiviticin A (1) is a C2-symmetric cytotoxin that contains two diazofluorene functional groups and which induces double-strand breaks (DSBs) in DNA. Evidence suggests DNA cleavage is initiated by hydrogen atom abstraction from the deoxyribose backbone. Here we demonstrate the formation of the vinyl radicals 1· and 2· from 1 by 1,7-addition of thiols to the diazofluorenes. These radicals can affect hydrogen atom abstraction from methanol and acetone. The first addition of thiol to 1 proceeds at a much greater rate than the second. The diazosulfide 5 formed en route to 1· has been detected at −50 °C and undergoes decomposition to 1· with a half-life of 110 min at −20 °C under air. These data, which constitute the first direct evidence for the generation of 1· and 2· from 1, provide insights into the mechanism of DNA cleavage by 1.
Co-reporter:Alan R. Healy, Herman Nikolayevskiy, Jaymin R. Patel, Jason M. Crawford, and Seth B. Herzon
Journal of the American Chemical Society 2016 Volume 138(Issue 48) pp:15563-15570
Publication Date(Web):November 11, 2016
DOI:10.1021/jacs.6b10354
Co-reporter:Alan R. Healy; Maria I. Vizcaino; Jason M. Crawford
Journal of the American Chemical Society 2016 Volume 138(Issue 16) pp:5426-5432
Publication Date(Web):March 30, 2016
DOI:10.1021/jacs.6b02276
The colibactins are hybrid polyketide–nonribosomal peptide natural products produced by certain strains of commensal and extraintestinal pathogenic Escherichia coli. The metabolites are encoded by the clb gene cluster as prodrugs termed precolibactins. clb+ E. coli induce DNA double-strand breaks in mammalian cells in vitro and in vivo and are found in 55–67% of colorectal cancer patients, suggesting that mature colibactins could initiate tumorigenesis. However, elucidation of their structures has been an arduous task as the metabolites are obtained in vanishingly small quantities (μg/L) from bacterial cultures and are believed to be unstable. Herein we describe a flexible and convergent synthetic route to prepare advanced precolibactins and derivatives. The synthesis proceeds by late-stage union of two complex precursors (e.g., 28 + 17 → 29a, 90%) followed by a base-induced double dehydrative cascade reaction to form two rings of the targets (e.g., 29a → 30a, 79%). The sequence has provided quantities of advanced candidate precolibactins that exceed those obtained by fermentation, and is envisioned to be readily scaled. These studies have guided a structural revision of the predicted metabolite precolibactin A (from 5a or 5b to 7) and have confirmed the structures of the isolated metabolites precolibactins B (3) and C (6). Synthetic precolibactin C (6) was converted to N-myristoyl-d-asparagine and its corresponding colibactin by colibactin peptidase ClbP. The synthetic strategy outlined herein will facilitate mechanism of action and structure–function studies of these fascinating metabolites, and is envisioned to accommodate the synthesis of additional (pre)colibactins as they are isolated.
Co-reporter:Xiaoshen Ma
Journal of the American Chemical Society 2016 Volume 138(Issue 28) pp:8718-8721
Publication Date(Web):July 6, 2016
DOI:10.1021/jacs.6b05271
A general method for the hydropyridylation of unactivated alkenes is described. The transformation connects metal-mediated hydrogen atom transfer to alkenes and Minisci addition reactions. The reaction proceeds under mild conditions with high site-selectivities and allows for the construction of tertiary and quaternary centers from simple alkene starting materials.
Co-reporter:Yulia V. Surovtseva; Vikram Jairam; Ahmed F. Salem; Ranjini K. Sundaram; Ranjit S. Bindra
Journal of the American Chemical Society 2016 Volume 138(Issue 11) pp:3844-3855
Publication Date(Web):February 29, 2016
DOI:10.1021/jacs.6b00162
Small-molecule inhibitors of DNA repair pathways are being intensively investigated as primary and adjuvant chemotherapies. We report the discovery that cardiac glycosides, natural products in clinical use for the treatment of heart failure and atrial arrhythmia, are potent inhibitors of DNA double-strand break (DSB) repair. Our data suggest that cardiac glycosides interact with phosphorylated mediator of DNA damage checkpoint protein 1 (phospho-MDC1) or E3 ubiquitin–protein ligase ring finger protein 8 (RNF8), two factors involved in DSB repair, and inhibit the retention of p53 binding protein 1 (53BP1) at the site of DSBs. These observations provide an explanation for the anticancer activity of this class of compounds, which has remained poorly understood for decades, and provide guidance for their clinical applications. This discovery was enabled by the development of the first high-throughput unbiased cellular assay to identify new small-molecule inhibitors of DSB repair. Our assay is based on the fully automated, time-resolved quantification of phospho-SER139-H2AX (γH2AX) and 53BP1 foci, two factors involved in the DNA damage response network, in cells treated with small molecules and ionizing radiation (IR). This primary assay is supplemented by robust secondary assays that establish lead compound potencies and provide further insights into their mechanisms of action. Although the cardiac glycosides were identified in an evaluation of 2366 small molecules, the assay is envisioned to be adaptable to larger compound libraries. The assay is shown to be compatible with small-molecule DNA cleaving agents, such as bleomycin, neocarzinostatin chromophore, and lomaiviticin A, in place of IR.
Co-reporter:Stephen K. Murphy, Mingshuo Zeng, and Seth B. Herzon
Organic Letters 2016 Volume 18(Issue 19) pp:4880-4883
Publication Date(Web):September 27, 2016
DOI:10.1021/acs.orglett.6b02320
A general strategy for conjugate addition–C-acylation that enables the synthesis of enantioenriched β-dicarbonyl compounds is described. A novel method for derivatizing these adducts by stereo- and site-selective zinc-catalyzed addition of alkyllithium reagents is also reported. These reactions can be performed in tandem to achieve an enantio- and diastereoselective four-component coupling. The in situ generation of weakly basic lithium zincate species is central to the success of all three transformations.
Co-reporter:Christina M. Woo;Zhenwu Li;Eric K. Paulson
PNAS 2016 113 (11 ) pp:2851-2856
Publication Date(Web):2016-03-15
DOI:10.1073/pnas.1519846113
(–)-Lomaiviticin A (1) is a complex antiproliferative metabolite that inhibits the growth of many cultured cancer cell lines
at low nanomolar–picomolar concentrations. (–)-Lomaiviticin A (1) possesses a C2-symmetric structure that contains two unusual diazotetrahydrobenzo[b]fluorene (diazofluorene) functional groups. Nucleophilic activation of each diazofluorene within 1 produces vinyl radical
intermediates that affect hydrogen atom abstraction from DNA, leading to the formation of DNA double-strand breaks (DSBs).
Certain DNA DSB repair-deficient cell lines are sensitized toward 1, and 1 is under evaluation in preclinical models of these
tumor types. However, the mode of binding of 1 to DNA had not been determined. Here we elucidate the structure of a 1:1 complex
between 1 and the duplex d(GCTATAGC)2 by NMR spectroscopy and computational modeling. Unexpectedly, we show that both diazofluorene residues of 1 penetrate the
duplex. This binding disrupts base pairing leading to ejection of the central AT bases, while placing the proreactive centers
of 1 in close proximity to each strand. DNA binding may also enhance the reactivity of 1 toward nucleophilic activation through
steric compression and conformational restriction (an example of shape-dependent catalysis). This study provides a structural
basis for the DNA cleavage activity of 1, will guide the design of synthetic DNA-activated DNA cleavage agents, and underscores
the utility of natural products to reveal novel modes of small molecule–DNA association.
Co-reporter:Xiaoshen Ma and Seth B. Herzon
The Journal of Organic Chemistry 2016 Volume 81(Issue 19) pp:8673-8695
Publication Date(Web):September 6, 2016
DOI:10.1021/acs.joc.6b01709
Cobalt bis(acetylacetonate) is shown to mediate hydrogen atom transfer to a broad range of functionalized alkenes; in situ oxidation of the resulting alkylradical intermediates, followed by hydrolysis, provides expedient access to ketones and esters. By modification of the alcohol solvent, different alkyl ester products may be obtained. The method is compatible with a number of functional groups including alkenyl halides, sulfides, triflates, and phosphonates and provides a mild and practical alternative to the Tamao–Fleming oxidation of vinylsilanes and the Arndt–Eistert homologation.
Co-reporter:Laureen C. Colis, Seth B. Herzon
Bioorganic & Medicinal Chemistry Letters 2016 26(13) pp: 3122-3126
Publication Date(Web):1 July 2016
DOI:10.1016/j.bmcl.2016.04.090
(−)-Lomaiviticin A (1) is a cytotoxic bacterial metabolite that induces double-strand breaks in DNA. Here we show that the cytotoxicity of (−)-lomaiviticin A (1) is synergistically potentiated in the presence of VE-821 (7), an inhibitor of ataxia telangiectasia and Rad3-related protein (ATR). While 0.5 nM 1 or 10 μM 7 alone are non-lethal to K562 cells, co-incubation of the two leads to high levels of cell kill (81% and 94% after 24 and 48 h, respectively). Mechanistic data indicate that cells treated with 1 and 7 suffer extensive DNA double-strand breaks and apoptosis. These data suggest combinations of 1 and 7 may be a valuable chemotherapeutic strategy.K562 cells treated with 500 pM (−)-lomaiviticin A and 10 μM of the ATR inhibitor VE-821 display pan-γH2AX staining and undergo apoptosis.Download high-res image (150KB)Download full-size image
Co-reporter:Laureen C. Colis; Denise C. Hegan; Miho Kaneko; Peter M. Glazer
Journal of the American Chemical Society 2015 Volume 137(Issue 17) pp:5741-5747
Publication Date(Web):April 7, 2015
DOI:10.1021/ja513117p
(−)-Lomaiviticin A (1) and the monomeric lomaiviticin aglycon [aka: (−)-MK7-206, (3)] are cytotoxic agents that induce double-strand breaks (DSBs) in DNA. Here we elucidate the cellular responses to these agents and identify synthetic lethal interactions with specific DNA repair factors. Toward this end, we first characterized the kinetics of DNA damage by 1 and 3 in human chronic myelogenous leukemia (K562) cells. DSBs are rapidly induced by 3, reaching a maximum at 15 min post addition and are resolved within 4 h. By comparison, DSB production by 1 requires 2–4 h to achieve maximal values and >8 h to achieve resolution. As evidenced by an alkaline comet unwinding assay, 3 induces extensive DNA damage, suggesting that the observed DSBs arise from closely spaced single-strand breaks (SSBs). Both 1 and 3 induce ataxia telangiectasia mutated- (ATM-) and DNA-dependent protein kinase- (DNA-PK-) dependent production of phospho-SER139-histone H2AX (γH2AX) and generation of p53 binding protein 1 (53BP1) foci in K562 cells within 1 h of exposure, which is indicative of activation of nonhomologous end joining (NHEJ) and homologous recombination (HR) repair. Both compounds also lead to ataxia telangiectasia and Rad3-related- (ATR-) dependent production of γH2AX at later time points (6 h post addition), which is indicative of replication stress. 3 is also shown to induce apoptosis. In accord with these data, 1 and 3 were found to be synthetic lethal with certain mutations in DNA DSB repair. 1 potently inhibits the growth of breast cancer type 2, early onset- (BRCA2-) deficient V79 Chinese hamster lung fibroblast cell line derivative (VC8), and phosphatase and tensin homologue deleted on chromosome ten- (PTEN-) deficient human glioblastoma (U251) cell lines, with LC50 values of 1.5 ± 0.5 and 2.0 ± 0.6 pM, respectively, and selectivities of >11.6 versus the isogenic cell lines transfected with and expressing functional BRCA2 and PTEN genes. 3 inhibits the growth of the same cell lines with LC50 values of 6.0 ± 0.5 and 11 ± 4 nM and selectivities of 84 and 5.1, for the BRCA2 and PTEN mutants, respectively. These data argue for the evaluation of these agents as treatments for tumors that are deficient in BRCA2 and PTEN, among other DSB repair factors.
Co-reporter:Xiaoshen Ma and Seth B. Herzon
Chemical Science 2015 vol. 6(Issue 11) pp:6250-6255
Publication Date(Web):21 Aug 2015
DOI:10.1039/C5SC02476E
Classical methods for alkene hydrogenation typically reduce less-substituted or more-strained alkenes, or those in proximity to a directing group, most rapidly. Here we describe a cobalt-mediated hydrogenation protocol that provides complementary selectivities in the reduction of several classes of olefins and alkynes. The selectivity of this reduction derives from a hydrogen atom transfer mechanism, which favors the generation of the more stable alkylradical intermediate. We also report the first alkene hydrobromination, hydroiodination, and hydroselenylation by a hydrogen atom transfer process.
Co-reporter:Roman Kats-Kagan and Seth B. Herzon
Organic Letters 2015 Volume 17(Issue 8) pp:2030-2033
Publication Date(Web):April 3, 2015
DOI:10.1021/acs.orglett.5b00841
A simple and general method for the synthesis of highly substituted α-tropolone ethers that allows rapid access to the bis(tropolone) core of the antiproliferative metabolites (−)-gukulenins A and F (3, 4) is described. The reaction proceeds by thermolytic opening of gem-dibromobicyclo[4.1.0]heptane intermediates, which are readily accessed from simple starting materials. Mechanistic studies suggest the reaction proceeds via an autocatalytic process mediated by methyl hypobromite. This synthetic sequence allows access to a broad array of highly substituted α-tropolones.
Co-reporter:Matthew Burk, Nolan Wilson, Seth B. Herzon
Tetrahedron Letters 2015 Volume 56(Issue 23) pp:3231-3234
Publication Date(Web):3 June 2015
DOI:10.1016/j.tetlet.2014.12.073
The synthesis of 1-O-acetyl-3-O-(4-methoxybenzyl)-4-N-(9-fluorenylmethoxycarbonyl)-4-N-methyl-l-pyrrolosamine (7), which constitutes a protected form of the N,N-dimethyl-l-pyrrolosamine residues found within the antiproliferative bacterial metabolites (−)-lomaiviticins A and B (1 and 2, respectively), is reported. The synthetic route to 7 proceeds in eight steps and 13% overall yield from (E)-crotyl alcohol. The protected carbohydrate 7 is envisioned to be a useful derivative for syntheses of 1 and 2.
Co-reporter:Mingshuo Zeng and Seth B. Herzon
The Journal of Organic Chemistry 2015 Volume 80(Issue 17) pp:8604-8618
Publication Date(Web):July 23, 2015
DOI:10.1021/acs.joc.5b01220
The half-sandwich ruthenium complexes 1–3 activate terminal alkynes toward anti-Markovnikov hydration and reductive hydration under mild conditions. These reactions are believed to proceed via addition of water to metal vinylidene intermediates (4). The functionalization of propargylic alcohols by metal vinylidene pathways is challenging owing to decomposition of the starting material and catalytic intermediates. Here we show that catalyst 2 can be employed to convert propargylic alcohols to 1,3-diols in high yield and with retention of stereochemistry at the propargylic position. The method is also amenable to propargylic amine derivatives, thereby establishing a route to enantioenriched 1,3-amino alcohol products. We also report the development of formal anti-Markovnikov reductive amination and oxidative hydration reactions to access linear amines and carboxylic acids, respectively, from terminal alkynes. This chemistry expands the scope of products that can be prepared from terminal alkynes by practical and high-yielding metal-catalyzed methods.
Co-reporter:Mingshuo Zeng ; Le Li
Journal of the American Chemical Society 2014 Volume 136(Issue 19) pp:7058-7067
Publication Date(Web):April 4, 2014
DOI:10.1021/ja501738a
The conversion of terminal alkynes to functionalized products by the direct addition of heteroatom-based nucleophiles is an important aim in catalysis. We report the design, synthesis, and mechanistic studies of the half-sandwich ruthenium complex 12, which is a highly active catalyst for the anti-Markovnikov reductive hydration of alkynes. The key design element of 12 involves a tridentate nitrogen-based ligand that contains a hemilabile 3-(dimethylamino)propyl substituent. Under neutral conditions, the dimethylamino substituent coordinates to the ruthenium center to generate an air-stable, 18-electron, κ3-complex. Mechanistic studies show that the dimethylamino substituent is partially dissociated from the ruthenium center (by protonation) in the reaction media, thereby generating a vacant coordination site for catalysis. These studies also show that this substituent increases hydrogenation activity by promoting activation of the reductant. At least three catalytic cycles, involving the decarboxylation of formic acid, hydration of the alkyne, and hydrogenation of the intermediate aldehyde, operate concurrently in reactions mediated by 12. A wide array of terminal alkynes are efficiently processed to linear alcohols using as little as 2 mol % of 12 at ambient temperature, and the complex 12 is stable for at least two weeks under air. The studies outlined herein establish 12 as the most active and practical catalyst for anti-Markovnikov reductive hydration discovered to date, define the structural parameters of 12 underlying its activity and stability, and delineate design strategies for synthesis of other multifunctional catalysts.
Co-reporter:Sandra M. King ; Xiaoshen Ma
Journal of the American Chemical Society 2014 Volume 136(Issue 19) pp:6884-6887
Publication Date(Web):April 30, 2014
DOI:10.1021/ja502885c
A general method for the selective hydrogenation of alkenyl halides to alkyl halides is described. Fluoro, chloro, bromo, iodo, and gem-dihaloalkenes are viable substrates for the transformation. The selectivity of the hydrogenation is consistent with reduction by a hydrogen atom transfer pathway.
Co-reporter:Miho Kaneko and Seth B. Herzon
Organic Letters 2014 Volume 16(Issue 10) pp:2776-2779
Publication Date(Web):May 1, 2014
DOI:10.1021/ol501101f
It is shown that 2-deoxy- and 2,6-dideoxyglycosyl bromides can be prepared in high yield (72–94%) and engaged in glycosylation reactions with β:α selectivities ≥6:1. Yields of product are 44–90%. Fully armed 2-deoxyglycoside donors are viable, while 2,6-dideoxyglycosides require one electron-withdrawing substituent for high efficiency and β-selectivity. Equatorial C-3 ester protecting groups decrease β-selectivity, and donors bearing an axial C-3 substituent are not suitable. The method is compatible with azide-containing donors and acid-sensitive functional groups.
Co-reporter:Dr. Christina M. Woo;Dr. Nihar Ranjan;Dr. Dev P. Arya;Dr. Seth B. Herzon
Angewandte Chemie International Edition 2014 Volume 53( Issue 35) pp:9325-9328
Publication Date(Web):
DOI:10.1002/anie.201404137
Abstract
The lomaiviticins and kinamycins are complex DNA damaging natural products that contain a diazofluorene functional group. Herein, we elucidate the influence of skeleton structure, ring and chain isomerization, D-ring oxidation state, and naphthoquinone substitution on DNA binding and damaging activity. We show that the electrophilicity of the diazofluorene appears to be a significant determinant of DNA damaging activity. These studies identify the monomeric diazofluorene 11 as a potent DNA cleavage agent in tissue culture. The simpler structure of 11 relative to the natural products establishes it as a useful lead for translational studies.
Co-reporter:Dr. Christina M. Woo;Dr. Nihar Ranjan;Dr. Dev P. Arya;Dr. Seth B. Herzon
Angewandte Chemie 2014 Volume 126( Issue 35) pp:9479-9482
Publication Date(Web):
DOI:10.1002/ange.201404137
Abstract
The lomaiviticins and kinamycins are complex DNA damaging natural products that contain a diazofluorene functional group. Herein, we elucidate the influence of skeleton structure, ring and chain isomerization, D-ring oxidation state, and naphthoquinone substitution on DNA binding and damaging activity. We show that the electrophilicity of the diazofluorene appears to be a significant determinant of DNA damaging activity. These studies identify the monomeric diazofluorene 11 as a potent DNA cleavage agent in tissue culture. The simpler structure of 11 relative to the natural products establishes it as a useful lead for translational studies.
Co-reporter:Sandra M. King and Seth B. Herzon
The Journal of Organic Chemistry 2014 Volume 79(Issue 19) pp:8937-8947
Publication Date(Web):August 7, 2014
DOI:10.1021/jo501516x
Functional group taxonomy provides a powerful conceptual framework to classify and predict the chemical reactivity of molecular structures. These principals are most effective in monofunctional settings, wherein individual functional groups can be analyzed without complications. In more complex settings, the predictive value of these analyses decreases as alternative reaction pathways, promoted by neighboring substituents and aggregate molecular properties, emerge. We refer to this phenomenon as substrate-modified functional group reactivity. In this Perspective, we explain how substrate-modified functional group reactivity molded our synthetic routes to the hasubanan and acutumine alkaloids. These investigations underscore the potential for discovery and insight that can only be gained by studying the reactivity of complex multifunctional structures.
