Co-reporter:Kehui Zhang, Wei Sun, Lihong Huang, Kaiyuan Zhu, Fen Pei, Longchao Zhu, Qian Wang, Yingying Lu, Hongmin Zhang, Hongwei Jin, Li-He Zhang, Liangren Zhang, and Jianbo Yue
Journal of the American Chemical Society 2016 Volume 139(Issue 1) pp:156-170
Publication Date(Web):December 12, 2016
DOI:10.1021/jacs.6b08088
Cyclic adenosine diphosphoribose (cADPR), an endogenous nucleotide derived from nicotinamide adenine dinucleotide (NAD+), mobilizes Ca2+ release from endoplasmic reticulum (ER) via ryanodine receptors (RyRs), yet the bridging protein(s) between cADPR and RyRs remain(s) unknown. Here we synthesized a novel photoaffinity labeling (PAL) cADPR agonist, PAL-cIDPRE, and subsequently applied it to purify its binding proteins in human Jurkat T cells. We identified glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as one of the cADPR binding protein(s), characterized the binding affinity between cADPR and GAPDH in vitro by surface plasmon resonance (SPR) assay, and mapped cADPR’s binding sites in GAPDH. We further demonstrated that cADPR induces the transient interaction between GAPDH and RyRs in vivo and that GAPDH knockdown abolished cADPR-induced Ca2+ release. However, GAPDH did not catalyze cADPR into any other known or novel compound(s). In summary, our data clearly indicate that GAPDH is the long-sought-after cADPR binding protein and is required for cADPR-mediated Ca2+ mobilization from ER via RyRs.
Co-reporter:Fufang Wu, Min Dong, Fang Fang, Yingxia Sang, Yun Li, Bin Cheng, Lihe Zhang, and Hongbin Zhai
Organic Letters 2013 Volume 15(Issue 4) pp:914-916
Publication Date(Web):February 4, 2013
DOI:10.1021/ol400070j
A novel one-step synthetic approach to tetrahydro[1,2]diazepinones via base-promoted rearrangement of α,β-epoxy-N-aziridinylimines, derived from α,β-epoxyketones and N-aminoaziridines, has been developed.
Co-reporter:Ye Huang, Zhuo Chen, Yue Chen, Hao Zhang, Yichao Zhang, Yilei Zhao, Zhenjun Yang, and Lihe Zhang
Bioconjugate Chemistry 2013 Volume 24(Issue 6) pp:951
Publication Date(Web):May 17, 2013
DOI:10.1021/bc300642u
We report here that all of the d- or l-isonucleoside (isoNA) modified siRNAs investigated showed the characteristic A-form conformation in the circular dichroism (CD) spectra compared to native siRNA. The d-isoNA modification had less influence on the thermal stability of siRNAs, but all l-isoNA modification displayed a significant tendency to decrease the thermal stability of siRNA. It was also found that the stabilities of d-/l-isoNA modified siMek1 in serum were different and d-isoNA modification was more potent, i.e., increase of serum stability of siRNA, than l-isoNA modification. When d-isoNA incorporated at position 4 and position 5 at antisense strand of siMek1 showed obvious improvement on serum stability, however, l-isoNA incorporated at positions 11 and 12 at antisense strand and position 9 at sense strand made the siMek1 duplex formed very unstable in serum. The silencing activities of modified siMek1s with d-/l-isoNA at position 1 of antisense strand also dropped dramatically; however, the modification at 3′-terminal of the sense strand with d- or l-isoNA significantly enhanced the silencing activity targeting the antisense strand as reporter and minimized the passenger strand-specific off-target effect. IsoNA modified in the seed area of siMek1, siMek1 A04D and siMek1 A05L, showed similar activity to the native one and better target selectivity. In the case of modification at the position near the cleavage area, it was found that d- or l-isoNA modified sense strand at position 8, 9, or 15 of siMek1 could retain the silencing activities targeting the antisense strand as reporter. Especially, both siMek1 S15D and siMek1 S15L showed good silencing activity and high target selectivity compared to native siMek1. The effects of conformational alteration of such isoNA modification of siRNA on their stability in serum and silencing activity are discussed based on computer simulation. Systematic investigation of the relationship between modified siRNA conformation and their physical and biological properties should provide a useful guideline for chemical modification and optimization of siRNA for further clinical application.
Co-reporter:Jun Zhang, Yue Chen, Ye Huang, Hong-Wei Jin, Ren-Ping Qiao, Lei Xing, Liang-Ren Zhang, Zhen-Jun Yang and Li-He Zhang
Organic & Biomolecular Chemistry 2012 vol. 10(Issue 37) pp:7566-7577
Publication Date(Web):02 Aug 2012
DOI:10.1039/C2OB26219C
Antisense oligonucleotides and siRNAs are potential therapeutic agents and their chemical modifications play an important role to improve the properties and activities of oligonucleotides. Isonucleoside is a type of nucleoside analogue, in which the nucleobase is moved from C-1 to other positions of ribose. In this report, a novel isonucleoside 5 containing a 5′-CH2-extended chain at the sugar moiety was synthesized, thus isoadenosine 5a and isothymidine 5b were incorporated into a DNA single strand and siRNA. It was found that isonucleoside 5 modified oligonucleotides can form stable double helical structures with their complementary DNA and RNA and the stability towards nuclease and ability to activate RNase H are more promising compared with the unmodified, natural analogues. In siRNA, passenger strand modified with isonucleoside (5a/b) at 3′ or 5′ terminal can retain the silencing activity and minimize the passenger strand specific off-target effect.
Co-reporter:Anna Ka Yee Kwong, Zhe Chen, HongMin Zhang, Fung Ping Leung, Connie Mo Ching Lam, Kai Yiu Ting, Liangren Zhang, Quan Hao, Li-He Zhang, and Hon Cheung Lee
Biochemistry 2012 Volume 51(Issue 1) pp:
Publication Date(Web):December 5, 2011
DOI:10.1021/bi201509f
