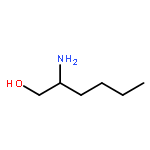
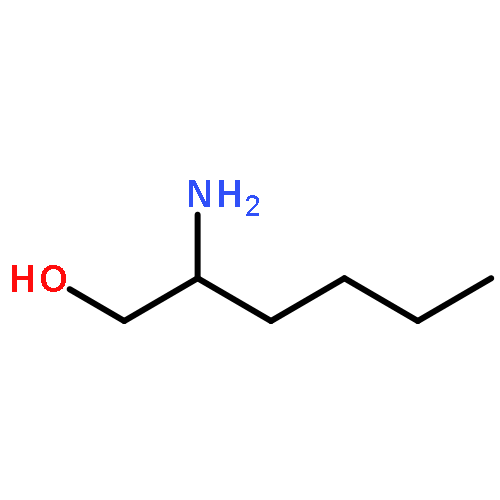
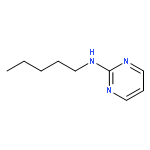
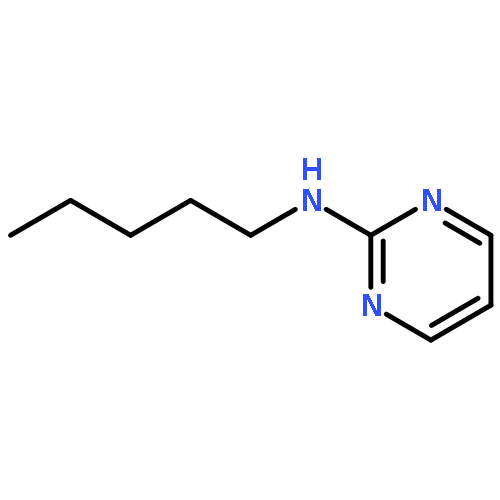
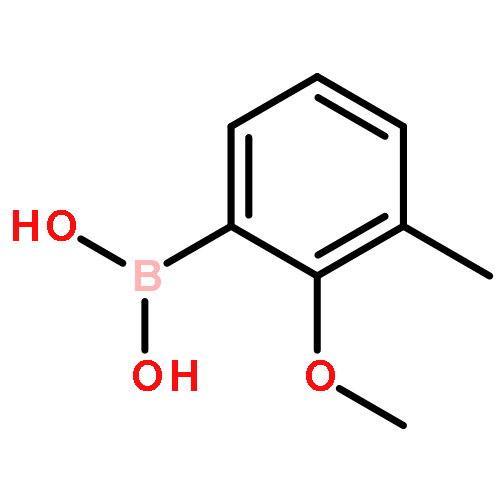
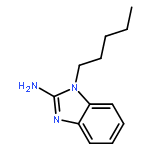
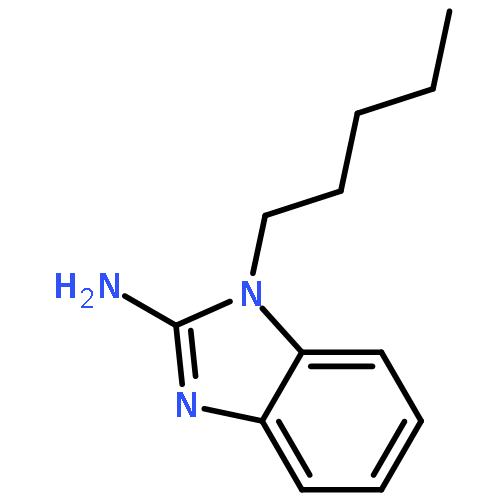
Co-reporter:Mallesh Beesu ; Subbalakshmi S. Malladi ; Lauren M. Fox ; Cassandra D. Jones ; Anshuman Dixit ;Sunil A. David
Journal of Medicinal Chemistry 2014 Volume 57(Issue 17) pp:7325-7341
Publication Date(Web):August 7, 2014
DOI:10.1021/jm500701q
Toll-like receptor (TLR)-8 agonists strongly induce the production of T helper 1-polarizing cytokines and may therefore serve as promising candidate vaccine adjuvants, especially for the very young and the elderly. Earlier structure-based ligand design led to the identification of 3-pentyl-quinoline-2-amine as a novel, human TLR8-specific agonist. Comprehensive structure–activity relationships in ring-contracted 1-alkyl-1H-benzimidazol-2-amines were undertaken, and the best-in-class compound, 4-methyl-1-pentyl-1H-benzo[d]imidazol-2-amine, was found to be a pure TLR8 agonist, evoking strong proinflammatory cytokine and Type II interferon responses in human PBMCs, with no attendant CD69 upregulation in natural lymphocytic subsets. The 1-alkyl-1H-benzimidazol-2-amines represent a novel, alternate chemotype with pure TLR8-agonistic activities and will likely prove useful not only in understanding TLR8 signaling but also perhaps as a candidate vaccine adjuvant.
Co-reporter:Euna Yoo ; Deepak B. Salunke ; Diptesh Sil ; Xiaoqiang Guo ; Alex C. D. Salyer ; Alec R. Hermanson ; Manoj Kumar ; Subbalakshmi S. Malladi ; Rajalakshmi Balakrishna ; Ward H. Thompson ; Hiromi Tanji ; Umeharu Ohto ; Toshiyuki Shimizu ;Sunil A. David
Journal of Medicinal Chemistry 2014 Volume 57(Issue 19) pp:7955-7970
Publication Date(Web):September 5, 2014
DOI:10.1021/jm500744f
Toll-like receptor (TLR) 7 and 8 agonists are potential vaccine adjuvants, since they directly activate APCs and enhance Th1-driven immune responses. Previous SAR investigations in several scaffolds of small molecule TLR7/8 activators pointed to the strict dependence of the selectivity for TLR7 vis-à-vis TLR8 on the electronic configurations of the heterocyclic systems, which we sought to examine quantitatively with the goal of developing “heuristics” to define structural requisites governing activity at TLR7 and/or TLR8. We undertook a scaffold-hopping approach, entailing the syntheses and biological evaluations of 13 different chemotypes. Crystal structures of TLR8 in complex with the two most active compounds confirmed important binding interactions playing a key role in ligand occupancy and biological activity. Density functional theory based quantum chemical calculations on these compounds followed by linear discriminant analyses permitted the classification of inactive, TLR8-active, and TLR7/8 dual-active compounds, confirming the critical role of partial charges in determining biological activity.