Co-reporter:Christina M. Woo, Shivajirao L. Gholap, and Seth B. Herzon
Journal of Natural Products 2013 Volume 76(Issue 7) pp:1238-1241
Publication Date(Web):June 26, 2013
DOI:10.1021/np400355h
The dimeric diazofluorenes known as the lomaiviticins are produced by the marine bacterium Salinispora pacifica DPJ-0019. Investigation of the fermentation broth of DPJ-0019 has yielded the first monomeric benzo[b]fluorene isolated from this species, (−)-homoseongomycin (13). (−)-Homoseongomycin (13) is related to the known natural product seongomycin (10), which is co-produced with the monomeric diazofluorenes known as the kinamycins. We describe the synthesis of the isotopically labeled derivative homoseongomycin-d5 (14), via the intermediacy of the diazofluorene “prelomaiviticin-d5” (12). Our studies establish that (−)-homoseongomycin (13) may be derived from prelomaiviticin (11) and suggest that 13 and 10 are shunt or detoxification metabolites in lomaiviticin and kinamycin biosynthesis, respectively.
Co-reporter:Sra M. King;Nicholas A. Calra ;Dr. Seth B. Herzon
Angewandte Chemie 2013 Volume 125( Issue 13) pp:3730-3733
Publication Date(Web):
DOI:10.1002/ange.201210076
Co-reporter:Dr. Jianbin Zheng;Dr. Kaveri Balan Urkalan;Dr. Seth B. Herzon
Angewandte Chemie 2013 Volume 125( Issue 23) pp:6184-6187
Publication Date(Web):
DOI:10.1002/ange.201301264
Co-reporter:Nicholas A. Calandra, Sandra M. King, and Seth B. Herzon
The Journal of Organic Chemistry 2013 Volume 78(Issue 20) pp:10031-10057
Publication Date(Web):September 13, 2013
DOI:10.1021/jo401889b
We describe a general strategy to prepare the hasubanan and acutumine alkaloids, a large family of botanical natural products that display antitumor, antiviral, and memory-enhancing effects. The absolute stereochemistry of the targets is established by an enantioselective Diels–Alder reaction between 5-(trimethylsilyl)cyclopentadiene (36) and 5-(2-azidoethyl)-2,3-dimethoxybenzoquinone (24). The Diels–Alder adduct 38 is transformed to the tetracyclic imine 39 by a Staudinger reduction–aza-Wittig sequence. The latter serves as a universal precursor to the targets. Key carbon–carbon bond constructions include highly diastereoselective acetylide additions to the N-methyliminium ion derived from 39 and Friedel–Crafts and Hosomi–Sakurai cyclizations to construct the carbocyclic skeleton of the targets. Initially, this strategy was applied to the syntheses of (−)-acutumine (4), (−)-dechloroacutumine (5), and four hasubanan alkaloids (1, 2, 3, and 8). Herein, the synthetic route is adapted to the syntheses of six additional hasubanan alkaloids (12, 13, 14, 15, 18, and 19). The strategic advantage of 5-(trimethylsilyl)cyclopentadiene Diels–Alder adducts is demonstrated by site-selective functionalization of distal carbon–carbon π-bonds in the presence of an otherwise reactive norbornene substructure. Evaluation of the antiproliferative properties of the synthetic metabolites revealed that four hasubanan alkaloids are submicromolar inhibitors of the N87 cell line.
Co-reporter:Sra M. King;Nicholas A. Calra ;Dr. Seth B. Herzon
Angewandte Chemie International Edition 2013 Volume 52( Issue 13) pp:3642-3645
Publication Date(Web):
DOI:10.1002/anie.201210076
Co-reporter:Dr. Jianbin Zheng;Dr. Kaveri Balan Urkalan;Dr. Seth B. Herzon
Angewandte Chemie International Edition 2013 Volume 52( Issue 23) pp:6068-6071
Publication Date(Web):
DOI:10.1002/anie.201301264
Co-reporter:Matthew J. Mitcheltree, Zef A. Konst, Seth B. Herzon
Tetrahedron 2013 69(27–28) pp: 5634-5639
Publication Date(Web):
DOI:10.1016/j.tet.2013.04.027
Co-reporter:YuMeng You ; Aaron Bloomfield ; Jian Liu ; Li Fu ; Seth B. Herzon ;Elsa C. Y. Yan
Journal of the American Chemical Society 2012 Volume 134(Issue 9) pp:4264-4268
Publication Date(Web):February 17, 2012
DOI:10.1021/ja2104608
Emulsions are widely used in industrial and environmental remediation applications. The breaking and reformulation of emulsions, which occur during their use, lead to changes in their surface composition as well as their physical and chemical properties. Hence, a fundamental understanding of the transfer of surfactant molecules between emulsion particles is required for optimization of their applications. However, such an understanding remains elusive because of the lack of in situ and real-time surface-specific techniques. To address this, we designed and synthesized the surfactant probe molecules MG-butyl-1 (2) and MG-octyl-1 (3), which contain an n-butyl and an n-octyl chain, respectively, and a charged headgroup similar to that in malachite green (MG, 1). MG is known to be effective in generating second harmonic generation (SHG) signals when adsorbed onto surfaces of colloidal microparticles. Making use of the coherent nature of SHG, we monitored in real-time the transfer of 2 and 3 between oil-in-water emulsion particles with diameters of ∼220 nm. We found that 3 is transferred ∼600 times slower than 2, suggesting that an increase in the hydrophobic chain length decreases the transfer rate. Our results show that SHG combined with molecular design and synthesis of surfactant probe molecules can be used to measure the rate of surfactant transfer between emulsion particles. This method provides an experimental framework for examining the factors controlling the kinetics of surfactant transfer between emulsion particles, which cannot be readily investigated in situ and in real-time using conventional methods.
Co-reporter:Christina M. Woo ; Nina E. Beizer ; Jeffrey E. Janso
Journal of the American Chemical Society 2012 Volume 134(Issue 37) pp:15285-15288
Publication Date(Web):September 10, 2012
DOI:10.1021/ja3074984
We describe the isolation of (–)-lomaiviticins C–E (6–8), elucidation of the complete absolute and relative stereochemistry of (–)-lomaiviticin A (1), the synthetic conversion of (–)-lomaiviticin C (6) to (–)-lomaiviticin A (1), and the first evidence that the dimeric diazofluorene of (–)-lomaiviticin A (1) plays a defining and critical role in antiproliferative activity.
Co-reporter:Christina M. Woo ; Shivajirao L. Gholap ; Liang Lu ; Miho Kaneko ; Zhenwu Li ; P. C. Ravikumar
Journal of the American Chemical Society 2012 Volume 134(Issue 41) pp:17262-17273
Publication Date(Web):October 3, 2012
DOI:10.1021/ja307497h
The development of enantioselective synthetic routes to (−)-kinamycin F (9) and (−)-lomaiviticin aglycon (6) are described. The diazotetrahydrobenzo[b]fluorene (diazofluorene) functional group of the targets was prepared by fluoride-mediated coupling of a β-trimethylsilylmethyl-α,β-unsaturated ketone (38) with an oxidized naphthoquinone (19), palladium-catalyzed cyclization (39→37), and diazo transfer (37→53). The D-ring precursors 60 and 68 were prepared from m-cresol and 3-ethylphenol, respectively. Coupling of the β-trimethylsilylmethyl-α,β-unsaturated ketone 60 with the juglone derivative 61, cyclization, and diazo transfer provided the advanced diazofluorene 63, which was elaborated to (−)-kinamycin F (9) in three steps. The diazofluorene 87 was converted to the C2-symmetric lomaiviticin aglycon precursor 91 by enoxysilane formation and oxidative dimerization with manganese tris(hexafluoroacetylacetonate) (94, 26%). The stereochemical outcome in the coupling is attributed to the steric bias engendered by the mesityl acetal of 87 and contact ion pairing of the intermediates. The coupling product 91 was deprotected (tert-butylhydrogen peroxide, trifluoroacetic acid–dichloromethane) to form mixtures of the chain isomer of lomaiviticin aglycon 98 and the ring isomer 6. These mixtures converged on purification or standing to the ring isomer 6 (39–41% overall). The scope of the fluoride-mediated coupling process is delineated (nine products, average yield = 72%); a related enoxysilane quinonylation reaction is also described (10 products, average yield = 77%). We establish that dimeric diazofluorenes undergo hydrodediazotization 2-fold faster than related monomeric diazofluorenes. This enhanced reactivity may underlie the cytotoxic effects of (–)-lomaiviticin A (1). The simple diazofluorene 103 is a potent inhibitor of ovarian cancer stem cells (IC50 = 500 nM).
Co-reporter:Le Li
Journal of the American Chemical Society 2012 Volume 134(Issue 42) pp:17376-17379
Publication Date(Web):October 16, 2012
DOI:10.1021/ja307145e
The regioselective reductive hydration of terminal alkynes using two complementary dual catalytic systems is described. Branched or linear alcohols are obtained in 75–96% yield with ≥25:1 regioselectivity from the same starting materials. The method is compatible with terminal, di-, and trisubstituted alkenes. This reductive hydration constitutes a strategic surrogate to alkene oxyfunctionalization and may be of utility in multistep settings.
Co-reporter:Seann P. Mulcahy, Christina M. Woo, Weidong Ding, George A. Ellestad and Seth B. Herzon
Chemical Science 2012 vol. 3(Issue 4) pp:1070-1074
Publication Date(Web):22 Dec 2011
DOI:10.1039/C2SC00854H
The lomaiviticins (1 and 2) and kinamycins (3–5) are bacterial metabolites with potent antimicrobial and antiproliferative activities. Herein we establish that 1–5 are capable of generating electrophilic acylfulvene intermediates (6) under mildly reducing conditions. These acylfulvenes 6 are formed by a multistep process comprising two-electron reduction and loss of dinitrogen to form an ortho-quinone methide, followed by elimination. Based on these studies, the structure of the product formed from 1 in DNA-cleavage assays is proposed (26). We also show that the bis(hydroxynaphthoquinone) substructures of the lomaiviticins activate the metabolites toward reduction. Finally, based on COMPARE and time-dependent cell response profiling analyses, we show that kinamycin C (4) and the monomeric lomaiviticin aglycon (24) operate by a mechanism of action that is distinct from simple diazofluorenes, such as 23.
Co-reporter:Aaron J. Bloomfield and Seth B. Herzon
Organic Letters 2012 Volume 14(Issue 17) pp:4370-4373
Publication Date(Web):August 20, 2012
DOI:10.1021/ol301831k
We show that a broad range of aryl iodides are efficiently coupled with secondary phosphine oxides using 1 mol % of a catalyst formed in situ from tris(dibenzylideneacetone)dipalladium and Xantphos (1). Scalemic (S)-methylphenylphosphine oxide [(S)-2e] is shown to undergo arylation without detectable stereoerosion. The application of this method to the synthesis of novel P-chiral phosphines and PCP ligands is demonstrated.
Co-reporter:Maung Kyaw Moe Tun and Seth B. Herzon
The Journal of Organic Chemistry 2012 Volume 77(Issue 20) pp:9422-9425
Publication Date(Web):October 3, 2012
DOI:10.1021/jo3017956
A three-step synthesis of (R)-(+)-4-methylcyclohex-2-ene-1-one (1) from (R)-(+)-pulegone (3), proceeding in 44% overall yield, is described. The sequence comprises vinyl triflate formation, site-selective ozonolysis, and reduction. The route requires only one chromatographic purification and provides a convenient method to access multigram quantities of (R)-(+)-4-methylcyclohex-2-ene-1-one (1).
Co-reporter:Seth B. Herzon ; Liang Lu ; Christina M. Woo ;Shivajirao L. Gholap
Journal of the American Chemical Society 2011 Volume 133(Issue 19) pp:7260-7263
Publication Date(Web):January 31, 2011
DOI:10.1021/ja200034b
Lomaiviticins A and B are complex antitumor antibiotics that were isolated from a strain of Micromonospora. A confluence of several unusual structural features renders the lomaiviticins exceedingly challenging targets for chemical synthesis. We report an 11-step, enantioselective synthetic route to lomaiviticin aglycon. Our route proceeds by late-stage, stereoselective dimerization of two equivalent monomeric intermediates, a transformation that may share parallels with the natural products' biosyntheses. The route we describe is scalable and convergent, and it lays the foundation for determination of the mode of action of these natural products.
Co-reporter:Maung Kyaw Moe Tun, Daniel-Joachim Wüstmann and Seth B. Herzon
Chemical Science 2011 vol. 2(Issue 11) pp:2251-2253
Publication Date(Web):25 Aug 2011
DOI:10.1039/C1SC00455G
(−)-Huperzine A (1) is a tricyclic alkaloid that is produced in low yield by the Chinese herb Huperzia serrata. There is intense contemporary interest in clinical application of (−)-huperzine A (1) for treating neurodegenerative diseases and protecting against the lethal effects of chemical warfare agents, such as sarin and VX. We report a robust, scalable, and efficient synthesis of (−)-huperzine A (1) from (R)-4-methyl-cyclohex-2-ene-1-one (5). Our route proceeds in 35–45% overall yield, delivers (−)-huperzine A (1) in only eight steps from cyclohexenone 5, requires only three chromatographic purifications, and can provide gram quantities of the target. This route represents a critical, enabling advance toward detailed evaluation of (−)-huperzine A (1) in clinical settings.
Co-reporter:Dr. Seth B. Herzon;Nicholas A. Calra ;Sra M. King
Angewandte Chemie 2011 Volume 123( Issue 38) pp:9025-9028
Publication Date(Web):
DOI:10.1002/ange.201102226
Co-reporter:Dr. Seth B. Herzon;Nicholas A. Calra ;Sra M. King
Angewandte Chemie 2011 Volume 123( Issue 38) pp:
Publication Date(Web):
DOI:10.1002/ange.201105017
Co-reporter:Dr. Seth B. Herzon;Nicholas A. Calra ;Sra M. King
Angewandte Chemie International Edition 2011 Volume 50( Issue 38) pp:8863-8866
Publication Date(Web):
DOI:10.1002/anie.201102226
Co-reporter:Dr. Seth B. Herzon;Nicholas A. Calra ;Sra M. King
Angewandte Chemie International Edition 2011 Volume 50( Issue 38) pp:
Publication Date(Web):
DOI:10.1002/anie.201105017
Co-reporter:Christina M. Woo ; Liang Lu ; Shivajirao L. Gholap ; Devin R. Smith
Journal of the American Chemical Society 2010 Volume 132(Issue 8) pp:2540-2541
Publication Date(Web):February 8, 2010
DOI:10.1021/ja910769j
We describe a 12-step enantioselective synthetic route to the complex anticancer antimicrobial agent kinamycin F (3). Key to the success of the route was the development of a three-step sequence for construction of the diazonapthoquinone (diazofluorene, blue in structure 3) function of the natural product. This sequence comprises fluoride-mediated coupling of a β-(trimethylsilylmethyl)-cyclohexenone and halonapthoquinone, palladium-mediated cyclization to construct the tetracyclic scaffold of the natural product, and mild diazo-transfer to a complex cyclopentadiene to introduce the diazo function. Ortho-quinone methide intermediates, formed by reduction and loss of dinitrogen from 3, have been postulated to form in vivo, and our approach provides a straightforward synthetic pathway to such compounds.
Co-reporter:Aaron J. Bloomfield, Jack M. Qian, and Seth B. Herzon
Organometallics 2010 Volume 29(Issue 18) pp:4193-4195
Publication Date(Web):August 31, 2010
DOI:10.1021/om100571w
We report that in the presence of trifluoroacetic acid primary phosphines undergo efficient addition to aldehydes to form the corresponding secondary phosphine oxides in 47−97% yield. This transformation is compatible with aryl and alkyl phosphines, as well as a broad range of aldehydes, including formaldehyde. By using 1,5-dialdehydes as reaction partners, the addition provides a straightforward route to bis(phosphine oxides), which are difficult to prepare by alternative methods. In the presence of boron trifluoride diethyl etherate as reagent, benzophenone was shown to couple to phenylphosphine and cyclohexylphosphine in 92% and 72% yield, respectively. Twenty-three examples are presented.
Co-reporter:Shivajirao L. Gholap, Christina M. Woo, P. C. Ravikumar and Seth B. Herzon
Organic Letters 2009 Volume 11(Issue 19) pp:4322-4325
Publication Date(Web):August 31, 2009
DOI:10.1021/ol901710b
We describe two four-step sequences for conversion of the inexpensive reagent ethyl sorbate to either O-allyl-N,N-dimethyl-d-pyrrolosamine or O-allyl-l-oleandrose, protected forms of the 2,6-dideoxy sugar residues found in the complex bacterial metabolite lomaiviticin A. We also report a gram-scale synthesis of the highly-oxygenated cyclohexenone ring of this metabolite, and show this may be coupled with the aforementioned donors to form the bis(glycoside) 6. The longest linear sequence to 6 is nine steps.
Co-reporter:Maung Kyaw Moe Tun, Daniel-Joachim Wüstmann and Seth B. Herzon
Chemical Science (2010-Present) 2011 - vol. 2(Issue 11) pp:NaN2253-2253
Publication Date(Web):2011/08/25
DOI:10.1039/C1SC00455G
(−)-Huperzine A (1) is a tricyclic alkaloid that is produced in low yield by the Chinese herb Huperzia serrata. There is intense contemporary interest in clinical application of (−)-huperzine A (1) for treating neurodegenerative diseases and protecting against the lethal effects of chemical warfare agents, such as sarin and VX. We report a robust, scalable, and efficient synthesis of (−)-huperzine A (1) from (R)-4-methyl-cyclohex-2-ene-1-one (5). Our route proceeds in 35–45% overall yield, delivers (−)-huperzine A (1) in only eight steps from cyclohexenone 5, requires only three chromatographic purifications, and can provide gram quantities of the target. This route represents a critical, enabling advance toward detailed evaluation of (−)-huperzine A (1) in clinical settings.
Co-reporter:Herman Nikolayevskiy, Maung Kyaw Moe Tun, Paul R. Rablen, Choukri Ben Mamoun and Seth B. Herzon
Chemical Science (2010-Present) 2017 - vol. 8(Issue 7) pp:NaN4871-4871
Publication Date(Web):2017/05/04
DOI:10.1039/C7SC01127J
Ocimicide A1 (1) and the semisynthetic derivative ocimicide A2 (2) are highly potent antimalarial agents efficacious against chloroquine-sensitive and -resistant Plasmodium falciparum strains with IC50 values in the nanomolar and picomolar range, respectively. Members of this family have demonstrated radical cure in rhesus monkeys, without detectable toxicity, but their structure–function relationships and mechanism of action are unknown. Herein we describe a twelve-step synthesis of an advanced N-acylated pentacyclic precursor to the proposed structure of 1 (11% overall yield). Instability and poor P. falciparum growth inhibition of the corresponding free donor–acceptor cyclopropylamine, and large discrepancies between reported and both experimental and DFT-calculated 13C chemical shifts and coupling constants, suggest that substantial revision of the proposed structures may be necessary.
Co-reporter:Seann P. Mulcahy, Christina M. Woo, Weidong Ding, George A. Ellestad and Seth B. Herzon
Chemical Science (2010-Present) 2012 - vol. 3(Issue 4) pp:NaN1074-1074
Publication Date(Web):2011/12/22
DOI:10.1039/C2SC00854H
The lomaiviticins (1 and 2) and kinamycins (3–5) are bacterial metabolites with potent antimicrobial and antiproliferative activities. Herein we establish that 1–5 are capable of generating electrophilic acylfulvene intermediates (6) under mildly reducing conditions. These acylfulvenes 6 are formed by a multistep process comprising two-electron reduction and loss of dinitrogen to form an ortho-quinone methide, followed by elimination. Based on these studies, the structure of the product formed from 1 in DNA-cleavage assays is proposed (26). We also show that the bis(hydroxynaphthoquinone) substructures of the lomaiviticins activate the metabolites toward reduction. Finally, based on COMPARE and time-dependent cell response profiling analyses, we show that kinamycin C (4) and the monomeric lomaiviticin aglycon (24) operate by a mechanism of action that is distinct from simple diazofluorenes, such as 23.
Co-reporter:Xiaoshen Ma and Seth B. Herzon
Chemical Science (2010-Present) 2015 - vol. 6(Issue 11) pp:NaN6255-6255
Publication Date(Web):2015/08/21
DOI:10.1039/C5SC02476E
Classical methods for alkene hydrogenation typically reduce less-substituted or more-strained alkenes, or those in proximity to a directing group, most rapidly. Here we describe a cobalt-mediated hydrogenation protocol that provides complementary selectivities in the reduction of several classes of olefins and alkynes. The selectivity of this reduction derives from a hydrogen atom transfer mechanism, which favors the generation of the more stable alkylradical intermediate. We also report the first alkene hydrobromination, hydroiodination, and hydroselenylation by a hydrogen atom transfer process.
.jpg)
![2-Methyl-2-(4-(3-methyl-2-oxo-8-(quinolin-3-yl)-2,3-dihydro-1H-imidazo[4,5-c]quinolin-1-yl)phenyl)propanenitrile](http://img.cochemist.com/ccimg/915100/915019-65-7.png)
![2-Methyl-2-(4-(3-methyl-2-oxo-8-(quinolin-3-yl)-2,3-dihydro-1H-imidazo[4,5-c]quinolin-1-yl)phenyl)propanenitrile](http://img.cochemist.com/ccimg/915100/915019-65-7_b.png)
![1(2H)-Pyridinecarboxylic acid,4-[(diphenoxyphosphinyl)oxy]-3,6-dihydro-, 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/856700/856684-31-6.png)
![1(2H)-Pyridinecarboxylic acid,4-[(diphenoxyphosphinyl)oxy]-3,6-dihydro-, 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/856700/856684-31-6_b.png)
![3-Amino-6-[4-(methylsulfonyl)phenyl]-N-phenyl-2-pyrazinecarboxamide](http://img.cochemist.com/ccimg/1232500/1232410-49-9.png)
![3-Amino-6-[4-(methylsulfonyl)phenyl]-N-phenyl-2-pyrazinecarboxamide](http://img.cochemist.com/ccimg/1232500/1232410-49-9_b.png)



