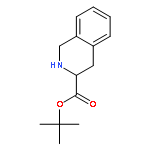
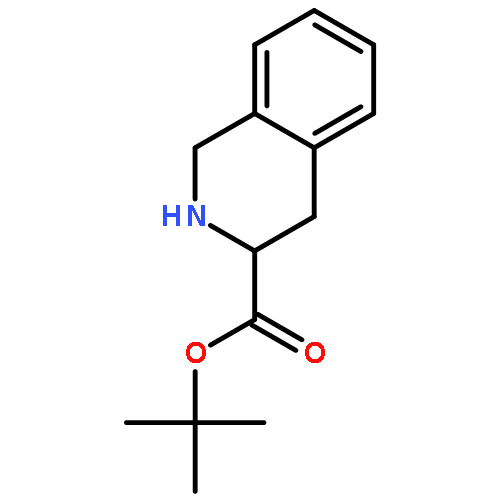
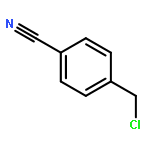
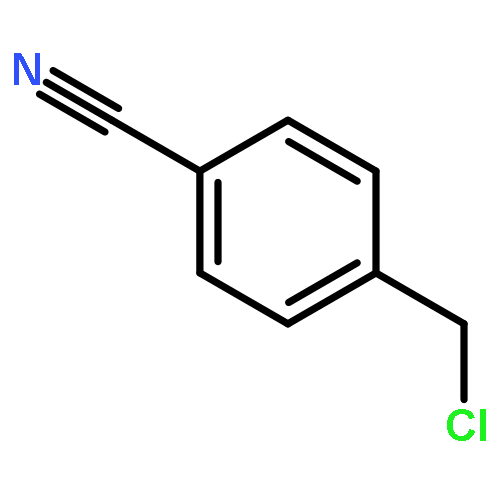
Co-reporter:Md. Ashraful Hoque, Toru Arai, Norikazu Nishino, Hyun-Jung Kim, Akihiro Ito, Minoru Yoshida
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 21) pp:6770-6772
Publication Date(Web):1 November 2012
DOI:10.1016/j.bmcl.2012.03.004
Two thioacetate tails were introduced to the chlamydocin- and CHAP31-related cyclic tetrapeptides. An intramolecular disulfide bridge could be formed in the CHAP31-related cyclic peptides. Both the thioacetate-tailed and disulfide-bridged peptides were potent histone deacetylase inhibitors in the presence of sulfhydryl compound. Potent p21 promoter inducing activity was also observed in vivo.
Co-reporter:Md. Shahidul Islam;Mohammed P. I. Bhuiyan;Md. Nurul Islam
Amino Acids 2012 Volume 42( Issue 6) pp:2103-2110
Publication Date(Web):2012 June
DOI:10.1007/s00726-011-0947-6
The naturally occurring cyclic tetrapeptide, chlamydocin, originally isolated from fungus Diheterospora chlamydosphoria, consists of α-aminoisobutyric acid, l-phenylalanine, d-proline and an unusual amino acid (S)-2-amino-8-((S)-oxiran-2-yl)-8-oxooctanoic acid (Aoe) and inhibits the histone deacetylases (HDACs), a class of regulatory enzymes. The epoxyketone moiety of Aoe is the key functional group for inhibition. The cyclic tetrapeptide scaffold is supposed to play important role for effective binding to the surface of enzymes. In place of the epoxyketone group, hydroxamic acid and sulfhydryl group have been applied to design inhibitor ligands to zinc atom in catalytic site of HDACs. In the research for more potent HDAC inhibitors, we replaced the epoxyketone moiety of Aoe with different functional groups and synthesized a series of chlamydocin analogs as HDAC inhibitors. Among the functional groups, methoxymethylketone moiety showed as potent inhibition as the hydroxamic acid. On the contrary, we confirmed that borate, trifruoromethylketone, and 2-aminoanilide are almost inactive in HDAC inhibition.