CD38 is a signaling enzyme responsible for catalyzing the synthesis of cyclic ADP ribose (cADPR) and nicotinic acid adenine dinucleotide phosphate; both are universal Ca2+ messenger molecules. Ablation of the CD38 gene in mice causes multiple physiological defects, including impaired oxytocin release, that result in altered social behavior. A series of catalysis-based inhibitors of CD38 were designed and synthesized, starting with arabinosyl-2′-fluoro-2′-deoxynicotinamide mononucleotide. Structure–function relationships were analyzed to assess the structural determinants important for inhibiting the NADase activity of CD38. X-ray crystallography was used to reveal the covalent intermediates that were formed with the catalytic residue, Glu226. Metabolically stable analogues that were resistant to inactivation by phosphatase and esterase were synthesized and shown to be effective in inhibiting intracellular cADPR production in human HL-60 cells during induction of differentiation by retinoic acid. The inhibition was species-independent, and the analogues were similarly effective in blocking the cyclization reaction of CD38 in rat ventricular tissue extracts, as well as inhibiting the α-agonist-induced constriction in rat mesentery arteries. These compounds thus represent the first generally applicable and catalysis-based inhibitors of the Ca2+ signaling function of CD38.
Co-reporter:Min Dong, Yuan-Qi Si, Shuang-Yong Sun, Xiao-Ping Pu, Zhen-Jun Yang, Liang-Ren Zhang, Li-He Zhang, Fung Ping Leung, Connie Mo Ching. Lam, Anna Ka Yee Kwong, Jianbo Yue, Yeyun Zhou, Irina A. Kriksunov, Quan Hao and Hon Cheung Lee
Organic & Biomolecular Chemistry 2011 vol. 9(Issue 9) pp:3246-3257
Publication Date(Web):17 Feb 2011
DOI:10.1039/C0OB00768D
Human CD38 is a novel multi-functional protein that acts not only as an antigen for B-lymphocyte activation, but also as an enzyme catalyzing the synthesis of a Ca2+ messenger molecule, cyclic ADP-ribose, from NAD+. It is well established that this novel Ca2+ signaling enzyme is responsible for regulating a wide range of physiological functions. Based on the crystal structure of the CD38/NAD+ complex, we synthesized a series of simplified N-substituted nicotinamide derivatives (Compound1–14). A number of these compounds exhibited moderate inhibition of the NAD+ utilizing activity of CD38, with Compound4 showing the highest potency. The crystal structure of CD38/Compound4 complex and computer simulation of Compound7 docking to CD38 show a significant role of the nicotinamide moiety and the distal aromatic group of the compounds for substrate recognition by the active site of CD38. Biologically, we showed that both Compounds4 and 7 effectively relaxed the agonist-induced contraction of muscle preparations from rats and guinea pigs. This study is a rational design of inhibitors for CD38 that exhibit important physiological effects, and can serve as a model for future drug development.
Co-reporter:Min Dong, Tanja Kirchberger, Xiangchen Huang, Zhen Jun Yang, Liang Ren Zhang, Andreas H. Guse and Li He Zhang
Organic & Biomolecular Chemistry 2010 vol. 8(Issue 20) pp:4705-4715
Publication Date(Web):25 Aug 2010
DOI:10.1039/C0OB00090F
A convenient trifluoromethylation method was firstly applied to the synthesis of 8- CF3-purine nucleosides. On the basis of this method, new protection and deprotection strategies were developed for the successful synthesis of the trifluoromethylated cyclic-ADP-ribose mimic, 8-CF3-cIDPRE 1. Using intact, fura-2-loaded Jurkat T cells compound 1 and 2′,3′-O-isopropylidene 8-CF3-cIDPRE 14 were characterized as membrane-permeant cADPR agonists. Contrary to the 8-substituted cADPR analogues that mainly act as antagonists of cADPR in cells, 8-substituted cIDPRE derivatives were shown to be Ca2+ mobilizing agonists. Here we report that even compound 1, the 8-substituted cIDPRE with the strong electron withdrawing CF3 group, behaves as an agonist in T cells. Interestingly, also the partially protected 2′,3′-O-isopropylidene 8-CF3-cIDPRE activated Ca2+ signaling indicating only a minor role for the hydroxyl groups of the southern ribose of cADPR for its biological activity. To our knowledge 8-CF3-cIDPRE 1 is the first reported fluoro substituted cADPR mimic and 8-CF3-cIDPRE 1 and compound 14 are promising molecular probes for elucidating the mode of action of cADPR.
Co-reporter:Lingjun Li, Cornelia C. Siebrands, Zhenjun Yang, Liangren Zhang, Andreas H. Guse and Lihe Zhang
Organic & Biomolecular Chemistry 2010 vol. 8(Issue 8) pp:1843-1848
Publication Date(Web):18 Feb 2010
DOI:10.1039/B925295A
A purine nucleobase-simplified cyclic ADP ribose (cADPR) analogue 6b was synthesized, in which a 1,2,3-triazole-4-amide was constructed, instead of a purine moiety, and the northern ribose was replaced by an ether strand. Compound 6b exhibits calcium release activity in intact T-lymphocytes and indicates that it is a membrane-permeable cADPR mimic. Thus, the cADPR analogue containing 1,2,3-triazole-4-amide provides a novel template for further designing cADPR analogues and elucidating their structure–activity relationships.
Co-reporter:Yanli Xu, Hongwei Jin, Zhenjun Yang, Liangren Zhang, Qi Wang, Manning Li, Lihe Zhang
Bioorganic & Medicinal Chemistry Letters 2009 Volume 19(Issue 8) pp:2103-2106
Publication Date(Web):15 April 2009
DOI:10.1016/j.bmcl.2009.03.021
Three new derivatives of neamine, 3 (NE), 6 (NEA) and 9 (NEL), were synthesized by connecting arginine or lysine to 5-hydroxyl group of neamine using ethylenediamine as a linker. The binding affinities of these derivatives to A site of 16S RNA and TAR RNA indicate that the modification on 5-hydroxyl of neamine by amino acid can enhance the binding affinity of neamine. Compound 9 (NEL) shows some antibacterial activities. These results demonstrate that modification on 5-hydroxyl group of neamine may provide a promising way for the development of potential candidates effectively targeting to RNAs.Modification on 5-hydroxyl group of neamine may provide a promising way for the development of potential candidates effectively targeting to RNAs.
Co-reporter:Yanli Xu, Hongwei Jin, Zhenjun Yang, Liangren Zhang, Lihe Zhang
Tetrahedron 2009 65(27) pp: 5228-5239
Publication Date(Web):
DOI:10.1016/j.tet.2009.04.084
Co-reporter:Peng Zhao, Hong-Wei Jin, Zhen-Jun Yang, Liang-Ren Zhang and Li-He Zhang