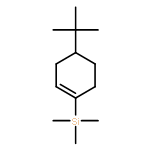
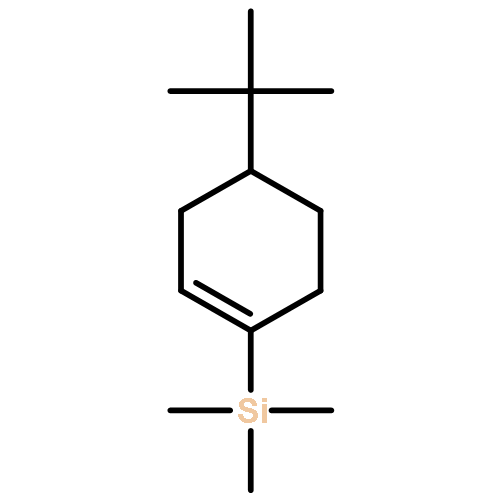
![Benzene, [[4-(1,1-dimethylethyl)-1-cyclohexen-1-yl]oxy]-](http://img.cochemist.com/ccimg/618900/618891-44-4.png)
![Benzene, [[4-(1,1-dimethylethyl)-1-cyclohexen-1-yl]oxy]-](http://img.cochemist.com/ccimg/618900/618891-44-4_b.png)