Co-reporter:Nurul M. Islam, Tamaki Kato, Norikazu Nishino, Hyun-Jung Kim, Akihiro Ito, Minoru Yoshida
Bioorganic & Medicinal Chemistry Letters 2010 Volume 20(Issue 3) pp:997-999
Publication Date(Web):1 February 2010
DOI:10.1016/j.bmcl.2009.12.054
Bicyclic tetrapeptide hydroxamic acids were prepared as histone deacetylase (HDAC) inhibitors, and the evaluated inhibitory activity shows that they are potent against HDAC1 and HDAC4. The in vivo activity depends on alkyl loop length.A series of bicyclic tetrapeptide hydroxamic acids were designed and synthesized as histone deacetylase (HDAC) inhibitors. Most of them were found to be potent inhibitors with remarkable selectivity among the HDACs. The in vivo activity depends on alkyl loop length.
Co-reporter:Norikazu Nishino, Gururaj M. Shivashimpi, Preeti B. Soni, Mohammed P.I. Bhuiyan, Tamaki Kato, Satoko Maeda, Tomonori G. Nishino, Minoru Yoshida
Bioorganic & Medicinal Chemistry 2008 Volume 16(Issue 1) pp:437-445
Publication Date(Web):1 January 2008
DOI:10.1016/j.bmc.2007.09.021
Inhibitors of histone deacetylases (HDACs) are a promising class of anticancer agents that effect gene regulation. To know the interaction of aliphatic cap groups with HDACs, cyclic tetrapeptide and bicyclic peptide disulfide hybrids were synthesized without aromatic ring in their macrocyclic framework. Benzene ring of l-Phe in chlamydocin was replaced with several aliphatic amino acids and also a fused bicyclic tetrapeptide was synthesized by ring closing metathesis using Grubb’s first generation catalyst. The inhibitory activities of the cyclic peptides against histone deacetylase enzymes were evaluated, which demonstrated most of them are interesting candidates as anticancer agents.
Co-reporter:Gururaj M. Shivashimpi, Satoshi Amagai, Tamaki Kato, Norikazu Nishino, Satoko Maeda, Tomonori G. Nishino, Minoru Yoshida
Bioorganic & Medicinal Chemistry 2007 Volume 15(Issue 24) pp:7830-7839
Publication Date(Web):15 December 2007
DOI:10.1016/j.bmc.2007.08.041
Chlamydocin, a cyclic tetrapeptide containing aminoisobutyric acid (Aib), l-phenylalanine (l-Phe), d-proline (d-Pro), and a unique amino acid l-2-amino-8-oxo-9,10-epoxydecanoic acid, inhibits the histone deacetylases (HDACs), a class of enzymes, which play important roles in regulation of gene expression. Sulfur containing amino acids can also inhibit potently, so zinc ligand, such as sulfhydryl group connected with a linker to the so-called capping group, corresponding to cyclic tetrapeptide framework in case of chlamydocin is supposed to interact with the surface of HDAC molecule. Various changes in amino acid residues in chlamydocin may afford specific inhibitors toward HDAC paralogs. To find out specific inhibitors, we focused on benzene ring of l-Phe in chlamydocin framework to shift to various parts of cyclic tetrapeptide. We prepared and introduced several aromatic amino acids into the cyclic tetrapeptides. By evaluating inhibitory activity of these macrocyclic peptides against HDACs, we could find potent inhibitors by shifting the aromatic ring to the Aib site.
Co-reporter:Keisuke Kobata, Junya Ogawa, Shyam S. Pandey, Hidetoshi Oshima, Toru Arai, Tamaki Kato, Norikazu Nishino
Synthetic Metals 2007 Volume 157(6–7) pp:311-317
Publication Date(Web):April 2007
DOI:10.1016/j.synthmet.2007.03.010
Amphiphilic l-lysine dendrons containing porphyrin and fullerene bearing amino acids with the ratio of 2:0, 2:1 and 1:1 were synthesized and characterized using UV–visible absorption spectroscopy, fluorescence emission spectroscopy and transmission electron microscopy. Photoluminescence spectra of these dendrimers showed photoluminescence quenching due to photoinduced electron transfer from porphyrin to fullerene moieties. Transmission electron microscopic observation evidenced the self-association of these dendrimers in water. Controllable and reversible dissociation and re-aggregation of some of the water soluble dendrimers have been investigated using electronic absorption and fluorescence spectra upon the addition of 2-hydroxypropyl-β-cyclodextrin and 1-adamantanecarboxylic acid in their aqueous dispersion. Enhanced ellipticity of dendritic porphyrin–fullerene conjugate compared to that only dendritic porphyrin indicates intramolecular porphyrin–fullerene interactions.