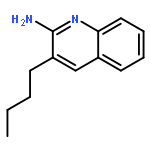
Co-reporter:Dr. Hari Prasad Kokatla;Dr. Diptesh Sil;Dr. Hiromi Tanji;Dr. Umeharu Ohto;Subbalakshmi S. Malladi;Lauren M. Fox; Toshiyoki Shimizu; Sunil A. David
ChemMedChem 2014 Volume 9( Issue 4) pp:719-723
Publication Date(Web):
DOI:10.1002/cmdc.201300573
Abstract
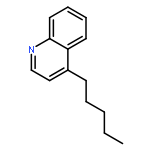
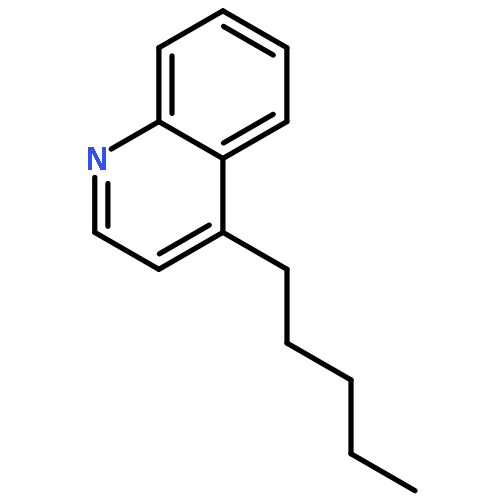
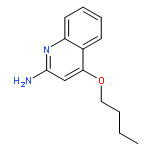
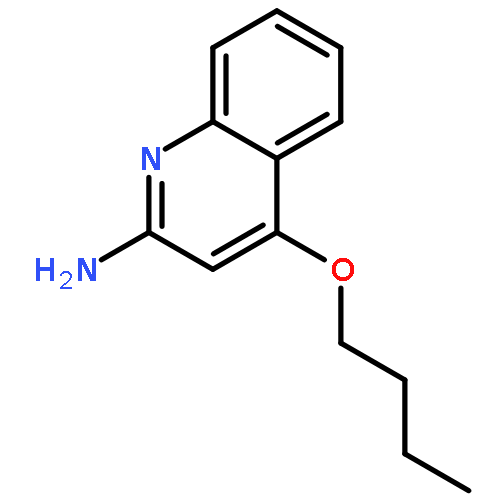
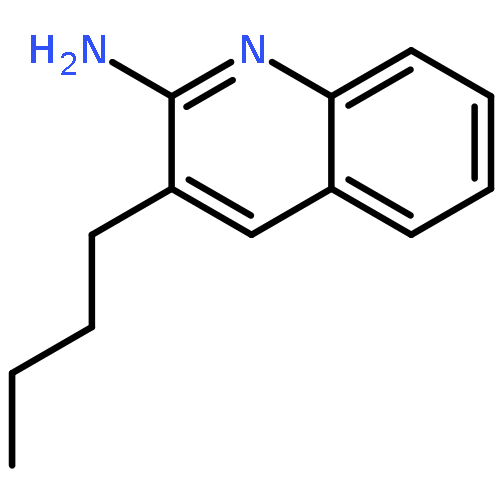
Toll-like receptor (TLR)-8 agonists activate adaptive immune responses by inducing robust production of T helper 1-polarizing cytokines, suggesting that TLR8-active compounds might be promising candidate vaccine adjuvants. Recently, a C2-butyl furo[2,3-c]quinoline was reported with purely TLR8 agonistic activity. This compound was successfully co-crystallized with the human TLR8 ectodomain, and the co-crystal structure revealed ligand-induced reorganization of the binding pocket of TLR8. The loss of a key hydrogen bond between the oxygen atom of the furanyl ring of the agonist and Thr 574 in TLR8 suggested that the furan ring is dispensable. Employing a disconnection strategy, 3- and 4-substituted aminoquinolines were investigated. Focused structure-based ligand design studies led to the identification of 3-pentyl-quinoline-2-amine as a novel, structurally simple, and highly potent human TLR8-specific agonist (EC50=0.2 μM). Preliminary evaluation of this compound in ex vivo human blood assay systems revealed that it retains prominent cytokine-inducing activity. Together, these results indicate the suitability of this compound as a novel vaccine adjuvant, warranting further investigation.
Co-reporter:Deepak B. Salunke ; Seth W. Connelly ; Nikunj M. Shukla ; Alec R. Hermanson ; Lauren M. Fox ;Sunil A. David
Journal of Medicinal Chemistry 2013 Volume 56(Issue 14) pp:5885-5900
Publication Date(Web):June 24, 2013
DOI:10.1021/jm400620g
Antigens in modern subunit vaccines are largely soluble and poorly immunogenic proteins inducing relatively short-lived immune responses. Appropriate adjuvants initiate early innate immune responses, amplifying subsequent adaptive immune responses. Agonists of Toll-like receptor 2 (TLR2) are devoid of significant proinflammatory activity in ex vivo human blood models and yet are potently adjuvantic, suggesting that this chemotype may be a safe and effective adjuvant. Our earlier work on the monoacyl lipopeptide class of TLR2 agonists led to the design of a highly potent lead but with negligible aqueous solubility, necessitating the reintroduction of aqueous solubility. We explored several strategies of introducing ionizable groups on the lipopeptide, as well as the systematic evaluation of chemically stable bioisosteres of the ester-linked palmitoyl group. These studies have led to a fully optimized, chemically stable, and highly water-soluble human TLR2-specific agonist, which was found to have an excellent safety profile and displayed prominent adjuvantic activities in rabbit models.
Co-reporter:Hari Prasad Kokatla ; Diptesh Sil ; Subbalakshmi S. Malladi ; Rajalakshmi Balakrishna ; Alec R. Hermanson ; Lauren M. Fox ; Xinkun Wang ; Anshuman Dixit ;Sunil A. David
Journal of Medicinal Chemistry 2013 Volume 56(Issue 17) pp:6871-6885
Publication Date(Web):July 30, 2013
DOI:10.1021/jm400694d
Toll-like receptor (TLR)-8 agonists activate adaptive immune responses by inducing robust production of T helper 1-polarizing cytokines, suggesting that TLR8-active compounds may be promising candidate adjuvants. We synthesized and evaluated hitherto unexplored furo[2,3-c]quinolines and regioisomeric furo[3,2-c]quinolines derived via a tandem, one-pot Sonogashira coupling and intramolecular 5-endo-dig cyclization strategy in a panel of primary screens. We observed a pure TLR8-agonistic activity profile in select furo[2,3-c]quinolines, with maximal potency conferred by a C2-butyl group (EC50 = 1.6 μM); shorter, longer, or substituted homologues as well as compounds bearing C1 substitutions were inactive, which was rationalized by docking studies using the recently described crystal structure of human TLR8. The best-in-class compound displayed prominent proinflammatory cytokine induction (including interleukin-12 and interleukin-18), but was bereft of interferon-α inducing properties, confirming its high selectivity for human TLR8.
Co-reporter:Euna Yoo, Breanna M. Crall, Rajalakshmi Balakrishna, Subbalakshmi S. Malladi, Lauren M. Fox, Alec R. Hermanson and Sunil A. David
Organic & Biomolecular Chemistry 2013 vol. 11(Issue 38) pp:6526-6545
Publication Date(Web):15 Aug 2013
DOI:10.1039/C3OB40816G
Engagement of TLR7 in plasmacytoid dendritic cells leads to the induction of IFN-α/β which plays essential functions in the control of adaptive immunity. We had previously examined structure–activity relationships (SAR) in TLR7/8-agonistic imidazoquinolines with a focus on substituents at the N1, C2, N3 and N4 positions, and we now report SAR on 1H-imidazo[4,5-c]pyridines. 1-Benzyl-2-butyl-1H-imidazo[4,5-c]pyridin-4-amine was found to be a pure TLR7-agonist with negligible activity on TLR8. Increase in potency was observed in N6-substituted analogues, especially in those compounds with electron-rich substituents. Direct aryl–aryl connections at C6 abrogated activity, but TLR7 agonism was reinstated in 6-benzyl and 6-phenethyl analogues. Consistent with the pure TLR7-agonistic behavior, prominent IFN-α induction in human PBMCs was observed with minimal proinflammatory cytokine induction. A benzologue of imidazoquinoline was also synthesized which showed substantial improvements in potency over the parent imidazopyridine. Distinct differences in N6-substituted analogues were observed with respect to IFN-α induction in human PBMCs on the one hand, and CD69 upregulation in lymphocytic subsets, on the other.