![Tert-butyl N-[(4-cyano-1,3-thiazol-2-yl)methyl]carbamate](http://img.cochemist.com/ccimg/251300/251294-64-1.png)
![Tert-butyl N-[(4-cyano-1,3-thiazol-2-yl)methyl]carbamate](http://img.cochemist.com/ccimg/251300/251294-64-1_b.png)

















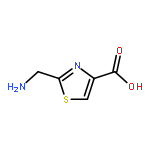
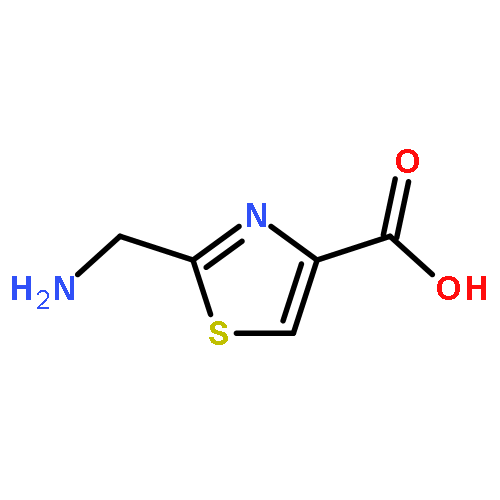
![2-[[(TERT-BUTOXYCARBONYL)AMINO]METHYL]THIAZOLE-4-CARBOXYLIC ACID](http://img.cochemist.com/ccimg/72000/71904-80-8.png)
![2-[[(TERT-BUTOXYCARBONYL)AMINO]METHYL]THIAZOLE-4-CARBOXYLIC ACID](http://img.cochemist.com/ccimg/72000/71904-80-8_b.png)