Organic & Biomolecular Chemistry 2008 vol. 6(Issue 20) pp:3741-3750
Publication Date(Web):13 Aug 2008
DOI:10.1039/B809598A
Four types of β-carboline–nucleoside conjugates were synthesized. The binding affinities of these β-carboline–nucleoside conjugates 4–11, 13 and 15 to TAR RNA were evaluated by affinity capillary electrophoresis. The data of binding affinities to TAR RNA show that conjugates 9 and 13 are stronger binders than the parent compound MC3. Computer modeling indicates that the β-carboline–nucleoside conjugate 13 can fit to the UCU three-nucleotide bulge region of TAR RNA.
Co-reporter:YingChun Liu;Yan Zhang;GuoZhu Ye;ZhenJun Yang
Science China Chemistry 2008 Volume 51( Issue 5) pp:
Publication Date(Web):2008 May
DOI:10.1007/s11426-008-0056-x
Aptamers that interact with various HIV-1 proteins, such as reverse transcriptase, Rev, Tat protein, and nuclear capsule protein, have been prepared through SELEX (systematic evolution of ligands by exponential enrichment) technique. However, there are few reports about the DNA or RNA aptamers that target HIV-1 integrase. In this investigation, we selected alternative RNA aptamers specific for the HIV-1 integrase by using a different binding buffer containing 10 mmol·L−1 MgCl2 and 100 mmol·L−1 KCl. Aptamer IN1, IN2, IN3 had similar and the highest Kd values from 145 to 239 nmol·L−1. Structural studies showed that they formed similar stem-loop structure. Deletion of any stem structure resulted in diminished affinity. In addition, structure probing study with antisense DNA indicated that the stem-loop structure in the random region was critical for integrase binding. Although aptamer IN1 failed to form G-quartet structure, it might directly interact with the DDE motif of integrase, which is the virus DNA-binding site, because G-quadruplex T40214 competitively inhibited the interaction between IN1 and integrase. Together, this study generated a novel RNA aptamer IN1, which could be useful in basic research and anti-HIV drug screening.
Co-reporter:Yuhang Wang, Xin-Shan Ye and Li-He Zhang
Organic & Biomolecular Chemistry 2007 vol. 5(Issue 14) pp:2189-2200
Publication Date(Web):22 May 2007
DOI:10.1039/B704586G
Saccharide synthesis is a formidable task for synthetic chemists. Although in recent years many advances have been made in this area, development of more convenient and efficient strategies for oligosaccharide synthesis is still in great demand. This review focuses on one of these new strategies—the one-pot sequential glycosylation approach as a potent tool for oligosaccharide assembly.
Co-reporter:Yi Liu, Tian-Xiang Han, Zhen-Jun Yang, Liang-Ren Zhang, Li-He Zhang
Tetrahedron: Asymmetry 2007 Volume 18(Issue 19) pp:2326-2331
Publication Date(Web):27 September 2007
DOI:10.1016/j.tetasy.2007.08.033
Our studies of the TIBAL-promoted Claisen rearrangement reaction and ring-closing metathesis (RCM) resulted in the development of an efficient synthetic route to polyfunctional seven-membered carbasugar synthons from d-arabinose. Moreover, the construction of 8-oxa-bicyclo[3.2.1]octane derivatives 10 and 13 was achieved by BCl3 or iodide-promoted intramolecular electrophilic addition reactions, which were regio- and stereoselective.2,5-Anhydro-3,4,6-tri-O-benzyl-1-iodo-5-vinyl-l-manno-hexitolC29H31IO4[α]D20=+18.1 (c 0.09, CH2Cl2)(1R,2R,3R)-4-Benzyloxymethyl-2,3-dibenzyloxycyclohepta-4-ene-1-olC29H32O4[α]D20=-63.1 (c 0.08, MeOH)Absolute configuration: (1R,2R,3R)(1S,2R,3R)-4-Benzyloxymethyl-2,3-dibenzyloxycyclohepta-4-ene-1-olC29H32O4[α]D20=-28.4 (c 0.12, MeOH)Absolute configuration: (1S,2R,3R)(1S,2S,3R,5R)-4-Methylene-8-oxa-bicyclo[3.2.1]octane-2,3-diolC8H12O3[α]D20=-16.7 (c 0.10, MeOH)Absolute configuration: (1S,2S,3R,5R)(1S,2S,3S,4R,5S)-2,3-Dibenzyloxy-1-benzyloxymethyl-4-iodo-8-oxa-bicyclo[3.2.1]octaneC29H31IO4[α]D20=-38.8 (c 0.37, CH2Cl2)Absolute configuration: (1S,2S,3S,4R,5S)
Co-reporter:Zong-Sheng Li, Ren-Ping Qiao, Zhen-Jun Yang, Liang-Ren Zhang, Li-He Zhang
Tetrahedron: Asymmetry 2006 Volume 17(Issue 7) pp:1056-1061
Publication Date(Web):3 April 2006
DOI:10.1016/j.tetasy.2006.03.034
Several tricyclic azido-isonucleosides were formed in high yields by the treatment of pyrimidine isonucleosides with triphenylphosphine, tetrabromomethane, and sodium azide. The regioselective ring opening of these tricyclic azido-isonucleosides was also investigated.6′-O-Benzoyl-1′-deoxy-1′-azido-4′-deoxy-4′-(thymin-1-yl)-2′,5′-anhydro-l-mannitolC18H19N5O6[α]D24=-39.2 (c 0.120, MeOH)Source of chirality: l-mannitol6′-O-Benzoyl-1′-deoxy-1′-azido-4′-deoxy-4′-(thymin-1-yl)-2,3′:2′,5′-dianhydro-l-altritolC18H17N5O5[α]D24=+28.9 (c 0.127, MeOH)Source of chirality: l-altriol6′-Deoxy-6′-azido-4′-deoxy-4′-(thymin-1-yl)-2′,5′-anhydro-l-mannofuranose dimethyl acetalC13H19N5O6[α]D24=-19.6 (c 0.160, MeOH)Source of chirality: l-mannofuranose6′-Deoxy-6′-azido-4′-deoxy-4′-(uracil-1-yl)-2′,5′-anhydro-l-mannofuranose dimethyl acetalC12H17N5O6[α]D24=-23.8 (c 0.080, MeOH)Source of chirality: l-mannofuranose6′-Deoxy-6′-azido-4′-deoxy-4′-(thymin-1-yl)-2,3′:2′,5′-dianhydro-l-altrofuranose dimethyl acetalC13H17N5O5[α]D24=+79.4 (c 0.152, MeOH)Source of chirality: l-altrofuranose6′-Deoxy-6′-azido-4′-deoxy-4′-(uracil-1-yl)-2,3′:2′,5′-dianhydro-l-altrofuranose dimethyl acetalC12H15N5O5[α]D24=+92.7 (c 0.070, MeOH)Source of chirality: l-altrofuranose1′- Deoxy-1′-azido-4′-deoxy-4′-(thymin-1-yl)-2,3′:2′,5′-dianhydro-l-altritolC11H13N5O4[α]D24=+45.0 (c 0.030, MeOH)Source of chirality: l-altriol1′-Deoxy-1′-azido-4′-deoxy-4′-(5-methyl-N2-methyl-iso-cytosin-1-yl)-2′,5′-anhydro-l-altritolC12H18N6O4[α]D24=-82.3 (c 0.038, MeOH)Source of chirality: l-altriol1′-Deoxy-1′-azido-4′-deoxy-4′-(thymin-1-yl)-2′,5′-anhydro-l-altritolC11H15N5O5[α]D24=-38.5 (c 0.040, MeOH)Source of chirality: l-altriol1′-Deoxy-1′-amino-4′-deoxy-4′-(thymin-1-yl)-2,1′:2′,5′-dianhydro-l-altritolC11H15N3O4[α]D24=+40.8 (c 0.105, MeOH)Source of chirality: l-altriol
Co-reporter:Min Dong, Yuan-Qi Si, Shuang-Yong Sun, Xiao-Ping Pu, Zhen-Jun Yang, Liang-Ren Zhang, Li-He Zhang, Fung Ping Leung, Connie Mo Ching. Lam, Anna Ka Yee Kwong, Jianbo Yue, Yeyun Zhou, Irina A. Kriksunov, Quan Hao and Hon Cheung Lee