Co-reporter:Mohammed P.I. Bhuiyan, Tamaki Kato, Tatsuo Okauchi, Norikazu Nishino, Satoko Maeda, Tomonori G. Nishino, Minoru Yoshida
Bioorganic & Medicinal Chemistry 2006 Volume 14(Issue 10) pp:3438-3446
Publication Date(Web):15 May 2006
DOI:10.1016/j.bmc.2005.12.063
A series of chlamydocin analogs with various carbonyl functionalities were designed and synthesized as histone deacetylase (HDAC) inhibitors. Chlamydocin is a cyclic tetrapeptide containing an epoxyketone surrogate in the side chain which makes it irreversible inhibitor of HDACs, whereas apicidins are a class of cyclic tetrapeptides that contain an ethylketone moiety as zinc ligand. We replaced the epoxyketone moiety of chlamydocin with several ketones and aldehyde to synthesize potent reversible and selective HDAC inhibitors. The inhibitory activity of the cyclic tetrapeptides against histone deacetylase enzymes were evaluated and the result showed most of them are potent inhibitors. Some of them have remarkable selectivity among the HDACs.
Co-reporter:Louis A. Watanabe, Saori Haranaka, Binoy Jose, Minoru Yoshida, Tamaki Kato, Mitsuaki Moriguchi, Kenji Soda, Norikazu Nishino
Tetrahedron: Asymmetry 2005 Volume 16(Issue 4) pp:903-908
Publication Date(Web):21 February 2005
DOI:10.1016/j.tetasy.2005.01.017
An efficient and convenient synthesis of both enantiomers of pipecolic acid has been developed using the intramolecular cyclization of 2-amino-6-bromohexanoic acid under mild conditions.l-Pipecolic acidC6H11NO2Ee > 99% (determined by chiral HPLC analysis)[α]D25=-26.3 (c 1, H2O)Source of chirality: enzymatic kinetic resolutiond-Pipecolic acidC6H11NO2Ee > 99% (determined by chiral HPLC analysis)[α]D25=+26.3 (c 1, H2O)Source of chirality: enzymatic kinetic resolutionBoc-l-2-amino-6-bromohexanoic acidC11H20BrNO4Ee > 99% (determined by chiral HPLC analysis)[α]D25=-2.8 (c 0.1, MeOH)Source of chirality: enzymatic kinetic resolutionBoc-d-2-amino-6-bromohexanoic acidC11H20BrNO4Ee > 99% (determined by chiral HPLC analysis)[α]D25=+2.8 (c 0.1, MeOH)Source of chirality: enzymatic kinetic resolution
Co-reporter:Norikazu Nishino, Binoy Jose, Ryuzo Shinta, Tamaki Kato, Yasuhiko Komatsu, Minoru Yoshida
Bioorganic & Medicinal Chemistry 2004 Volume 12(Issue 22) pp:5777-5784
Publication Date(Web):15 November 2004
DOI:10.1016/j.bmc.2004.08.041
Chlamydocin–hydroxamic acid analogues were designed and synthesized as histone deacetylase (HDAC) inhibitors based on the structure and HDAC inhibitory activity of chlamydocin and trichostatin A. Chlamydocin is a cyclic tetrapeptide containing an epoxyketone moiety in the side chain that makes it an irreversible inhibitor of HDAC. We replaced the epoxyketone moiety of chlamydocin with hydroxamic acid to design potent and reversible inhibitors of HDAC. In addition, a number of amino-cycloalkanecarboxylic acids (Acc) are introduced instead of the simple amino-isobutric acid (Aib) for a variety of the series of chlamydocin analogues. The compounds synthesized were tested for HDAC inhibitory activity and the results showed that many of them are potent inhibitors of HDAC. The replacement of Aib residue of chlamydocin with an aromatic amino acid enhances the in vivo and in vitro inhibitory activity. We have carried out circular dichroism and molecular modeling studies on chlamydocin–hydroxamic acid analogue and compared it with the solution structure of chlamydocin.