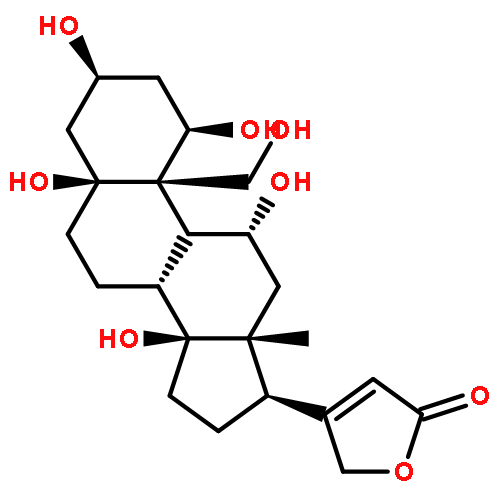
![Card-20(22)-enolide,3-[(O-b-D-glucopyranosyl-(1®4)-O-3-O-acetyl-2,6-dideoxy-b-D-ribo-hexopyranosyl-(1®4)-O-2,6-dideoxy-b-D-ribo-hexopyranosyl-(1®4)-2,6-dideoxy-b-D-ribo-hexopyranosyl)oxy]-12,14-dihydroxy-,(3b,5b,12b)-](http://img.cochemist.com/ccimg/17600/17575-22-3.png)
![Card-20(22)-enolide,3-[(O-b-D-glucopyranosyl-(1®4)-O-3-O-acetyl-2,6-dideoxy-b-D-ribo-hexopyranosyl-(1®4)-O-2,6-dideoxy-b-D-ribo-hexopyranosyl-(1®4)-2,6-dideoxy-b-D-ribo-hexopyranosyl)oxy]-12,14-dihydroxy-,(3b,5b,12b)-](http://img.cochemist.com/ccimg/17600/17575-22-3_b.png)