Organic & Biomolecular Chemistry 2011 - vol. 9(Issue 9) pp:NaN3257-3257
Publication Date(Web):2011/02/17
DOI:10.1039/C0OB00768D
Human CD38 is a novel multi-functional protein that acts not only as an antigen for B-lymphocyte activation, but also as an enzyme catalyzing the synthesis of a Ca2+ messenger molecule, cyclic ADP-ribose, from NAD+. It is well established that this novel Ca2+ signaling enzyme is responsible for regulating a wide range of physiological functions. Based on the crystal structure of the CD38/NAD+ complex, we synthesized a series of simplified N-substituted nicotinamide derivatives (Compound1–14). A number of these compounds exhibited moderate inhibition of the NAD+ utilizing activity of CD38, with Compound4 showing the highest potency. The crystal structure of CD38/Compound4 complex and computer simulation of Compound7 docking to CD38 show a significant role of the nicotinamide moiety and the distal aromatic group of the compounds for substrate recognition by the active site of CD38. Biologically, we showed that both Compounds4 and 7 effectively relaxed the agonist-induced contraction of muscle preparations from rats and guinea pigs. This study is a rational design of inhibitors for CD38 that exhibit important physiological effects, and can serve as a model for future drug development.
Co-reporter:Peng Zhao, Hong-Wei Jin, Zhen-Jun Yang, Liang-Ren Zhang and Li-He Zhang
Organic & Biomolecular Chemistry 2008 - vol. 6(Issue 20) pp:NaN3750-3750
Publication Date(Web):2008/08/13
DOI:10.1039/B809598A
Four types of β-carboline–nucleoside conjugates were synthesized. The binding affinities of these β-carboline–nucleoside conjugates 4–11, 13 and 15 to TAR RNA were evaluated by affinity capillary electrophoresis. The data of binding affinities to TAR RNA show that conjugates 9 and 13 are stronger binders than the parent compound MC3. Computer modeling indicates that the β-carboline–nucleoside conjugate 13 can fit to the UCU three-nucleotide bulge region of TAR RNA.
Co-reporter:Jun Zhang, Yue Chen, Ye Huang, Hong-Wei Jin, Ren-Ping Qiao, Lei Xing, Liang-Ren Zhang, Zhen-Jun Yang and Li-He Zhang
Organic & Biomolecular Chemistry 2012 - vol. 10(Issue 37) pp:NaN7577-7577
Publication Date(Web):2012/08/02
DOI:10.1039/C2OB26219C
Antisense oligonucleotides and siRNAs are potential therapeutic agents and their chemical modifications play an important role to improve the properties and activities of oligonucleotides. Isonucleoside is a type of nucleoside analogue, in which the nucleobase is moved from C-1 to other positions of ribose. In this report, a novel isonucleoside 5 containing a 5′-CH2-extended chain at the sugar moiety was synthesized, thus isoadenosine 5a and isothymidine 5b were incorporated into a DNA single strand and siRNA. It was found that isonucleoside 5 modified oligonucleotides can form stable double helical structures with their complementary DNA and RNA and the stability towards nuclease and ability to activate RNase H are more promising compared with the unmodified, natural analogues. In siRNA, passenger strand modified with isonucleoside (5a/b) at 3′ or 5′ terminal can retain the silencing activity and minimize the passenger strand specific off-target effect.
Co-reporter:Yuhang Wang, Xin-Shan Ye and Li-He Zhang
Organic & Biomolecular Chemistry 2007 - vol. 5(Issue 14) pp:NaN2200-2200
Publication Date(Web):2007/05/22
DOI:10.1039/B704586G
Saccharide synthesis is a formidable task for synthetic chemists. Although in recent years many advances have been made in this area, development of more convenient and efficient strategies for oligosaccharide synthesis is still in great demand. This review focuses on one of these new strategies—the one-pot sequential glycosylation approach as a potent tool for oligosaccharide assembly.
Co-reporter:Lingjun Li, Cornelia C. Siebrands, Zhenjun Yang, Liangren Zhang, Andreas H. Guse and Lihe Zhang
Organic & Biomolecular Chemistry 2010 - vol. 8(Issue 8) pp:NaN1848-1848
Publication Date(Web):2010/02/18
DOI:10.1039/B925295A
A purine nucleobase-simplified cyclic ADP ribose (cADPR) analogue 6b was synthesized, in which a 1,2,3-triazole-4-amide was constructed, instead of a purine moiety, and the northern ribose was replaced by an ether strand. Compound 6b exhibits calcium release activity in intact T-lymphocytes and indicates that it is a membrane-permeable cADPR mimic. Thus, the cADPR analogue containing 1,2,3-triazole-4-amide provides a novel template for further designing cADPR analogues and elucidating their structure–activity relationships.
Co-reporter:Min Dong, Tanja Kirchberger, Xiangchen Huang, Zhen Jun Yang, Liang Ren Zhang, Andreas H. Guse and Li He Zhang
Organic & Biomolecular Chemistry 2010 - vol. 8(Issue 20) pp:NaN4715-4715
Publication Date(Web):2010/08/25
DOI:10.1039/C0OB00090F
A convenient trifluoromethylation method was firstly applied to the synthesis of 8- CF3-purine nucleosides. On the basis of this method, new protection and deprotection strategies were developed for the successful synthesis of the trifluoromethylated cyclic-ADP-ribose mimic, 8-CF3-cIDPRE 1. Using intact, fura-2-loaded Jurkat T cells compound 1 and 2′,3′-O-isopropylidene 8-CF3-cIDPRE 14 were characterized as membrane-permeant cADPR agonists. Contrary to the 8-substituted cADPR analogues that mainly act as antagonists of cADPR in cells, 8-substituted cIDPRE derivatives were shown to be Ca2+ mobilizing agonists. Here we report that even compound 1, the 8-substituted cIDPRE with the strong electron withdrawing CF3 group, behaves as an agonist in T cells. Interestingly, also the partially protected 2′,3′-O-isopropylidene 8-CF3-cIDPRE activated Ca2+ signaling indicating only a minor role for the hydroxyl groups of the southern ribose of cADPR for its biological activity. To our knowledge 8-CF3-cIDPRE 1 is the first reported fluoro substituted cADPR mimic and 8-CF3-cIDPRE 1 and compound 14 are promising molecular probes for elucidating the mode of action of cADPR.

