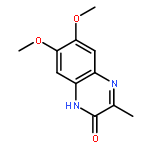
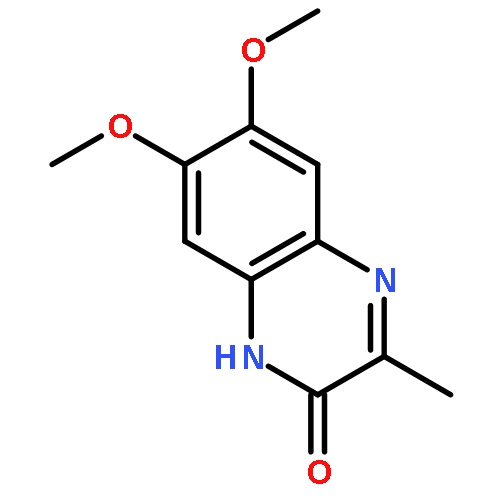
Co-reporter:Qiao-Hong Xia, Wei Hu, Chen Li, Ji-Feng Wu, Liang Yang, Xue-Mei Han, Yue-Mao Shen, Zhi-Yu Li, Xun Li
European Journal of Medicinal Chemistry 2016 Volume 124() pp:311-325
Publication Date(Web):29 November 2016
DOI:10.1016/j.ejmech.2016.08.010
•Quinoxaline peptidomimetic analogues derived from XK469 were proposed.•These derivatives tend to have similar mechanism of action with XK469.•Molecular docking studies provided visual evidences of binding modes of target compounds.•Two compounds standed out from bioevaluation and might be promising leads.XK469 is identified as a potent quinoxaline antineoplastic agent based on its significant clinical efficacy. It probably exerts its activity via DNA topoisomerase II (topo II) inhibition. To obtain more effective antineoplastic agents, a spectrum of peptidomimetic-type quinoxaline analogues of XK469 was herein designed, synthesized, and evaluated. Few compounds (e.g. 13a and 13b) exhibited obvious cytotoxicity indicated by in vitro anti-proliferative assay. SAR investigation revealed that introducing of hydrophobic tert-butylamine or dodecylamine moiety at the 3-position of quinoxaline core is favorable for achieving a better anti-proliferative potency, while peptidomimetic derivatives only yielded moderate cytotoxicity. Compounds with improved anti-proliferative activities also demonstrated decent anti-metastatic potencies comparable with that of doxorubicin (Doxo) based on in vivo mouse model study. The topo II-mediated kinetoplast DNA (kDNA) decatenation assay as well as molecular docking studies implicated that these compounds tend to be potent topo II inhibitors. Overall, compounds 13a and 13b, 13b in particular, standed out from various assessments and might be promising candidates for further chemical optimization.
Co-reporter:Leilei Shi, Jianfeng Zhou, Jifeng Wu, Junya Cao, Yuemao Shen, Hua Zhou, Xun Li
Bioorganic & Medicinal Chemistry 2016 Volume 24(Issue 8) pp:1840-1852
Publication Date(Web):15 April 2016
DOI:10.1016/j.bmc.2016.03.008
Inhibition of VEGFR-2 kinase has been highlighted as one of the well-defined strategies to suppress tumor growth via blockade of angiogenesis. Guided by the principles of bioisosteric replacement and pharmacophoric fragment migration, a series of novel quinoxalinone derivates were designed, synthesized and evaluated for their VEGFR-2 inhibitory potencies. Among them, compounds 7c, 8b, 8c, 8e and 10b displayed antiangiogenic abilities via the in vitro tube formation assay (cellular level) and ex vivo rat aortic ring assay (tissue level) at a low concentration (0.1 μM). By means of in vivo zebrafish embryo model, two (Z)-3-(2-(pyridin-4-yl)vinyl)quinoxalinone derivates 8c and 8e showed significant antiangiogenesis effects, suggesting they have potentials to be developed into antiangiogenesis agents via further structural optimization. Moreover, these two compounds also demonstrated potent inhibition toward VEGFR-2 and B-raf kinases in a low concentration (1 μM). A possible interpretation of our evaluation result has been presented by a molecular docking study by docking representative compound 8c with VEGFR-2.