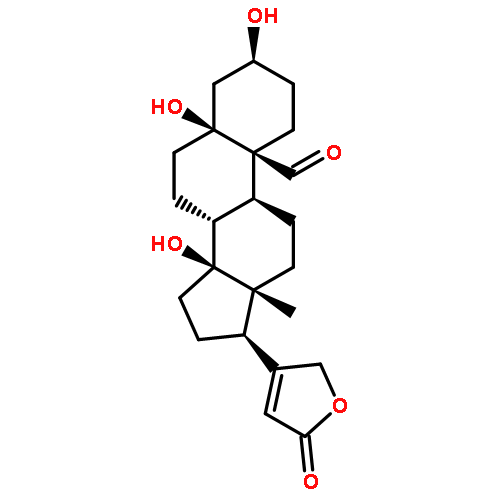
![Card-20(22)-enolide,3-[(6-deoxy-a-L-mannopyranosyl)oxy]-1,5,11,14,19-pentahydroxy-,(1b,3b,5b,11a)-](http://img.cochemist.com/ccimg/700/630-60-4.png)
![Card-20(22)-enolide,3-[(6-deoxy-a-L-mannopyranosyl)oxy]-1,5,11,14,19-pentahydroxy-,(1b,3b,5b,11a)-](http://img.cochemist.com/ccimg/700/630-60-4_b.png)