Co-reporter:Binoy Jose, Yusuke Oniki, Tamaki Kato, Norikazu Nishino, Yuko Sumida, Minoru Yoshida
Bioorganic & Medicinal Chemistry Letters 2004 Volume 14(Issue 21) pp:5343-5346
Publication Date(Web):1 November 2004
DOI:10.1016/j.bmcl.2004.08.016
Cyclic tetrapeptides containing trifluoromethyl and pentafluoroethyl ketone as zinc binding functional group were synthesized as potent HDAC inhibitors. Evaluation by human HDAC inhibition assay and p21 promoter assay showed that these inhibitors are promising anticancer agents.





![(1R,4R)-tert-Butyl 3-oxo-2-oxa-5-azabicyclo[2.2.1]heptane-5-carboxylate](http://img.cochemist.com/ccimg/848500/848488-70-0.png)
![(1R,4R)-tert-Butyl 3-oxo-2-oxa-5-azabicyclo[2.2.1]heptane-5-carboxylate](http://img.cochemist.com/ccimg/848500/848488-70-0_b.png)
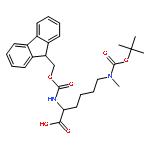
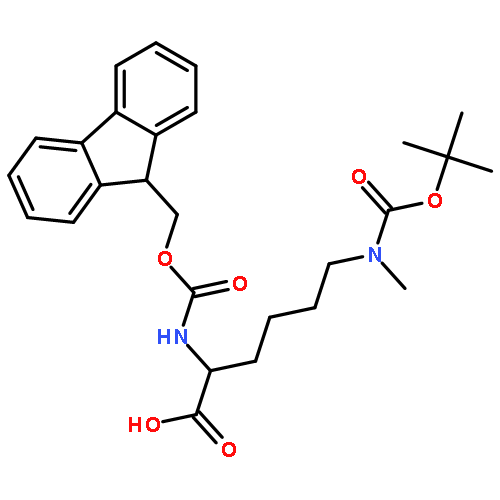
![L-Lysine, N2-[(9H-fluoren-9-ylmethoxy)carbonyl]-N6,N6-dimethyl-](http://img.cochemist.com/ccimg/378900/378802-00-7.png)