Co-reporter:Liang Yang, Ping Wang, Ji-Feng Wu, Liu-Meng Yang, Rui-Rui Wang, Wei Pang, Yong-Gang Li, Yue-Mao Shen, Yong-Tang Zheng, Xun Li
Bioorganic & Medicinal Chemistry 2016 Volume 24(Issue 9) pp:2125-2136
Publication Date(Web):1 May 2016
DOI:10.1016/j.bmc.2016.03.043
As our ongoing work on research of gelatinase inhibitors, an array of hydrazide-containing peptidomimetic derivatives bearing quinoxalinone as well as spiro-heterocyclic backbones were designed, synthesized, and assayed for their in vitro enzymatic inhibitory effects. The results demonstrated that both the quinoxalinone (series I and II) and 1,4-dithia-7-azaspiro[4,4]nonane-based hydrazide peptidomimetics (series III) displayed remarkably selectivity towards gelatinase A as compared to APN, with IC50 values in the micromole range. Structure–activity relationships were herein briefly discussed. Given evidences have validated that gelatinase inhibition may be contributable to the therapy of HIV-1 infection, all the target compounds were also submitted to the preliminary in vitro anti-HIV-1 evaluation. It resulted that gelatinase inhibition really has positive correlation with anti-HIV-1 activity, especially compounds 4m and 7h, which gave enhanced gelatinase inhibition in comparison with the positive control LY52, and also decent anti-HIV-1 potencies. The FlexX docking results provided a straightforward insight into the binding pattern between inhibitors and gelatinase, as well as the selective inhibition towards gelatinase over APN. Collectively, our research encouraged potent gelatinase inhibitors might be used in the development of anti-HIV-1 agents. And else, compounds 4m and 7h might be promising candidates to be considered for further chemical optimization.
Co-reporter:Leilei Shi, Dongmei Zhang, Riyuan Lin, Chun Zhang, Xun Li, Ning Jiao
Tetrahedron Letters 2014 Volume 55(Issue 14) pp:2243-2245
Publication Date(Web):2 April 2014
DOI:10.1016/j.tetlet.2014.02.071
A novel and efficient transition metal-free C–H bond halogenation of indole derivatives has been developed. 3-Halogenated (3-Br, 3-I) indoles are highly regioselectively produced by this protocol. Simple and readily available halide salts (TBAB, KI) are employed as the halogen source. The transition metal-free and the mild conditions make this protocol very easy to handle and practical.Graphical abstractA novel and efficient transition metal-free C–H bond halogenation of indole derivatives has been developed. 3-Halogenated (3-Br, 3-I) indoles are highly regioselectively produced by this protocol. Simple and readily available halide salts (TBAB, KI) are employed as the halogen source. The transition metal-free and the mild conditions make this protocol very easy to handle and practical.
Co-reporter:Leilei Shi, Qiang Wang, Haina Wang, Hua Zhou, Yonggang Li, Xun Li
Bioorganic & Medicinal Chemistry 2013 Volume 21(Issue 24) pp:7752-7762
Publication Date(Web):15 December 2013
DOI:10.1016/j.bmc.2013.10.016
Gelatinase A, a zinc-containing endopeptidase, has been shown to be an essential therapeutic target for tumor intervention owing to its participation in almost all types of solid tumors. Based on our previous work with respect to quinoxalinone peptidomimetics, a novel series of sulphonamide-containing 1,4-dithia-7-azaspiro[4,4]nonane (DAN) derivatives have been synthesized and evaluated as potential gelatinase A inhibitors hereby. The results revealed that the majority of tested compounds displayed satisfactory inhibition activity against gelatinase A. Among the tested compounds, 2b, 3a, 4a–d, 6a, 6d, 7a–d exhibited more potent gelatinase A inhibition than the positive control LY52. Furthermore, two test compounds 2b and 6a demonstrated moderate anti-proliferative in vitro and anti-metastatic activities in vivo, which might be utilized as potential leads in future chemical optimization.A novel series of sulphonamide-containing 1,4-dithia-7-azaspiro[4,4]nonane (DAN) derivatives have been synthesized and evaluated as potential gelatinase A inhibitors. It revealed that the majority of tested compounds displayed satisfactory inhibition activity against gelatinase A. Compounds 2b, 3a, 4a–d, 6a, 6d, 7a–d exhibited more potent gelatinase A inhibition than the positive control LY52. Two test compounds 2b and 6a demonstrated moderate anti-proliferative in vitro and anti-metastatic activities in vivo.
Co-reporter:Leilei Shi, Wei Jia, Xun Li, Ning Jiao
Tetrahedron Letters 2013 Volume 54(Issue 15) pp:1951-1955
Publication Date(Web):10 April 2013
DOI:10.1016/j.tetlet.2013.01.118
A mild procedure of Cu-catalyzed decarboxylative cross-coupling of aryl- and alkynyl-boronic acids for construction of unsymmetrical substituted alkynes has been developed. The usage of inexpensive copper chloride as catalyst, and employing stable alkynl carboxylic acids and boronic acids as the substrates under oxidative conditions for sp–sp2 coupling, make this method very easy to operate.A mild procedure of Cu-catalyzed decarboxylative cross-coupling of aryl- and alkynyl-boronic acids for construction of unsymmetrical substituted alkynes has been developed. The usage of inexpensive copper chloride as catalyst, and employing stable alkynl carboxylic acids and boronic acids as the substrates under oxidative conditions for sp–sp2 coupling, make this method very easy to operate.