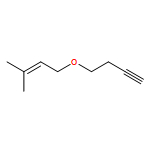
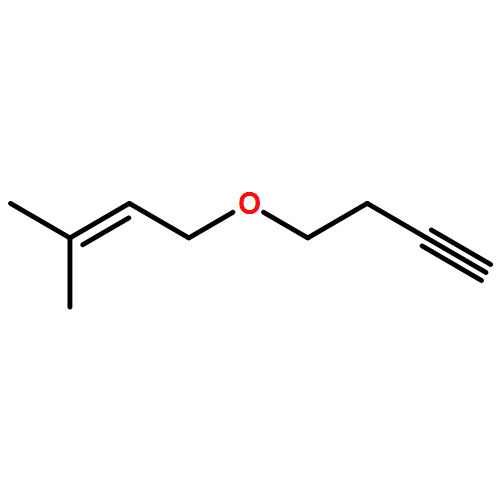


![1-BUTANOL, 4-[[(2E)-3-PHENYL-2-PROPENYL]OXY]-](http://img.cochemist.com/ccimg/700900/700865-83-4.png)
![1-BUTANOL, 4-[[(2E)-3-PHENYL-2-PROPENYL]OXY]-](http://img.cochemist.com/ccimg/700900/700865-83-4_b.png)
![Silane, trimethyl[[1-(3,4,5-trimethoxyphenyl)ethenyl]oxy]-](http://img.cochemist.com/ccimg/405600/405555-15-9.png)
![Silane, trimethyl[[1-(3,4,5-trimethoxyphenyl)ethenyl]oxy]-](http://img.cochemist.com/ccimg/405600/405555-15-9_b.png)


![1(2H)-Pyridinecarboxylic acid,3,6-dihydro-4-[[(trifluoromethyl)sulfonyl]oxy]-, phenylmethyl ester](http://img.cochemist.com/ccimg/287000/286961-24-8.png)
![1(2H)-Pyridinecarboxylic acid,3,6-dihydro-4-[[(trifluoromethyl)sulfonyl]oxy]-, phenylmethyl ester](http://img.cochemist.com/ccimg/287000/286961-24-8_b.png)




![Propanal, 2-[(4-methoxyphenyl)methoxy]-, (2R)-](http://img.cochemist.com/ccimg/218500/218435-12-2.png)
![Propanal, 2-[(4-methoxyphenyl)methoxy]-, (2R)-](http://img.cochemist.com/ccimg/218500/218435-12-2_b.png)











![9-[1-amino-4-({4-[(diaminomethylidene)amino]butoxy}carbonyl)-3,5,6,7-tetrahydropyrrolo[1,2-c]pyrimidin-3-yl]nonyl (7S)-7-heptyl-4-methyl-2,2a,5,7,8,8a-hexahydro-1H-5,6,8b-triazaacenaphthylene-3-carboxylate](http://img.cochemist.com/ccimg/161600/161503-23-7.png)
![9-[1-amino-4-({4-[(diaminomethylidene)amino]butoxy}carbonyl)-3,5,6,7-tetrahydropyrrolo[1,2-c]pyrimidin-3-yl]nonyl (7S)-7-heptyl-4-methyl-2,2a,5,7,8,8a-hexahydro-1H-5,6,8b-triazaacenaphthylene-3-carboxylate](http://img.cochemist.com/ccimg/161600/161503-23-7_b.png)


![Ferrocene,1-[(1R)-1-(dicyclohexylphosphino)ethyl]-2-(diphenylphosphino)-, (2R)-](http://img.cochemist.com/ccimg/155900/155806-35-2.png)
![Ferrocene,1-[(1R)-1-(dicyclohexylphosphino)ethyl]-2-(diphenylphosphino)-, (2R)-](http://img.cochemist.com/ccimg/155900/155806-35-2_b.png)





![2-CYCLOHEXEN-1-ONE, 4-[(4-METHOXYPHENYL)METHOXY]-, (S)-](http://img.cochemist.com/ccimg/118300/118207-44-6.png)
![2-CYCLOHEXEN-1-ONE, 4-[(4-METHOXYPHENYL)METHOXY]-, (S)-](http://img.cochemist.com/ccimg/118300/118207-44-6_b.png)
![L-arabino-Hex-1-enitol, 1,5-anhydro-2,6-dideoxy-4-O-[(1,1-dimethylethyl)dimethylsilyl]-, 3-benzoate](/data/chemimg/3739800/117452-73-0.png)
![L-arabino-Hex-1-enitol, 1,5-anhydro-2,6-dideoxy-4-O-[(1,1-dimethylethyl)dimethylsilyl]-, 3-benzoate](/data/chemimg/3739800/117452-73-0_b.png)
![Cyclohexanemethanol, α-[2-(trimethylsilyl)ethynyl]-](/data/chemimg/123900/112497-25-3.png)
![Cyclohexanemethanol, α-[2-(trimethylsilyl)ethynyl]-](/data/chemimg/123900/112497-25-3_b.png)
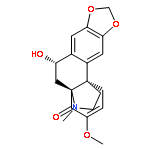
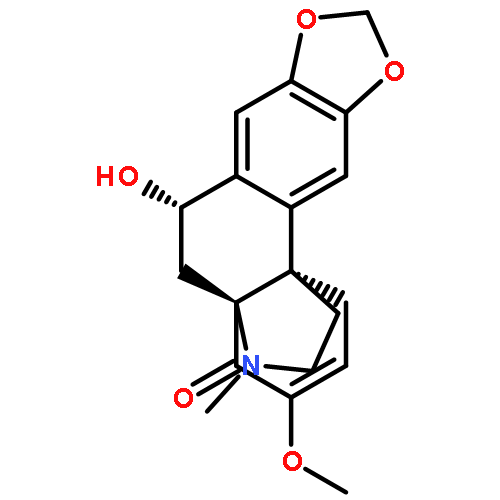



![Cyclohexene, 3-ethenyl-3-methyl-1-[(trimethylsilyl)oxy]-](/data/chemimg/125400/102308-02-1.png)
![Cyclohexene, 3-ethenyl-3-methyl-1-[(trimethylsilyl)oxy]-](/data/chemimg/125400/102308-02-1_b.png)
![L-arabino-Hex-1-enitol, 1,5-anhydro-2,6-dideoxy-3-O-[(1,1-dimethylethyl)dimethylsilyl]-](http://img.cochemist.com/ccimg/101900/101856-96-6.png)
![L-arabino-Hex-1-enitol, 1,5-anhydro-2,6-dideoxy-3-O-[(1,1-dimethylethyl)dimethylsilyl]-](http://img.cochemist.com/ccimg/101900/101856-96-6_b.png)