Co-reporter:Hari Prasad Kokatla, Euna Yoo, Deepak B. Salunke, Diptesh Sil, Cameron F. Ng, Rajalakshmi Balakrishna, Subbalakshmi S. Malladi, Lauren M. Fox and Sunil A. David
Organic & Biomolecular Chemistry 2013 vol. 11(Issue 7) pp:1179-1198
Publication Date(Web):21 Dec 2012
DOI:10.1039/C2OB26705E
Toll-like receptor (TLR)-8 agonists typified by the 2-alkylthiazolo[4,5-c]quinolin-4-amine (CL075) chemotype are uniquely potent in activating adaptive immune responses by inducing robust production of T helper 1-polarizing cytokines, suggesting that TLR8-active compounds could be promising candidate vaccine adjuvants, especially for neonatal vaccines. Alkylthiazoloquinolines with methyl, ethyl, propyl and butyl groups at C2 displayed comparable TLR8-agonistic potencies; activity diminished precipitously in the C2-pentyl compound, and higher homologues were inactive. The C2-butyl compound was unique in possessing substantial TLR7-agonistic activity. Analogues with branched alkyl groups at C2 displayed poor tolerance of terminal steric bulk. Virtually all modifications at C8 led to abrogation of agonistic activity. Alkylation on the C4-amine was not tolerated, whereas N-acyl analogues with short acyl groups (other than acetyl) retained TLR8 agonistic activity, but were substantially less water-soluble. Immunization in rabbits with a model subunit antigen adjuvanted with the lead C2-butyl thiazoloquinoline showed enhancements of antigen-specific antibody titers.
Co-reporter:Deepak B. Salunke ; Nikunj M. Shukla ; Euna Yoo ; Breanna M. Crall ; Rajalakshmi Balakrishna ; Subbalakshmi S. Malladi ;Sunil A. David
Journal of Medicinal Chemistry 2012 Volume 55(Issue 7) pp:3353-3363
Publication Date(Web):March 2, 2012
DOI:10.1021/jm3000533
Toll-like receptor 2-agonistic lipopeptides typified by S-[2,3-bis(palmitoyloxy)-(2RS)-propyl]-R-cysteinyl-S-serine (PAM2CS) compounds are potential vaccine adjuvants. We had previously determined that at least one acyl group of optimal length (C16) and an appropriately orientated ester carbonyl group is essential for TLR2-agonistic activity. We now show that these structurally simpler analogues display agonistic activities with human, but not murine, TLR2. SAR studies on the monoacyl derivatives show that the optimal acyl chain length is C16, and aryl substituents are not tolerated. A variety of alkyl and acyl substituents on the cysteine amine were examined. All N-alkyl derivatives were inactive. In contradistinction, short-chain N-acyl analogues were found to be highly active, with a clear dependence on the chain length. A cysteine N-acetyl analogue was found to be the most potent (EC50: 1 nM), followed by the N-butyryl analogue. The N-acetyl analogue is human TLR2-specific, with its potency comparable to that of PAM2CS.
Co-reporter:Deepak B. Salunke ; Euna Yoo ; Nikunj M. Shukla ; Rajalakshmi Balakrishna ; Subbalakshmi S. Malladi ; Katelyn J. Serafin ; Victor W. Day ; Xinkun Wang ;Sunil A. David
Journal of Medicinal Chemistry 2012 Volume 55(Issue 18) pp:8137-8151
Publication Date(Web):August 27, 2012
DOI:10.1021/jm301066h
In our ongoing search toward identifying novel and synthetically simpler candidate vaccine adjuvants, we hypothesized that the imidazo[1,2-a]pyrazines, readily accessible via the Groebke–Blackburn–Bienaymé multicomponent reaction, would possess sufficient structural similarity with TLR7/8-agonistic imidazoquinolines. With pyridoxal as the aldehyde component, furo[2,3-c]pyridines, rather than the expected imidazo[1,2-a]pyridines, were obtained, which were characterized by NMR spectroscopy and crystallography. Several analogues were found to activate TLR8-dependent NF-κB signaling. In a focused library of furo[2,3-c]pyridines, a distinct SAR was observed with varying substituents at C2. In human PBMCs, none of the furo[2,3-c]pyridines showed any proinflammatory cytokine induction but upregulated several chemokine ligand genes. In immunization studies in rabbits, the most active compound showed prominent adjuvantic effects. The complete lack of proinflammatory cytokine induction coupled with strong adjuvantic activity of the novel furo[2,3-c]pyridines render this hitherto unknown chemotype an attractive class of compounds which are expected to be devoid of local or systemic reactogenicity.
Co-reporter:Nikunj M. Shukla ; Cole A. Mutz ; Subbalakshmi S. Malladi ; Hemamali J. Warshakoon ; Rajalakshmi Balakrishna ;Sunil A. David
Journal of Medicinal Chemistry 2012 Volume 55(Issue 3) pp:1106-1116
Publication Date(Web):January 12, 2012
DOI:10.1021/jm2010207
Toll-like receptors (TLRs) are pattern recognition receptors that recognize specific molecular patterns present in molecules that are broadly shared by pathogens but are structurally distinct from host molecules. The TLR7-agonistic imidazoquinolines are of interest as vaccine adjuvants given their ability to induce pronounced Th1-skewed humoral responses. Minor modifications on the imidazoquinoline scaffold result in TLR7-antagonistic compounds which may be of value in addressing innate immune activation-driven immune exhaustion observed in HIV. We describe the syntheses and evaluation of TLR7 and TLR8 modulatory activities of dimeric constructs of imidazoquinoline linked at the C2, C4, C8, and N1-aryl positions. Dimers linked at the C4, C8, and N1-aryl positions were agonistic at TLR7; only the N1-aryl dimer with a 12-carbon linker was dual TLR7/8 agonistic. Dimers linked at C2 position showed antagonistic activities at TLR7 and TLR8; the C2 dimer with a propylene spacer was maximally antagonistic at both TLR7 and TLR8.