Co-reporter:Xun Li;Junli Wang;Lei Zhang ;Wenfang Xu
Archiv der Pharmazie 2011 Volume 344( Issue 8) pp:494-504
Publication Date(Web):
DOI:10.1002/ardp.201100109
Abstract
The synthesis of a series of novel N-α-galloylated isoglutamic acid γ-amide peptidomimetics is described. Their enzymatic inhibition against aminopeptidase N (APN/CD13) and matrix metalloproteinase-2 (MMP-2) was tested. The preliminary activity assay revealed that most of the compounds displayed selective inhibition against APN as compared with MMP-2, with IC50 values in a micromolar range. Within this series, compound 4 (IC50 = 10.2 ± 0.9 µM) demonstrated comparable APN inhibition as compared with the positive control bestatin (IC50 = 13.1 ± 0.7 µM), which might be a promising lead for further molecular optimizations.
Co-reporter:Yonggang Li, Jian Zhang, Wenfang Xu, Huawei Zhu, Xun Li
Bioorganic & Medicinal Chemistry 2010 Volume 18(Issue 4) pp:1516-1525
Publication Date(Web):15 February 2010
DOI:10.1016/j.bmc.2010.01.008
A series of quinoxalinone peptidomimetic derivatives was designed, synthesized, and assayed for their inhibitory activities on metalloproteinase-2 (MMP-2) and aminopeptidase N (APN). The results showed that all of these quinoxalinone derivatives displayed highly selective inhibition against MMP-2 as compared with APN, with IC50 values in the micromole range. Compound A3 showed comparable MMP-2 inhibitory activities than the positive control LY52, which might be used as a potential lead in future research on anticancer agents.A series of novel quinoxalinone peptidomimetic derivatives (A1–A19) were synthesized and evaluated for their in vitro enzymatic inhibitory activities against aminopeptidase N (APN/CD13) and MMP-2.
Co-reporter:Xun Li, Yazhou Wang, Jifeng Wu, Yonggang Li, Qiang Wang, Wenfang Xu
Bioorganic & Medicinal Chemistry 2009 Volume 17(Issue 8) pp:3061-3071
Publication Date(Web):15 April 2009
DOI:10.1016/j.bmc.2009.03.017
As our ongoing work, a series of peptidomimetic l-iso-glutamine derivatives derived from antineoplaston AS2–5 scaffold were prepared and their APN/CD13 and MMP-2 inhibitory activities were evaluated hereby. The results displayed that these compounds exhibited selective inhibition against APN as compared with MMP-2, with IC50 values in micromole range. Compounds A1 and A2 showed comparable APN inhibitory activities than the positive control bestatin.A series of novel l-iso-glutamine derivatives (A1–A18 and B1–B7) based on the antineoplaston AS2–5 scaffold were synthesized and evaluated for their in vitro enzymatic inhibitory activities against aminopeptidase N (APN, CD13) and MMP-2.
Co-reporter:Xun Li, Junli Wang, Jinpei Li, Jifeng Wu, Yonggang Li, Huawei Zhu, Ruifang Fan, Wenfang Xu
Bioorganic & Medicinal Chemistry 2009 Volume 17(Issue 8) pp:3053-3060
Publication Date(Web):15 April 2009
DOI:10.1016/j.bmc.2009.02.063
Overexpression of zinc-dependent metalloproteinase, aminopeptidase N (APN/CD13), is considered to be involved in the process of tumor invasion and metastasis. Herein we describe the synthesis and in vitro enzymatic inhibition assay of antineoplaston AS2–5 scaffold peptidomimetic compounds. The results demonstrated that most of these l-iso-glutamine derivatives displayed selective inhibitory activity against APN as compared with MMP-2, with IC50 values in the micromole range. The structure–activity relationships were also briefly discussed.A series of novel l-iso-glutamine derivatives (6a–u) based on the antineoplaston AS2-5 scaffold were synthesized and evaluated for their in vitro enzymatic inhibitory activities against aminopeptidase N (APN) and MMP-2.