![(6Z)-6-{[2-(1,1-dimethylprop-2-en-1-yl)-1H-indol-3-yl]methylidene}-3-hydroxy-3-methylpiperazine-2,5-dione](http://img.cochemist.com/ccimg/1256000/1255929-51-1.png)
![(6Z)-6-{[2-(1,1-dimethylprop-2-en-1-yl)-1H-indol-3-yl]methylidene}-3-hydroxy-3-methylpiperazine-2,5-dione](http://img.cochemist.com/ccimg/1256000/1255929-51-1_b.png)


























![Benzene, [[2,3-bis(hexadecyloxy)propoxy]methyl]-](http://img.cochemist.com/ccimg/96900/96868-38-1.png)
![Benzene, [[2,3-bis(hexadecyloxy)propoxy]methyl]-](http://img.cochemist.com/ccimg/96900/96868-38-1_b.png)


![2-amino-9-[(2r,4s,5r)-5-[[bis(4-methoxyphenyl)-phenylmethoxy]methyl]-4-hydroxyoxolan-2-yl]-3h-purin-6-one](http://img.cochemist.com/ccimg/81200/81144-43-6.png)
![2-amino-9-[(2r,4s,5r)-5-[[bis(4-methoxyphenyl)-phenylmethoxy]methyl]-4-hydroxyoxolan-2-yl]-3h-purin-6-one](http://img.cochemist.com/ccimg/81200/81144-43-6_b.png)