Co-reporter:Nikunj M. Shukla, Deepak B. Salunke, Euna Yoo, Cole A. Mutz, Rajalakshmi Balakrishna, Sunil A. David
Bioorganic & Medicinal Chemistry 2012 Volume 20(Issue 19) pp:5850-5863
Publication Date(Web):1 October 2012
DOI:10.1016/j.bmc.2012.07.052
We sought to explore the imidazo[1,2-a]pyridin-3-amines for TLR7 (or 8)-modulatory activities. This chemotype, readily accessed via the Groebke–Blackburn–Bienaymé multi-component reaction, resulted in compounds that were TLR7/8-inactive, but exhibited bacteriostatic activity against Gram-positive bacteria, including methicillin-resistant Staphylococcus aureus (MRSA). To investigate the mechanism of antibacterial activity of this new chemotype, a resistant strain of S. aureus was generated by serially passaging the organism in escalating doses of the most active analogue. A comparison of minimum inhibitory concentrations (MICs) of known bacteriostatic agents in wild-type and resistant strains indicates a novel mechanism of action. Structure–activity relationship studies have led to the identification of positions on the scaffold for additional structural modifications that should allow for the introduction of probes designed to examine cognate binding partners and molecular targets, while not significantly compromising antibacterial potency.
Co-reporter:Rehman Ukani, Tyler C. Lewis, Timothy P. Day, Wenyan Wu, Subbalakshmi S. Malladi, Hemamali J. Warshakoon, Sunil A. David
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 1) pp:293-295
Publication Date(Web):1 January 2012
DOI:10.1016/j.bmcl.2011.11.014
A bis-quinoline compound, (7-chloro-N-(4-(7-chloroquinolin-4-ylamino)butyl)quinolin-4-amine; RE-660) was found to have C–C chemokine receptor type 1 (CCR1)-agonistic properties. RE-660 displayed strong adjuvantic activity in mice when co-administered with bovine α-lactalbumin used as a model subunit protein antigen. RE-660 evoked a balanced Th1 (IgG2)/Th2 (IgG1) antibody profile, and the quality of antibodies elicited by the bis-quinoline was found to be superior to that evoked by glucopyranosyl lipid A by surface plasmon resonance experiments. No evidence of proinflammatory activity was observed in human blood ex vivo models. In preliminary acute toxicity studies, the compound was found to be of lower toxicity than chloroquine in mice, and was non-mutagenic in an Ames screen.
Co-reporter:Geetanjali Agnihotri ; Rehman Ukani ; Subbalakshmi S. Malladi ; Hemamali J. Warshakoon ; Rajalakshmi Balakrishna ; Xinkun Wang ;Sunil A. David
Journal of Medicinal Chemistry 2011 Volume 54(Issue 5) pp:1490-1510
Publication Date(Web):February 7, 2011
DOI:10.1021/jm101535e
N-Acyl-γ-glutamyldiaminopimelic acid is a prototype ligand for Nod1. We report a detailed SAR of C12-γ-d-Glu-DAP. Analogues with glutaric or γ-aminobutyric acid replacing the glutamic acid show greatly attenuated Nod1-agonistic activity. Substitution of the meso-diaminopimelic (DAP) acid component with monoaminopimelic acid, l- or d-lysine, or cadaverine also results in reduced activity. The free amine on DAP is crucial. However, the N-acyl group on the d-glutamyl residue can be substituted with N-alkyl groups with full preservation of activity. The free carboxylates on the DAP and Glu components can also be esterified, resulting in more lipophilic but active analogues. Transcriptomal profiling showed a dominant up-regulation of IL-19, IL-20, IL-22, and IL-24, which may explain the pronounced Th2-polarizing activity of these compounds and also implicate cell signaling mediated by TREM-1. These results may explain the hitherto unknown mechanism of synergy between Nod1 and TLR agonists and are likely to be useful in designing vaccine adjuvants.
Co-reporter:Geetanjali Agnihotri ; Breanna M. Crall ; Tyler C. Lewis ; Timothy P. Day ; Rajalakshmi Balakrishna ; Hemamali J. Warshakoon ; Subbalakshmi S. Malladi ;Sunil A. David
Journal of Medicinal Chemistry 2011 Volume 54(Issue 23) pp:8148-8160
Publication Date(Web):October 18, 2011
DOI:10.1021/jm201071e
Toll-like receptor 2-agonistic lipopeptides typified by S-[2,3-bis(palmitoyloxy)-(2RS)-propyl]-R-cysteinyl-S-serine (PAM2CS) compounds are potential vaccine adjuvants. In continuation of previously reported structure–activity relationships on this chemotype, we have determined that at least one acyl group of optimal length (C16) and an appropriately oriented ester carbonyl group is essential for TLR2-agonistic activity. The spacing between one of the palmitoyl ester carbonyl and the thioether is crucial to allow for an important H-bond, which observed in the crystal structure of the lipopeptide:TLR2 complex; consequently, activity is lost in homologated compounds. Penicillamine-derived analogues are also inactive, likely due to unfavorable steric interactions with the carbonyl of Ser 12 in TLR2. The thioether in this chemotype can be replaced with a selenoether. Importantly, the thioglycerol motif can be dispensed with altogether and can be replaced with a thioethanol bridge. These results have led to a structurally simpler, synthetically more accessible, and water-soluble analogue possessing strong TLR2-agonistic activities in human blood.
Co-reporter:Timothy P. Day, Diptesh Sil, Nikunj M. Shukla, Asokan Anbanandam, Victor W. Day, and Sunil A. David
Molecular Pharmaceutics 2011 Volume 8(Issue 1) pp:297-301
Publication Date(Web):December 8, 2010
DOI:10.1021/mp100363f
Aqueous solubilities of many drugs in current clinical use are very low, necessitating formulations that often present problems for parenteral administration, including toxicities due to the excipients used. Recognizing that pharmacologically active compounds frequently possess amines, we asked whether pyridoxal phosphate (PLP), an inoccuous, water-soluble vitamin, could be utilized to form prodrug-like complexes via the formation of imine or iminium adducts and whether the vitamin would impart solubilizing properties to such complexes. Direct spectroscopic and crystallographic data obtained using model primary and secondary amines showed that PLP forms stable imine adducts with primary amines under entirely aqueous conditions and at physiologic pH, while no reaction was observed for secondary amines; the basis of the exceptional stability appears to be a consequence of favorable H-bond interactions of the imine nitrogen with the 5-OH group of PLP. Amphotericin B and nystatin in their native forms display marked aqueous insolubility and possess lone primary amines. We were able to utilize PLP in achieving excellent solubilization of both of these antifungal agents, surpassing aqueous solubilities of 100 mg/mL. In in vitro bioassays, both polyenes in their PLP-adducted form display attenuated antifungal potencies which are attributable to “prodrug-like” complexes. These results point to the utility of excipient-free, entirely aqueous formulations of amphotericin B for parenteral use, and may also be extended to other primary amine-bearing compounds exhibiting poor aqueous solubility.Keywords: Amphotericin B; aqueous solubilization; nystatin; pyridoxal phosphate;
Co-reporter:Nikunj M. Shukla, Subbalakshmi S. Malladi, Victor Day, Sunil A. David
Bioorganic & Medicinal Chemistry 2011 Volume 19(Issue 12) pp:3801-3811
Publication Date(Web):15 June 2011
DOI:10.1016/j.bmc.2011.04.052
Toll-like receptors (TLR) -7 and -8 are thought to play an important role in immune activation processes underlying the pathophysiology of HIV and several clinically important autoimmune diseases. Based on our earlier findings of TLR7-antagonistic activity in a 3H imidazoquinoline, we sought to examine a pilot library of 3H imidazoquinolines for dual TLR7/8 antagonists, since they remain a poorly explored chemotype. 2D-NOE experiments were employed to unequivocally characterize the compounds. A quinolinium compound 12, bearing p-methoxybenzyl substituents on N3 and N5 positions was identified as a lead. Compound 12 was found to inhibit both TLR7 and TLR8 at low micromolar concentrations. Our preliminary results suggest that alkylation with electron-rich substituents on the quinoline N5, or conversely, elimination of the fixed charge of the resultant quaternary amine on the quinolinium may yield more active compounds.