![L-Lysine, N2-[(9H-fluoren-9-ylmethoxy)carbonyl]-N6,N6-dimethyl-](http://img.cochemist.com/ccimg/378900/378802-00-7_b.png)
![L-Homoserine, N-[(1,1-dimethylethoxy)carbonyl]-O-2-propenyl-](http://img.cochemist.com/ccimg/350500/350498-77-0.png)
![L-Homoserine, N-[(1,1-dimethylethoxy)carbonyl]-O-2-propenyl-](http://img.cochemist.com/ccimg/350500/350498-77-0_b.png)
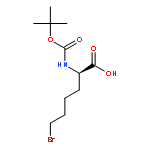
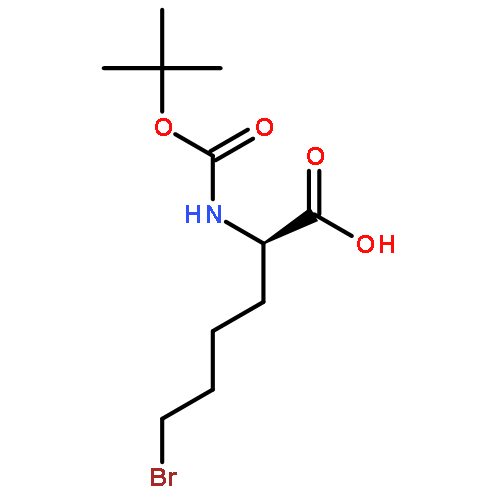
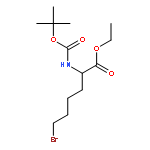
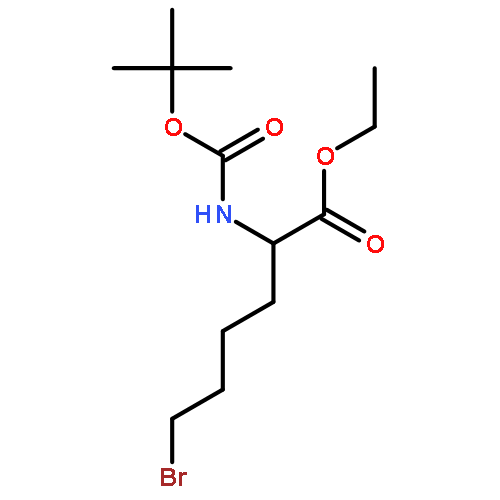
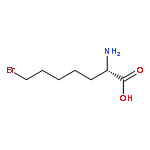
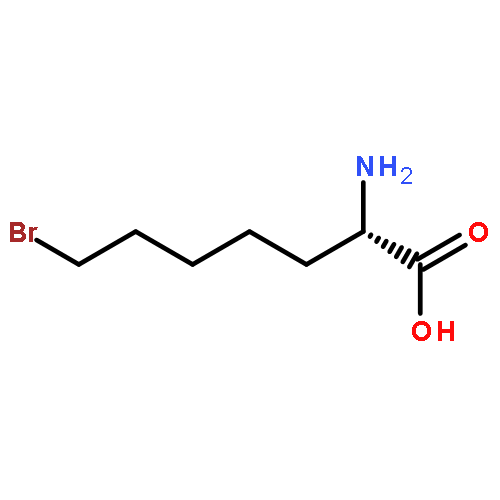
![L-Norleucine, 6-bromo-N-[(1,1-dimethylethoxy)carbonyl]-](http://img.cochemist.com/ccimg/153000/152922-78-6.png)
![L-Norleucine, 6-bromo-N-[(1,1-dimethylethoxy)carbonyl]-](http://img.cochemist.com/ccimg/153000/152922-78-6_b.png)