![(2S,3S,6S)-2-hydroxy-3,4-dimethoxy-14-methyl-1,2,3,4,5,6-hexahydro-4a,11b-(epiminoethano)-4,6-epoxyphenanthro[2,3-d][1,3]dioxol-13-one](http://img.cochemist.com/ccimg/100000/99964-47-3.png)
![(2S,3S,6S)-2-hydroxy-3,4-dimethoxy-14-methyl-1,2,3,4,5,6-hexahydro-4a,11b-(epiminoethano)-4,6-epoxyphenanthro[2,3-d][1,3]dioxol-13-one](http://img.cochemist.com/ccimg/100000/99964-47-3_b.png)
![Hasubanan-16-one, 6-(benzoyloxy)-8,10-epoxy-7,8-dimethoxy-17-methyl-2,3-[methylenebis(oxy)]-, (6β,7β,8β,10β)- (9CI)](/data/chemimg/2080000/99964-46-2.png)
![Hasubanan-16-one, 6-(benzoyloxy)-8,10-epoxy-7,8-dimethoxy-17-methyl-2,3-[methylenebis(oxy)]-, (6β,7β,8β,10β)- (9CI)](/data/chemimg/2080000/99964-46-2_b.png)







![SILANE, [(3-ETHENYL-1-CYCLOHEXEN-1-YL)OXY]TRIMETHYL-](http://img.cochemist.com/ccimg/78900/78828-42-9.png)
![SILANE, [(3-ETHENYL-1-CYCLOHEXEN-1-YL)OXY]TRIMETHYL-](http://img.cochemist.com/ccimg/78900/78828-42-9_b.png)


![Phosphine oxide, 1,1'-[1,3-phenylenebis(methylene)]bis[1,1-diphenyl-](/data/chemimg/1952500/73207-71-3.png)
![Phosphine oxide, 1,1'-[1,3-phenylenebis(methylene)]bis[1,1-diphenyl-](/data/chemimg/1952500/73207-71-3_b.png)


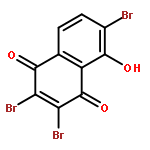
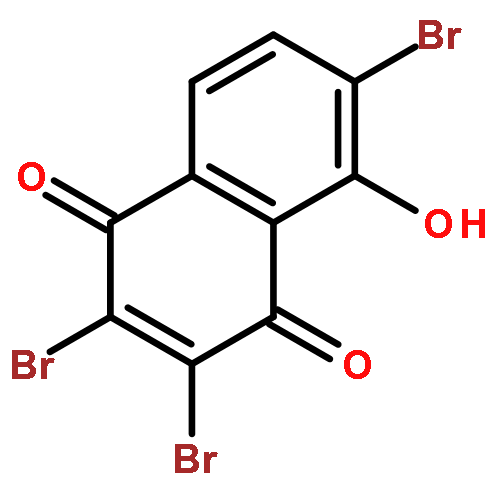














![(1R,2R,3R,4S)-1,2,3-tris(acetyloxy)-11-diazonio-4,10-dihydroxy-2-methyl-9-oxo-2,3,4,9-tetrahydro-1H-benzo[b]fluoren-5-olate](http://img.cochemist.com/ccimg/35400/35303-12-9.png)
![(1R,2R,3R,4S)-1,2,3-tris(acetyloxy)-11-diazonio-4,10-dihydroxy-2-methyl-9-oxo-2,3,4,9-tetrahydro-1H-benzo[b]fluoren-5-olate](http://img.cochemist.com/ccimg/35400/35303-12-9_b.png)








![L-Phenylalanine, N-[(1,1-dimethylethoxy)carbonyl]-L-valyl-, methyl ester](/data/chemimg/1786900/20902-47-0.png)
![L-Phenylalanine, N-[(1,1-dimethylethoxy)carbonyl]-L-valyl-, methyl ester](/data/chemimg/1786900/20902-47-0_b.png)





![Benzene, 1-methoxy-4-[(2-propenyloxy)methyl]-](http://img.cochemist.com/ccimg/14300/14289-62-4.png)
![Benzene, 1-methoxy-4-[(2-propenyloxy)methyl]-](http://img.cochemist.com/ccimg/14300/14289-62-4_b.png)






![1,4-Dioxaspiro[4.5]dec-6-en-8-one](http://img.cochemist.com/ccimg/4700/4683-24-3.png)
![1,4-Dioxaspiro[4.5]dec-6-en-8-one](http://img.cochemist.com/ccimg/4700/4683-24-3_b.png)

















![ACETONITRILEBIS[2-DIPHENYLPHOSPHINO-6-T-BUTYLPYRIDINE]CYCLOPENTADIENYLRUTHENIUM (II) HEXAFLUOROPHOSPHATE](http://img.cochemist.com/ccimg/776300/776230-17-2.png)
![ACETONITRILEBIS[2-DIPHENYLPHOSPHINO-6-T-BUTYLPYRIDINE]CYCLOPENTADIENYLRUTHENIUM (II) HEXAFLUOROPHOSPHATE](http://img.cochemist.com/ccimg/776300/776230-17-2_b.png)
![(3aR,6aR)-2,2-Dimethyl-3aH-cyclopenta[d][1,3]dioxol-4(6aH)-one](http://img.cochemist.com/ccimg/115600/115509-13-2.png)
![(3aR,6aR)-2,2-Dimethyl-3aH-cyclopenta[d][1,3]dioxol-4(6aH)-one](http://img.cochemist.com/ccimg/115600/115509-13-2_b.png)


![2-Cyclohexen-1-one, 3-[(trimethylsilyl)methyl]-](http://img.cochemist.com/ccimg/101800/101773-30-2.png)
![2-Cyclohexen-1-one, 3-[(trimethylsilyl)methyl]-](http://img.cochemist.com/ccimg/101800/101773-30-2_b.png)








![Silane, trimethyl[(2-methyl-1-cyclohexen-1-yl)oxy]-](http://img.cochemist.com/ccimg/20000/19980-35-9.png)
![Silane, trimethyl[(2-methyl-1-cyclohexen-1-yl)oxy]-](http://img.cochemist.com/ccimg/20000/19980-35-9_b.png)




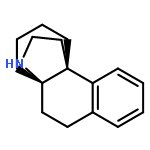
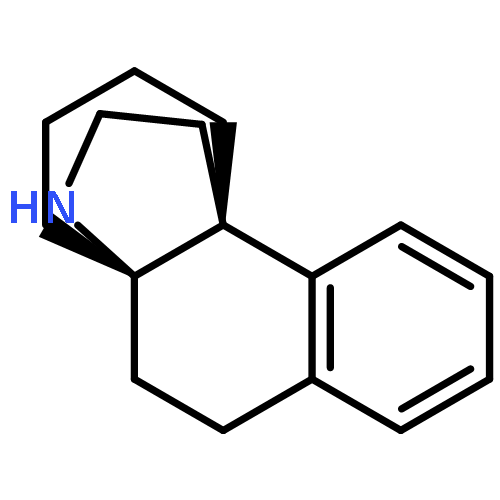
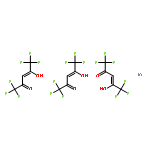
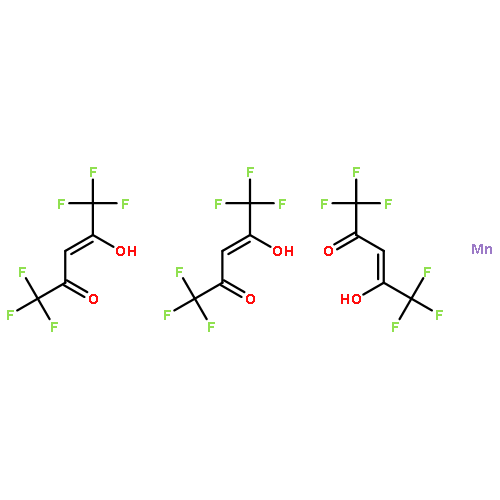








![(1R,2S,3R,4S)-11-diazonio-1,2,3,4,10-pentahydroxy-2-methyl-9-oxo-2,3,4,9-tetrahydro-1H-benzo[b]fluoren-5-olate](http://img.cochemist.com/ccimg/50600/50556-18-8.png)
![(1R,2S,3R,4S)-11-diazonio-1,2,3,4,10-pentahydroxy-2-methyl-9-oxo-2,3,4,9-tetrahydro-1H-benzo[b]fluoren-5-olate](http://img.cochemist.com/ccimg/50600/50556-18-8_b.png)