Co-reporter:Nikunj M. Shukla, Tyler C. Lewis, Timothy P. Day, Cole A. Mutz, Rehman Ukani, Chase D. Hamilton, Rajalakshmi Balakrishna, Sunil A. David
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 11) pp:3232-3236
Publication Date(Web):1 June 2011
DOI:10.1016/j.bmcl.2011.04.050
Toll-like receptor (TLR)-7 agonists show prominent Th1-biased immunostimulatory activities. A TLR7-active N1-(4-aminomethyl)benzyl substituted imidazoquinoline 1 served as a convenient precursor for the syntheses of isothiocyanate and maleimide derivatives for covalent attachment to free amine and thiol groups of peptides and proteins. 1 was also amenable to direct reductive amination with maltoheptaose without significant loss of activity. Covalent conjugation of the isothiocyanate derivative 2 to α-lactalbumin could be achieved under mild, non-denaturing conditions, in a controlled manner and with full preservation of antigenicity. The self-adjuvanting α-lactalbumin construct induced robust, high-affinity immunoglobulin titers in murine models. The premise of covalently decorating protein antigens with adjuvants offers the possibility of drastically reducing systemic exposure of the adjuvant, and yet eliciting strong, Th1-biased immune responses.
Co-reporter:Wenyan Wu ; Rongti Li ; Subbalakshmi S. Malladi ; Hemamali J. Warshakoon ; Matthew R. Kimbrell ; Michael W. Amolins ; Rehman Ukani ; Apurba Datta ;Sunil A. David
Journal of Medicinal Chemistry 2010 Volume 53(Issue 8) pp:3198-3213
Publication Date(Web):March 19, 2010
DOI:10.1021/jm901839g
The N-termini of bacterial lipoproteins are acylated with a (S)-(2,3-bisacyloxypropyl)cysteinyl residue. Lipopeptides derived from lipoproteins activate innate immune responses by engaging Toll-like receptor 2 (TLR2) and are highly immunostimulatory and yet without apparent toxicity in animal models. The lipopeptides may therefore be useful as potential immunotherapeutic agents. Previous structure−activity relationships in such lipopeptides have largely been obtained using murine cells, and it is now clear that significant species-specific differences exist between human and murine TLR responses. We have examined in detail the role of the highly conserved Cys residue as well as the geometry and stereochemistry of the Cys-Ser dipeptide unit. (R)-Diacylthioglycerol analogues are maximally active in reporter gene assays using human TLR2. The Cys-Ser dipeptide unit represents the minimal part-structure, but its stereochemistry was found not to be a critical determinant of activity. The thioether bridge between the diacyl and dipeptide units is crucial, and replacement by an oxoether bridge results in a dramatic decrease in activity.
Co-reporter:Nikunj M. Shukla ; Subbalakshmi S. Malladi ; Cole A. Mutz ; Rajalakshmi Balakrishna ;Sunil A. David
Journal of Medicinal Chemistry 2010 Volume 53(Issue 11) pp:4450-4465
Publication Date(Web):May 18, 2010
DOI:10.1021/jm100358c
Engagement of toll-like receptors serve to link innate immune responses with adaptive immunity and can be exploited as powerful vaccine adjuvants for eliciting both primary and anamnestic immune responses. TLR7 agonists are highly immunostimulatory without inducing dominant proinflammatory cytokine responses. A structure−activity study was conducted on the TLR7-agonistic imidazoquinolines, starting with 1-(4-amino-2-((ethylamino)methyl)-1H-imidazo[4,5-c]quinolin-1-yl)-2-methylpropan-2-ol as a lead. Modifications of the secondary amine of the C2 ethylaminomethylene side chain are poorly tolerated. The 4-amino group must be retained for activity. Replacement of the imidazole ring of the scaffold with triazole or cyclic urea led to complete loss of activity. A systematic exploration of N1-benzyl-C2-alkyl substituents showed a very distinct relationship between alkyl length and TLR7-agonistic potency with the optimal compound bearing a C2-n-butyl group. Transposition of the N1 and C2 substituents led to the identification of an extremely active TLR7-agonistic compound with an EC50 value of 8.6 nM. The relative potencies in human TLR7-based primary reporter gene assays were paralleled by interferon-α induction activities in whole human blood models.
Co-reporter:Nikunj M. Shukla, Cole A. Mutz, Rehman Ukani, Hemamali J. Warshakoon, David S. Moore, Sunil A. David
Bioorganic & Medicinal Chemistry Letters 2010 Volume 20(Issue 22) pp:6384-6386
Publication Date(Web):15 November 2010
DOI:10.1016/j.bmcl.2010.09.093
Toll-like receptor (TLR)-7 agonists show prominent immunostimulatory activities. The synthesis of a TLR7-active N1-(4-aminomethyl)benzyl substituted imidazoquinoline 5d served as a convenient precursor for the covalent attachment of fluorophores without significant loss of activity. Fluorescence microscopy experiments show that the fluorescent analogues are internalized and distributed in the endosomal compartment. Flow cytometry experiments using whole human blood show differential partitioning into B, T, and natural killer (NK) lymphocytic subsets, which correlate with the degree of activation in these subsets. These fluorescently-labeled imidazoquinolines will likely be useful in examining the trafficking of TLR7 in immunological synapses.
Co-reporter:Samusi A. Adediran, Timothy P. Day, Diptesh Sil, Matthew R. Kimbrell, Hemamali J. Warshakoon, Subbalakshmi S. Malladi and Sunil A. David
Molecular Pharmaceutics 2009 Volume 6(Issue 5) pp:1582-1590
Publication Date(Web):August 7, 2009
DOI:10.1021/mp9001602
Amphotericin B (AmB), a well-known polyene antifungal agent, displays a marked tendency to self-associate and, as a consequence, exhibits very poor solubility in water. The therapeutic index of AmB is low and is associated with significant dose-related nephrotoxicity, as well as acute, infusion-related febrile reactions. Reports in the literature indicate that the toxicity of AmB may be related to the physical state of the drug. Reaction of AmB in dimethylformamide with bis(dimethylaminopropyl)carbodiimide yielded an unexpected N-alkylguanidine/N-acylurea bis-adduct of AmB which was highly water-soluble. The absorption spectrum of the AmB derivative in water indicated excellent monomerization, and the antifungal activities of reference AmB and its water-soluble derivative against Candida albicans were found to be virtually identical. Furthermore, the water-soluble adduct is significantly less active in engaging TLR4, which would suggest that the adduct may be less proinflammatory.Keywords: aggregation; Amphotericin B; antifungal; carbodiimide; solubility; toll-like receptors; toxicity;
Co-reporter:Wenyan Wu, Diptesh Sil, Michal L. Szostak, Subbalakshmi S. Malladi, Hemamali J. Warshakoon, Matthew R. Kimbrell, Jens R. Cromer, Sunil A. David
Bioorganic & Medicinal Chemistry 2009 Volume 17(Issue 2) pp:709-715
Publication Date(Web):15 January 2009
DOI:10.1016/j.bmc.2008.11.051
The toxicity of Gram-negative bacterial endotoxin (lipopolysaccharide, LPS) resides in its structurally highly conserved glycolipid component called lipid A. Our major goal has been to develop small-molecules that would sequester LPS by binding to the lipid A moiety, so that it could be useful for the prophylaxis or adjunctive therapy of Gram-negative sepsis. We had previously identified in rapid-throughput screens several guanylhydrazones as potent LPS binders. We were desirous of examining if the presence of the guanylhydrazone (rather than an amine) functionality would afford greater LPS sequestration potency. In evaluating a congeneric set of guanylhydrazone analogues, we find that C16 alkyl substitution is optimal in the N-alkylguanylhydrazone series; a homospermine analogue with the terminal amine N-alkylated with a C16 chain with the other terminus of the molecule bearing an unsubstituted guanylhydrazone moiety is marginally more active, suggesting very slight, if any, steric effects. Neither C16 analogue is significantly more active than the N-C16-alkyl or N-C16-acyl compounds that we had characterized earlier, indicating that basicity of the phosphate-recognizing cationic group, is not a determinant of LPS sequestration activity.