![Cyclo[(aS,2S)-a-amino-h-oxo-2-oxiraneoctanoyl-L-phenylalanyl-L-phenylalanyl-D-prolyl]](http://img.cochemist.com/ccimg/133200/133155-90-5.png)
![Cyclo[(aS,2S)-a-amino-h-oxo-2-oxiraneoctanoyl-L-phenylalanyl-L-phenylalanyl-D-prolyl]](http://img.cochemist.com/ccimg/133200/133155-90-5_b.png)
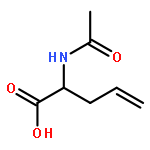
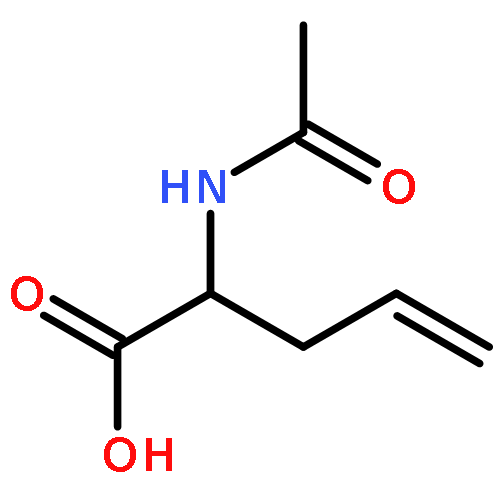
![4-Pentenoic acid,2-[[(1,1-dimethylethoxy)carbonyl]amino]-, phenylmethyl ester, (2S)-](http://img.cochemist.com/ccimg/108700/108634-99-7.png)
![4-Pentenoic acid,2-[[(1,1-dimethylethoxy)carbonyl]amino]-, phenylmethyl ester, (2S)-](http://img.cochemist.com/ccimg/108700/108634-99-7_b.png)
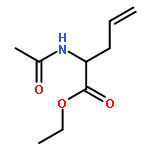
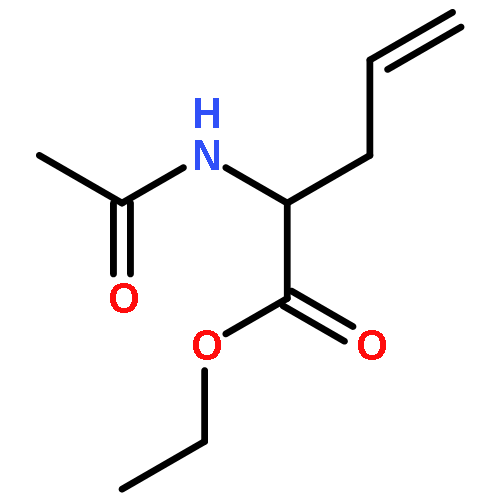


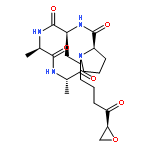
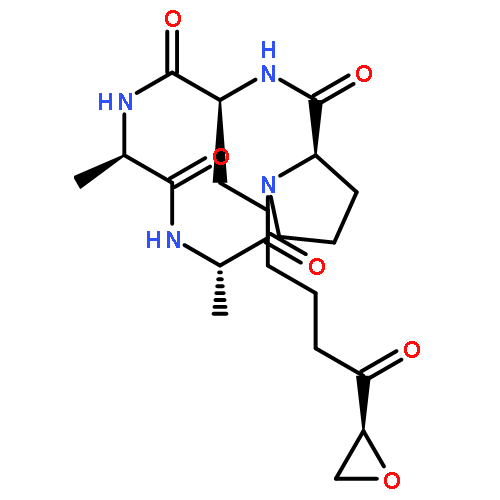
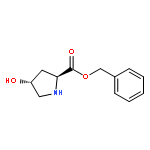
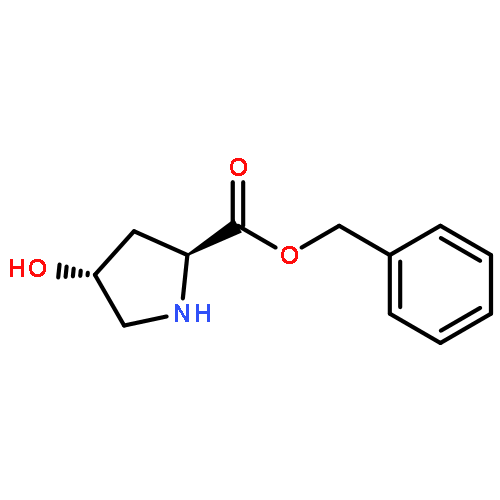
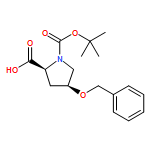
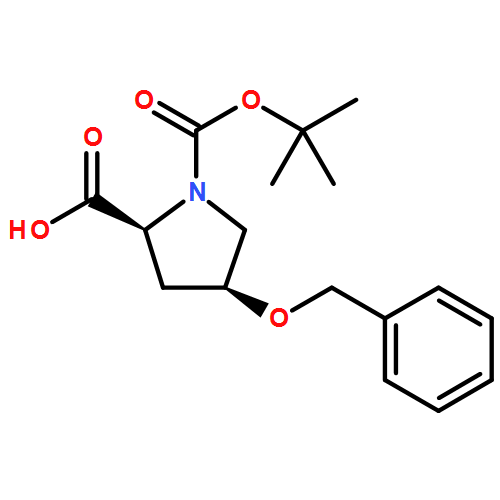