![3-[2-(1,1-dimethyl-allyl)-5-(3-methyl-but-2-enyl)-indol-3-ylmethylene]-6-methyl-piperazine-2,5-dione](http://img.cochemist.com/ccimg/60500/60422-87-9.png)
![3-[2-(1,1-dimethyl-allyl)-5-(3-methyl-but-2-enyl)-indol-3-ylmethylene]-6-methyl-piperazine-2,5-dione](http://img.cochemist.com/ccimg/60500/60422-87-9_b.png)

![6-[2-(1,1-dimethyl-allyl)-indol-3-ylmethylene]-piperazine-2,3,5-trione](http://img.cochemist.com/ccimg/58000/57944-03-3.png)
![6-[2-(1,1-dimethyl-allyl)-indol-3-ylmethylene]-piperazine-2,3,5-trione](http://img.cochemist.com/ccimg/58000/57944-03-3_b.png)


![3-[2-(1,1-dimethyl-allyl)-6-(3-methyl-but-2-enyl)-indol-3-ylmethylene]-6-methylene-piperazine-2,5-dione](http://img.cochemist.com/ccimg/55200/55179-54-9.png)
![3-[2-(1,1-dimethyl-allyl)-6-(3-methyl-but-2-enyl)-indol-3-ylmethylene]-6-methylene-piperazine-2,5-dione](http://img.cochemist.com/ccimg/55200/55179-54-9_b.png)
![3-[2-(1,1-dimethyl-allyl)-indol-3-ylmethylene]-6-methylene-piperazine-2,5-dione](http://img.cochemist.com/ccimg/55200/55179-53-8.png)
![3-[2-(1,1-dimethyl-allyl)-indol-3-ylmethylene]-6-methylene-piperazine-2,5-dione](http://img.cochemist.com/ccimg/55200/55179-53-8_b.png)


![2-[4-(aminoiminomethyl)phenyl]-1H-Indole-6-carboximidamide](http://img.cochemist.com/ccimg/47200/47165-04-8.png)
![2-[4-(aminoiminomethyl)phenyl]-1H-Indole-6-carboximidamide](http://img.cochemist.com/ccimg/47200/47165-04-8_b.png)
![3H-Indolium,2-[3-(1,3-dihydro-3,3- dimethyl-1-octadecyl-2H-indol-2-ylidene)- 1-propenyl]-3,3-dimethyl-1-octadecyl-,perchlorate](http://img.cochemist.com/ccimg/41100/41085-99-8.png)
![3H-Indolium,2-[3-(1,3-dihydro-3,3- dimethyl-1-octadecyl-2H-indol-2-ylidene)- 1-propenyl]-3,3-dimethyl-1-octadecyl-,perchlorate](http://img.cochemist.com/ccimg/41100/41085-99-8_b.png)



![2,3,5-Piperazinetrione,6-[[2-(1,1-dimethyl-2-propen-1-yl)-6-(3-methyl-2-buten-1-yl)-1H-indol-3-yl]methylene]-,(6Z)-](http://img.cochemist.com/ccimg/25700/25644-25-1.png)
![2,3,5-Piperazinetrione,6-[[2-(1,1-dimethyl-2-propen-1-yl)-6-(3-methyl-2-buten-1-yl)-1H-indol-3-yl]methylene]-,(6Z)-](http://img.cochemist.com/ccimg/25700/25644-25-1_b.png)















![Adenosine,5'-O-[bis(4-methoxyphenyl)phenylmethyl]-2'-deoxy- (9CI)](http://img.cochemist.com/ccimg/17400/17331-22-5.png)
![Adenosine,5'-O-[bis(4-methoxyphenyl)phenylmethyl]-2'-deoxy- (9CI)](http://img.cochemist.com/ccimg/17400/17331-22-5_b.png)



















![1-Propanol, 2,3-bis[(9Z)-9-octadecenyloxy]-](http://img.cochemist.com/ccimg/6100/6076-41-1.png)
![1-Propanol, 2,3-bis[(9Z)-9-octadecenyloxy]-](http://img.cochemist.com/ccimg/6100/6076-41-1_b.png)