Co-reporter:Anurupa Shrestha, Diptesh Sil, Subbalakshmi S. Malladi, Hemamali J. Warshakoon, Sunil A. David
Bioorganic & Medicinal Chemistry Letters 2009 Volume 19(Issue 9) pp:2478-2481
Publication Date(Web):1 May 2009
DOI:10.1016/j.bmcl.2009.03.055
We have previously shown that simple N-acyl or N-alkyl polyamines bind to and sequester Gram-negative bacterial lipopolysaccharide, affording protection against lethality in animal models of endotoxicosis. Several iterative design-and-test cycles of SAR studies, including high-throughput screens, had converged on compounds with polyamine scaffolds which have been investigated extensively with reference to the number, position, and length of acyl or alkyl appendages. However, the polyamine backbone itself had not been explored sufficiently, and it was not known if incremental variations on the polymethylene spacing would affect LPS-binding and neutralization properties. We have now systematically explored the relationship between variously elongated spermidine [NH2–(CH2)3–NH–(CH2)4–NH2] and norspermidine [NH2–(CH2)3–NH–(CH2)3–NH2] backbones, with the N-alkyl group being held constant at C16 in order to examine if changing the spacing between the inner secondary amines may yield additional SAR information. We find that the norspermine-type compounds consistently showed higher activity compared to corresponding spermine homologues.In exploring the relationship between variously elongated N-alkyl spermidine and norspermidine, norspermine-type compounds consistently showed higher lipopolysaccharide-sequestering activities.
Co-reporter:Nikunj M. Shukla, Matthew R. Kimbrell, Subbalakshmi S. Malladi, Sunil A. David
Bioorganic & Medicinal Chemistry Letters 2009 Volume 19(Issue 8) pp:2211-2214
Publication Date(Web):15 April 2009
DOI:10.1016/j.bmcl.2009.02.100
Chronic immune activation is a hallmark of progressive HIV infection. Recent reports point to the engagement of toll-like receptor 7 (TLR7) and -9 by viral RNA as contributing to the activation of innate immune responses, which drive viral replication leading to immune exhaustion. The only known class of TLR7 antagonists is single-stranded phosphorothioate oligonucleotides, which has been demonstrated to inhibit immune activation in human and Rhesus macaque in vitro models. The availability of a selective and potent small-molecule TLR7 antagonist should allow the evaluation of potential benefits of suppression of TLR7-mediated immune activation in HIV/AIDS. Gardiquimod is a known N1-substituted 1H-imidazoquinoline TLR7 agonist, the synthesis of which has not been published. We show that the 3H regioisomer is completely inactive as a TLR7 agonist and is weakly antagonistic. A des-amino precursor of the 3H regioisomer is more potent as a TLR7 antagonist, with an IC50 value of 7.5 μM. This class of compound may serve as a starting point for the development of small-molecule inhibitors of TLR7.A regioisomer of a known TLR7 agonist was found to have TLR7 antagonistic activity.Figure optionsDownload full-size imageDownload as PowerPoint slide
Co-reporter:Thuan B. Nguyen, E. V. K. Suresh Kumar, Diptesh Sil, Stewart J. Wood, Kelly A. Miller, Hemamali J. Warshakoon, Apurba Datta and Sunil A. David
Molecular Pharmaceutics 2008 Volume 5(Issue 6) pp:1131-1137
Publication Date(Web):October 7, 2008
DOI:10.1021/mp8001123
Hydrophobically substituted polyamine compounds, particularly N-acyl or N-alkyl derivatives of homospermine, are potent endotoxin (lipopolysaccharide) sequestrants. Despite their polycationic nature, the aqueous solubilites are limited owing to the considerable overall hydrophobicity contributed by the long-chain aliphatic substituent, but solubilization is readily achieved in the presence of human serum albumin (HSA). We desired first to delineate the structural basis of lipopolyamine−albumin interactions and, second, to explore possible structure−activity correlates in a well-defined, congeneric series of N-alkyl and -acyl homospermine lead compounds. Fluorescence spectroscopic and isothermal titration calorimetry (ITC) results indicate that these compounds appear to bind to HSA via occupancy of the fatty-acid binding sites on the protein. The acyl and carbamate compounds bind HSA the strongest; the ureido and N-alkyl analogues are significantly weaker, and the branched alkyl compound is weaker still. ITC-derived dissociation constants are weighted almost in their entirety by enthalpic ΔH terms, which is suggestive that the polarizability of the carbonyl groups facilitate, at least in large part, their interactions with HSA. The relative affinities of these lipopolyamines toward HSA is reflected in discernible differences in apparent potencies of LPS-sequestering activity under experimental conditions requiring physiological concentrations of HSA, and also of in vivo pharmacodynamic behavior. These results are likely to be useful in designing analogues with varying pharmacokinetic profiles.Keywords: Albumin; endotoxin sequestrants; lipopolyamines; plasma protein binding;
Co-reporter:Anurupa Shrestha;Rongti Li;Diptesh Sil;Neha N. Pardeshi;Nancy Schwarting;Karl S. Schorno;Roger A. Rajewski;Apurba Datta;Sunil A. David
Journal of Pharmaceutical Sciences 2008 Volume 97( Issue 12) pp:5376-5385
Publication Date(Web):
DOI:10.1002/jps.21361
Abstract
The pharmacokinetics of DS-96, an N-alkylhomospermine analog designed to sequester bacterial lipopolysaccharides, has been determined in rodent species. The elimination half-life in mice and rats are about 400 and 500 min, respectively, with other PK parameters being quite similar in the two rodent species. Interestingly, the mouse intravenous plasma concentration time curves exhibit an apparent absorption phase. While the rat intravenous data did not exhibit a pronounced apparent absorption phase immediately following injection, plasma levels did increase between 10 and 30 min following an expected drop from time 0 to 5 min. The data are consistent with first-pass uptake, possibly by the lung, with back diffusion as a function of time. The observed Cmax values of 1.36 µg/mL in the mouse intraperitoneal model suggest that a plasma concentration of 0.5–1 µg/mL corresponds to complete protection for a 200 ng/animal dose of intraperitoneally administered LPS in the D-galactosamine-primed model of endotoxin-induced lethality. © 2008 Wiley-Liss, Inc. and the American Pharmacists Association J Pharm Sci 97:5376–5385, 2008
Co-reporter:Jian-Xin Guo, Stewart J. Wood, Sunil A. David, Gerald H. Lushington
Bioorganic & Medicinal Chemistry Letters 2006 Volume 16(Issue 3) pp:714-717
Publication Date(Web):1 February 2006
DOI:10.1016/j.bmcl.2005.10.025