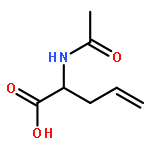
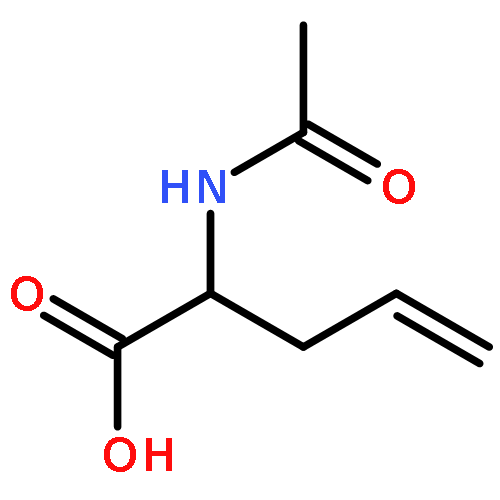
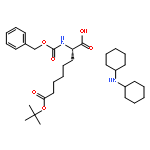
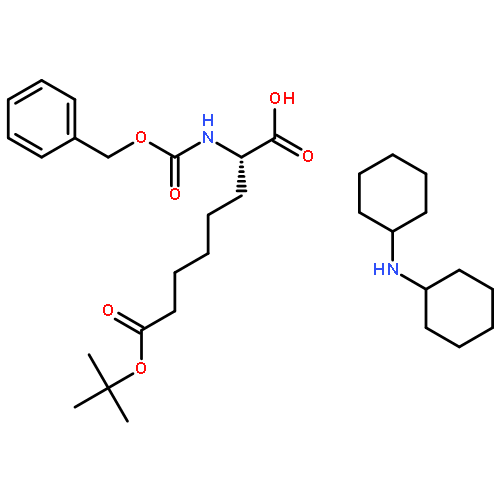
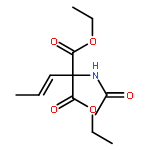
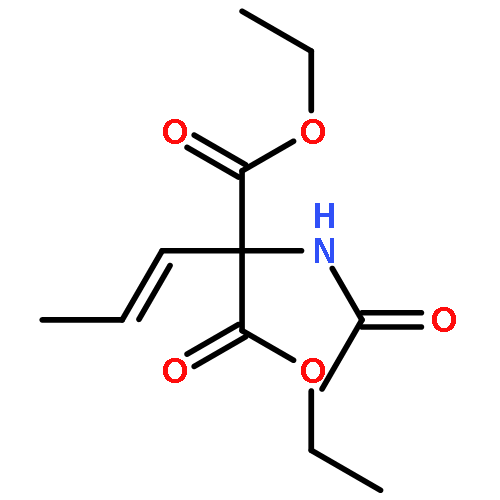
![10H-3,10a-Epidithiopyrazino[1,2-a]indole-1,4-dione,2,3,5a,6-tetrahydro-6-hydroxy-3-(hydroxymethyl)-2-methyl-, (3R,5aS,6S,10aR)-](http://img.cochemist.com/ccimg/100/67-99-2.png)
![10H-3,10a-Epidithiopyrazino[1,2-a]indole-1,4-dione,2,3,5a,6-tetrahydro-6-hydroxy-3-(hydroxymethyl)-2-methyl-, (3R,5aS,6S,10aR)-](http://img.cochemist.com/ccimg/100/67-99-2_b.png)
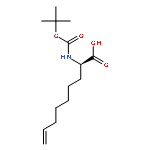
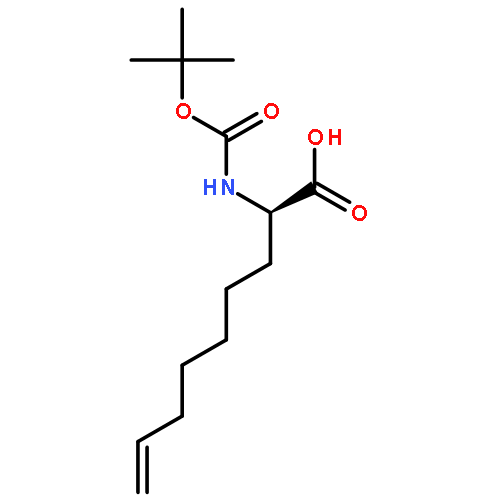
![7-Octenoic acid, 2-[[(1,1-dimethylethoxy)carbonyl]amino]-, (2S)-](http://img.cochemist.com/ccimg/552400/552335-71-4.png)
![7-Octenoic acid, 2-[[(1,1-dimethylethoxy)carbonyl]amino]-, (2S)-](http://img.cochemist.com/ccimg/552400/552335-71-4_b.png)