![9-Octadecenoic acid(9Z)-,1,1'-[(1R)-1-[[[(2-aminoethoxy)hydroxyphosphinyl]oxy]methyl]-1,2-ethanediyl]ester](http://img.cochemist.com/ccimg/4100/4004-05-1.png)
![9-Octadecenoic acid(9Z)-,1,1'-[(1R)-1-[[[(2-aminoethoxy)hydroxyphosphinyl]oxy]methyl]-1,2-ethanediyl]ester](http://img.cochemist.com/ccimg/4100/4004-05-1_b.png)
![2,5-Piperazinedione,3-[[2-(1,1-dimethyl-2-propen-1-yl)-5,7-bis(3-methyl-2-buten-1-yl)-1H-indol-3-yl]methyl]-6-methyl-,(3S,6S)-](http://img.cochemist.com/ccimg/1900/1859-87-6.png)
![2,5-Piperazinedione,3-[[2-(1,1-dimethyl-2-propen-1-yl)-5,7-bis(3-methyl-2-buten-1-yl)-1H-indol-3-yl]methyl]-6-methyl-,(3S,6S)-](http://img.cochemist.com/ccimg/1900/1859-87-6_b.png)




![8-Oxa-3-azabicyclo[3.2.1]octane](http://img.cochemist.com/ccimg/300/280-13-7.png)
![8-Oxa-3-azabicyclo[3.2.1]octane](http://img.cochemist.com/ccimg/300/280-13-7_b.png)








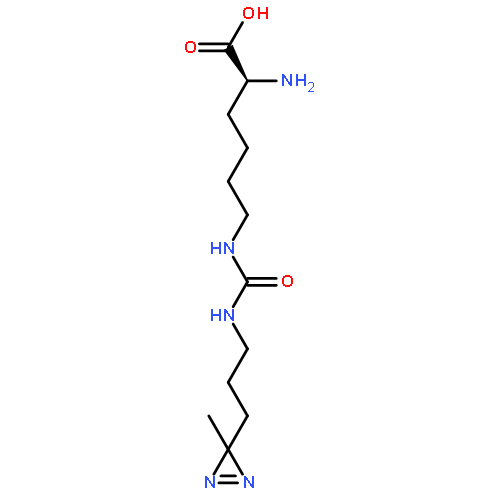
![L-Lysine, N6-[(2-azidoethoxy)carbonyl]-](/data/chemimg/3722800/1167421-25-1.png)
![L-Lysine, N6-[(2-azidoethoxy)carbonyl]-](/data/chemimg/3722800/1167421-25-1_b.png)


















![5-[(2-PHENOXYPHENYL)METHYLIDENE]-1,3-THIAZOLIDINE-2,4-DIONE](/data/chemimg/384200/1162262-40-9.png)
![5-[(2-PHENOXYPHENYL)METHYLIDENE]-1,3-THIAZOLIDINE-2,4-DIONE](/data/chemimg/384200/1162262-40-9_b.png)



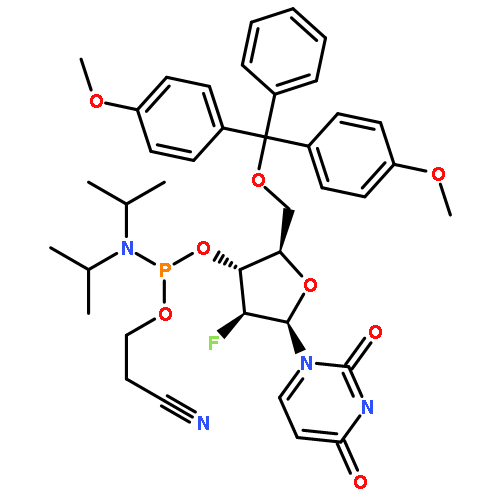
![Phosphoramidous acid, N,N-bis(1-methylethyl)-, 2-cyanoethyl [1-(2-nitrophenyl)ethyl] ester](/data/chemimg/3726900/461425-49-0.png)
![Phosphoramidous acid, N,N-bis(1-methylethyl)-, 2-cyanoethyl [1-(2-nitrophenyl)ethyl] ester](/data/chemimg/3726900/461425-49-0_b.png)
![2-Propylthiazolo[4,5-c]quinolin-4-amine](http://img.cochemist.com/ccimg/257000/256922-53-9.png)
![2-Propylthiazolo[4,5-c]quinolin-4-amine](http://img.cochemist.com/ccimg/257000/256922-53-9_b.png)


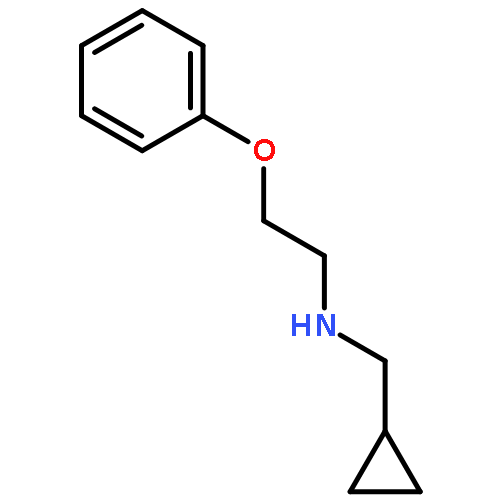
![Cyclopropanecarboxamide, N-[3-(4-benzofuranyl)propyl]-](http://img.cochemist.com/ccimg/199400/199391-16-7.png)
![Cyclopropanecarboxamide, N-[3-(4-benzofuranyl)propyl]-](http://img.cochemist.com/ccimg/199400/199391-16-7_b.png)
![Acetamide, N-[2-(2-chloro-6-methoxy-4-quinolinyl)ethyl]-](http://img.cochemist.com/ccimg/186900/186887-41-2.png)
![Acetamide, N-[2-(2-chloro-6-methoxy-4-quinolinyl)ethyl]-](http://img.cochemist.com/ccimg/186900/186887-41-2_b.png)
![Acetamide, N-[(7-methoxy-1-naphthalenyl)methoxy]-](http://img.cochemist.com/ccimg/178400/178366-47-7.png)
![Acetamide, N-[(7-methoxy-1-naphthalenyl)methoxy]-](http://img.cochemist.com/ccimg/178400/178366-47-7_b.png)