Lipopolysaccharides (LPS), otherwise termed ‘endotoxins’, are outer-membrane constituents of Gram-negative bacteria and play a key role in the pathogenesis of ‘septic shock’, a major cause of mortality in the critically ill patient. We have shown that the pharmacophore necessary for optimal recognition and neutralization of LPS by small molecules requires an interaction between two protonatable positive charges separated by a distance of ∼14 Å, which corresponds to the distance between two anionic phosphates on the glycolipid component of LPS called lipid A. The in silico binding of a diverse set of compounds with bis-amino, -amidino, -guanidino, and -aminoguanidino functionalities, identified as potential lead scaffolds in a high-throughput screen, with lipid A was explored using molecular docking simulations. A weighted expression for binding affinity was trained relative to experimental ED50 measurements, attaining a correlation of R2 = 0.66. Our docking results showed that the electrostatic interaction between ligands and lipid A phosphates dominates the expression and varies little across the series, and other ligand–receptor interactions seem to play a secondary role in governing the observed variations in the relative ligand binding affinity. Further, it appears that the ligand internal energy plays the primary role in differentiating between compound binding affinities which also correlated well with experimental ED50 data (R = 0.77). Application of this strategy would be useful in the de novo design of highly active endotoxin-sequestering agents.The lipopolysaccharide pharmacophore, dominated by cationic phosphate centers but also comprising hydrophobic and H-bond acceptor and donor sites.
Co-reporter:Mark R. Burns, Scott A. Jenkins, Nicolas M. Vermeulen, Rajalakshmi Balakrishna, Thuan B. Nguyen, Matthew R. Kimbrell, Sunil A. David
Bioorganic & Medicinal Chemistry Letters 2006 Volume 16(Issue 24) pp:6209-6212
Publication Date(Web):15 December 2006
DOI:10.1016/j.bmcl.2006.09.026
Lipopolysaccharides (LPS), otherwise termed ‘endotoxins’, are outer-membrane constituents of Gram-negative bacteria, and play a key role in the pathogenesis of ‘Septic Shock’, a major cause of mortality in the critically ill patient. We had previously defined the pharmacophore necessary for small molecules to specifically bind and neutralize this complex carbohydrate. A series of aryl and aliphatic spermine-sulfonamide analogs were synthesized and tested in a series of binding and cell-based assays in order to probe the effect of lipophilicity on sequestration ability. A strong correlation was indeed found, supporting the hypothesis that endotoxin-neutralizing ability involves a lipophilic or membrane attachment event. The research discussed herein may be useful for the design of additional carbohydrate recognizing molecules and endotoxin-neutralizing drugs.SAR in a series of aryl and aliphatic spermine-sulfonamide analogs showed a strong correlation between hydrophobicity of the substituent and lipopolysaccharide-sequestration activity.
Co-reporter:Mark R. Burns, Stewart J. Wood, Kelly A. Miller, Thuan Nguyen, Jens R. Cromer, Sunil A. David
Bioorganic & Medicinal Chemistry 2005 Volume 13(Issue 7) pp:2523-2536
Publication Date(Web):1 April 2005
DOI:10.1016/j.bmc.2005.01.038
Lipopolysaccharides (LPS), otherwise termed ‘endotoxins’, are outer-membrane constituents of Gram-negative bacteria. Lipopolysaccharides play a key role in the pathogenesis of ‘Septic Shock’, a major cause of mortality in the critically ill patient. Therapeutic options aimed at limiting downstream systemic inflammatory processes by targeting lipopolysaccharide do not exist at the present time. We have defined the pharmacophore necessary for small molecules to specifically bind and neutralize LPS and, using animal models of sepsis, have shown that the sequestration of circulatory LPS by small molecules is a therapeutically viable strategy. In this paper, the interactions of a focused library of lysine–spermine conjugates with lipopolysaccharide (LPS) have been characterized. Lysine–spermine conjugates with the ε-amino terminus of the lysinyl moiety derivatized with long-chain aliphatic hydrophobic substituents in acyl or alkyl linkage bind and neutralize bacterial lipopolysaccharides, and may be of use in the prevention or treatment of endotoxic shock states.Lysine–spermine conjugates with a long-chain aliphatic (C16–C20) substituent at R1 bind and neutralize bacterial lipopolysaccharides. These compounds reduce lethality in a murine model of lipopolysaccharide-induced shock, and may serve as novel leads for developing novel anti-lipopolysaccharide agents for the therapy of Gram-negative sepsis.
Co-reporter:Sunil David, Lourdes Pérez, M.Rosa Infante
Bioorganic & Medicinal Chemistry Letters 2002 Volume 12(Issue 3) pp:357-360
Publication Date(Web):11 February 2002
DOI:10.1016/S0960-894X(01)00749-1
Gemini compounds of the type Nα,Nω-bis(Nα-lauroyl arginine)α,ω-alkylenediamides or bis(Args) bind bacterial lipopolysaccharide and neutralize endotoxic activity in in vitro tumor necrosis factor-α and nitric oxide release assays. Sequestration of lipopolysaccharide results in protection in a murine model of endotoxemia. However, the bis(Args) compounds are cytotoxic by virtue of being highly membrane-active. The development of less surface-active analogues may yield potentially therapeutically useful compounds for the treatment of Gram-negative sepsis.Graphic
Co-reporter:Sunil A. David
Journal of Molecular Recognition 2001 Volume 14(Issue 6) pp:370-387
Publication Date(Web):20 DEC 2001
DOI:10.1002/jmr.549
Endotoxins, or lipopolysaccharides (LPS), present on the surface of Gram-negative bacteria, play a key role in the pathogenesis of septic shock, a common clinical problem and a leading cause of mortality in critically ill patients, for which no specific therapeutic modalities are available at the present time. The toxic moiety of LPS is a glycolipid called ‘lipid A’, which is composed of a bisphosphorylated diglucosamine backbone bearing up to seven acyl chains in ester and amide linkages. Lipid A is structurally highly conserved in Gram-negative bacteria, and is therefore an attractive target for developing anti-endotoxin molecules designed to sequester, and thereby neutralize, the deleterious effects of endotoxins. The anionic and amphipathic nature of lipid A enables the interaction of a wide variety of cationic amphiphiles with the toxin. This review describes the systematic evaluation of several structural classes of cationic amphiphiles, both peptides and non-peptidic small molecules, in the broader context of recent efforts aimed at developing novel anti-endotoxin strategies. The derivation of a pharmacophore for LPS recognition has led to the identification of novel, nontoxic, structurally simple small molecules, the lipopolyamines. The lipopolyamines bind and neutralize LPS in in vitro experiments as well as in animal models of endotoxicity, and thus present novel and exciting leads for rational, structure-based development of LPS-sequestering agents of potential clinical value. Copyright © 2001 John Wiley & Sons, Ltd.