![Thiourea, N-[2-(5-methoxy-1H-indol-3-yl)ethyl]-N'-propyl-](http://img.cochemist.com/ccimg/147700/147621-54-3.png)
![Thiourea, N-[2-(5-methoxy-1H-indol-3-yl)ethyl]-N'-propyl-](http://img.cochemist.com/ccimg/147700/147621-54-3_b.png)








![Acetamide,N-[(2R)-2-(6-chloro-5-methoxy-1H-indol-3-yl)propyl]-](http://img.cochemist.com/ccimg/118800/118702-11-7.png)
![Acetamide,N-[(2R)-2-(6-chloro-5-methoxy-1H-indol-3-yl)propyl]-](http://img.cochemist.com/ccimg/118800/118702-11-7_b.png)




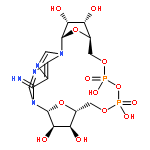
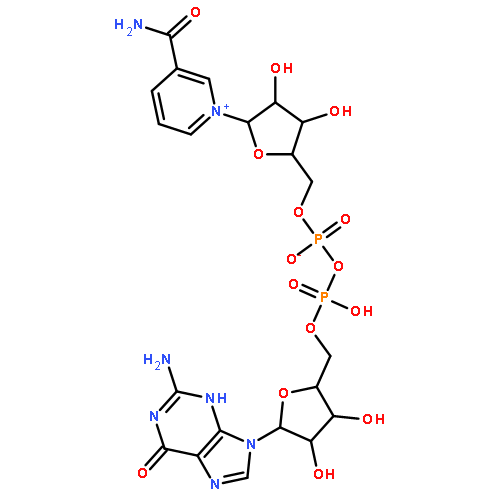
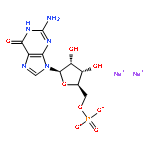
![Adenosine5'-(trihydrogen diphosphate),P'-[[(1R,2R,3S,4R)-4-[3-(aminocarbonyl)pyridinio]-2,3-dihydroxycyclopentyl]methyl]ester, inner salt](http://img.cochemist.com/ccimg/112400/112345-60-5.png)
![Adenosine5'-(trihydrogen diphosphate),P'-[[(1R,2R,3S,4R)-4-[3-(aminocarbonyl)pyridinio]-2,3-dihydroxycyclopentyl]methyl]ester, inner salt](http://img.cochemist.com/ccimg/112400/112345-60-5_b.png)
![N-Benzoyl-5'-O-[bis(4-methoxyphenyl)phenylmethyl]-2'-O-methyl-adenosine](http://img.cochemist.com/ccimg/110800/110764-72-2.png)
![N-Benzoyl-5'-O-[bis(4-methoxyphenyl)phenylmethyl]-2'-O-methyl-adenosine](http://img.cochemist.com/ccimg/110800/110764-72-2_b.png)
![Phosphoramidous acid, N,N-bis(1-methylethyl)-, 2-[[2-[bis(4-methoxyphenyl)phenylmethoxy]ethyl]sulfonyl]ethyl 2-cyanoethyl ester](/data/chemimg/3726900/108783-02-4.png)
![Phosphoramidous acid, N,N-bis(1-methylethyl)-, 2-[[2-[bis(4-methoxyphenyl)phenylmethoxy]ethyl]sulfonyl]ethyl 2-cyanoethyl ester](/data/chemimg/3726900/108783-02-4_b.png)












![Pyridinium, 1-[2-(2-naphthalenyl)-2-oxoethyl]-, bromide](http://img.cochemist.com/ccimg/26100/26031-78-7.png)
![Pyridinium, 1-[2-(2-naphthalenyl)-2-oxoethyl]-, bromide](http://img.cochemist.com/ccimg/26100/26031-78-7_b.png)

![Pyridinium, 1-[2-(4-chlorophenyl)-2-oxoethyl]-, bromide](http://img.cochemist.com/ccimg/26100/26031-66-3.png)
![Pyridinium, 1-[2-(4-chlorophenyl)-2-oxoethyl]-, bromide](http://img.cochemist.com/ccimg/26100/26031-66-3_b.png)





















![Acetamide,N-[2-(6-hydroxy-5-methoxy-1H-indol-3-yl)ethyl]-](http://img.cochemist.com/ccimg/2300/2208-41-5.png)
![Acetamide,N-[2-(6-hydroxy-5-methoxy-1H-indol-3-yl)ethyl]-](http://img.cochemist.com/ccimg/2300/2208-41-5_b.png)


![(8s,9s,10r,13s,14s,17s)-17-acetyl-10,13-dimethyl-6,7,8,9,11,12,14,15,16,17-decahydrocyclopenta[a]phenanthren-3-one](http://img.cochemist.com/ccimg/1200/1162-54-5.png)
![(8s,9s,10r,13s,14s,17s)-17-acetyl-10,13-dimethyl-6,7,8,9,11,12,14,15,16,17-decahydrocyclopenta[a]phenanthren-3-one](http://img.cochemist.com/ccimg/1200/1162-54-5_b.png)






![6H-Isoindolo[2,1-a]indole](http://img.cochemist.com/ccimg/300/248-71-5.png)
![6H-Isoindolo[2,1-a]indole](http://img.cochemist.com/ccimg/300/248-71-5_b.png)
![3H-Naphtho[2,1-b]pyran](http://img.cochemist.com/ccimg/300/229-80-1.png)
![3H-Naphtho[2,1-b]pyran](http://img.cochemist.com/ccimg/300/229-80-1_b.png)