Co-reporter:Matthew R. Kimbrell, Hemamali Warshakoon, Jens R. Cromer, Subbalakshmi Malladi, ... Sunil A. David
Immunology Letters (30 June 2008) Volume 118(Issue 2) pp:132-141
Publication Date(Web):30 June 2008
DOI:10.1016/j.imlet.2008.03.009
The role of lipopolysaccharide (LPS) in the pathogenesis of Gram-negative septic shock is well established. The corresponding proinflammatory and immunostimulatory molecule(s) on the Gram-positive bacteria is less well understood, and its identification and characterization would be a key prerequisite in designing specific sequestrants of the Gram-positive endotoxin(s). We report in this paper the comparison of NF-κB-, cytokine- and chemokine-inducing activities of the TLR2 ligands, lipoteichoic acid (LTA), peptidoglycan (PGN), and lipopeptides, to LPS, a prototype TLR4 agonist, in murine macrophage cell-lines as well as in human blood. In murine cells, di- and triacyl liopopeptides are equipotent in their NF-κB inducing activity relative to LPS, but elicit much lower proinflammatory cytokines. However, both LPS and the lipopeptides potently induce the secretion of a pattern of chemokines that is suggestive of the engagement of a TLR4-independent TRIF pathway. In human blood, although the lipopeptides induce p38 MAP kinase phosphorylation and CD11b upregulation in granulocytes at ng/ml concentrations, they do not elicit proinflammatory cytokine production even at very high doses; LTA, however, activates neutrophils and induces cytokine secretion, although its potency is considerably lower than that of LPS, presumably due to its binding to plasma proteins. We conclude that, in human blood, the pattern of immunostimulation and proinflammatory mediator production elicited by LTA parallels that of LPS.
Co-reporter:Hari Prasad Kokatla, Euna Yoo, Deepak B. Salunke, Diptesh Sil, Cameron F. Ng, Rajalakshmi Balakrishna, Subbalakshmi S. Malladi, Lauren M. Fox and Sunil A. David
Organic & Biomolecular Chemistry 2013 - vol. 11(Issue 7) pp:NaN1198-1198
Publication Date(Web):2012/12/21
DOI:10.1039/C2OB26705E
Toll-like receptor (TLR)-8 agonists typified by the 2-alkylthiazolo[4,5-c]quinolin-4-amine (CL075) chemotype are uniquely potent in activating adaptive immune responses by inducing robust production of T helper 1-polarizing cytokines, suggesting that TLR8-active compounds could be promising candidate vaccine adjuvants, especially for neonatal vaccines. Alkylthiazoloquinolines with methyl, ethyl, propyl and butyl groups at C2 displayed comparable TLR8-agonistic potencies; activity diminished precipitously in the C2-pentyl compound, and higher homologues were inactive. The C2-butyl compound was unique in possessing substantial TLR7-agonistic activity. Analogues with branched alkyl groups at C2 displayed poor tolerance of terminal steric bulk. Virtually all modifications at C8 led to abrogation of agonistic activity. Alkylation on the C4-amine was not tolerated, whereas N-acyl analogues with short acyl groups (other than acetyl) retained TLR8 agonistic activity, but were substantially less water-soluble. Immunization in rabbits with a model subunit antigen adjuvanted with the lead C2-butyl thiazoloquinoline showed enhancements of antigen-specific antibody titers.
Co-reporter:Euna Yoo, Breanna M. Crall, Rajalakshmi Balakrishna, Subbalakshmi S. Malladi, Lauren M. Fox, Alec R. Hermanson and Sunil A. David
Organic & Biomolecular Chemistry 2013 - vol. 11(Issue 38) pp:NaN6545-6545
Publication Date(Web):2013/08/15
DOI:10.1039/C3OB40816G
Engagement of TLR7 in plasmacytoid dendritic cells leads to the induction of IFN-α/β which plays essential functions in the control of adaptive immunity. We had previously examined structure–activity relationships (SAR) in TLR7/8-agonistic imidazoquinolines with a focus on substituents at the N1, C2, N3 and N4 positions, and we now report SAR on 1H-imidazo[4,5-c]pyridines. 1-Benzyl-2-butyl-1H-imidazo[4,5-c]pyridin-4-amine was found to be a pure TLR7-agonist with negligible activity on TLR8. Increase in potency was observed in N6-substituted analogues, especially in those compounds with electron-rich substituents. Direct aryl–aryl connections at C6 abrogated activity, but TLR7 agonism was reinstated in 6-benzyl and 6-phenethyl analogues. Consistent with the pure TLR7-agonistic behavior, prominent IFN-α induction in human PBMCs was observed with minimal proinflammatory cytokine induction. A benzologue of imidazoquinoline was also synthesized which showed substantial improvements in potency over the parent imidazopyridine. Distinct differences in N6-substituted analogues were observed with respect to IFN-α induction in human PBMCs on the one hand, and CD69 upregulation in lymphocytic subsets, on the other.



![Thiazolo[4,5-c]quinolin-4-amine, 2-butyl-](http://img.cochemist.com/ccimg/257000/256922-56-2.png)
![Thiazolo[4,5-c]quinolin-4-amine, 2-butyl-](http://img.cochemist.com/ccimg/257000/256922-56-2_b.png)
![2-Propylthiazolo[4,5-c]quinolin-4-amine](http://img.cochemist.com/ccimg/257000/256922-53-9.png)
![2-Propylthiazolo[4,5-c]quinolin-4-amine](http://img.cochemist.com/ccimg/257000/256922-53-9_b.png)
![Thiazolo[4,5-c]quinolin-2-amine](http://img.cochemist.com/ccimg/143700/143667-61-2.png)
![Thiazolo[4,5-c]quinolin-2-amine](http://img.cochemist.com/ccimg/143700/143667-61-2_b.png)


![[1,2,4]Triazolo[4,3-a]quinoxaline, 1-butyl-4-chloro-](http://img.cochemist.com/ccimg/113200/113181-23-0.png)
![[1,2,4]Triazolo[4,3-a]quinoxaline, 1-butyl-4-chloro-](http://img.cochemist.com/ccimg/113200/113181-23-0_b.png)
![1H-Naphth[2,3-d]imidazol-2-amine(9CI)](http://img.cochemist.com/ccimg/102500/102408-31-1.png)
![1H-Naphth[2,3-d]imidazol-2-amine(9CI)](http://img.cochemist.com/ccimg/102500/102408-31-1_b.png)



![1-Butyl-1H-benzo[d]imidazol-2-amine](http://img.cochemist.com/ccimg/91400/91337-45-0.png)
![1-Butyl-1H-benzo[d]imidazol-2-amine](http://img.cochemist.com/ccimg/91400/91337-45-0_b.png)