Co-reporter:Hongyu Yuan;Xianjun Qu;Lirui Sun;Wenfang Xu
Medicinal Chemistry Research 2009 Volume 18( Issue 8) pp:
Publication Date(Web):2009 November
DOI:10.1007/s00044-008-9159-3
P-glycoprotein-mediated drug efflux from cells is believed to be an important mechanism in multidrug resistance (MDR) in cancer chemotherapy. The identification and development of P-glycoprotein inhibitors with high potency and low cytotoxicity holds great promise for overcoming MDR. A series of quinoxalinone derivatives were synthesized and evaluated primarily for their antiproliferative effect and MDR reversal activity in in vitro assay systems. Biological assays demonstrated that the compounds were, in general, endowed with good activity as P-glycoprotein inhibitors. Among them, compounds 10 and 16, which showed the highest MDR reversal activity without significant cytotoxicity, displayed potent P-glycoprotein inhibition activities and may be worthy of further research as potential adjunctive agents for tumor chemotherapy.

![(2r)-2-[4-(7-chloroquinoxalin-2-yl)oxyphenoxy]propanoic Acid](http://img.cochemist.com/ccimg/157600/157542-91-1.png)
![(2r)-2-[4-(7-chloroquinoxalin-2-yl)oxyphenoxy]propanoic Acid](http://img.cochemist.com/ccimg/157600/157542-91-1_b.png)


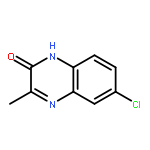
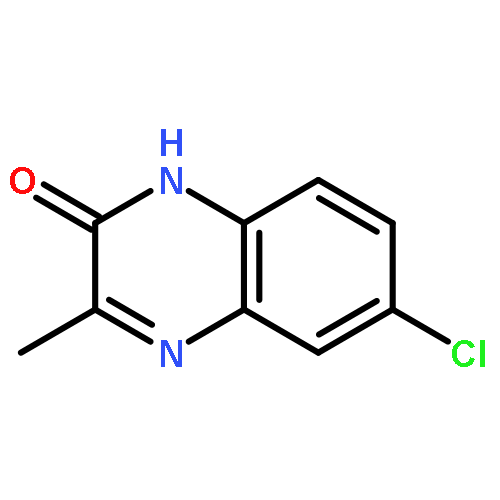
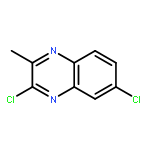
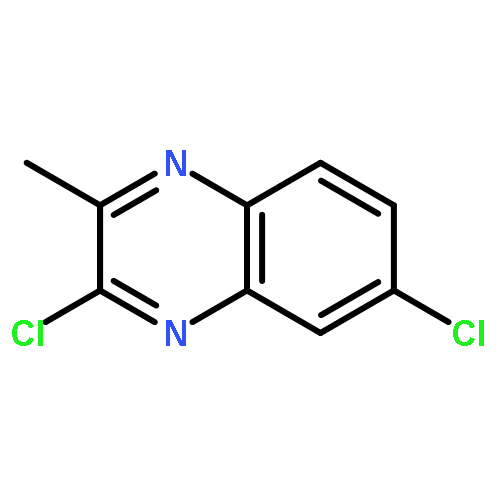
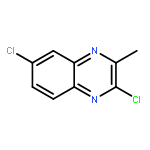
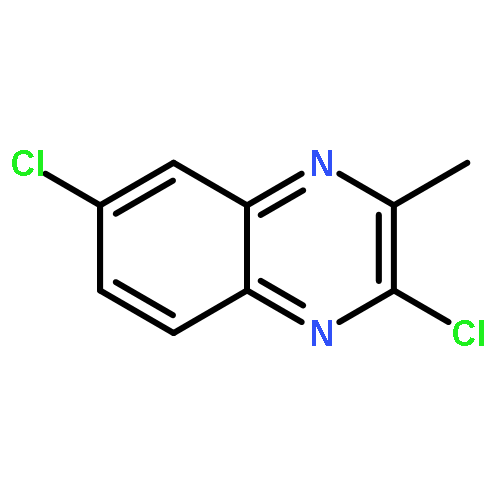



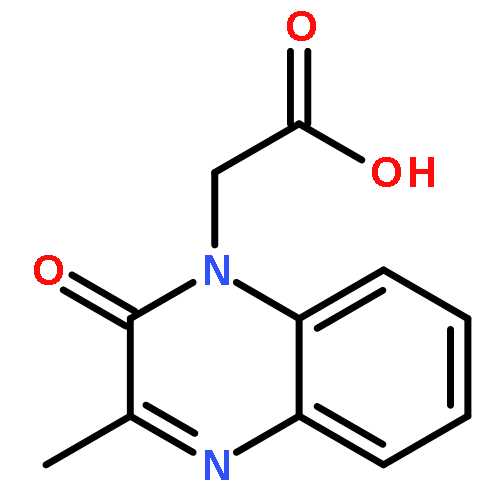


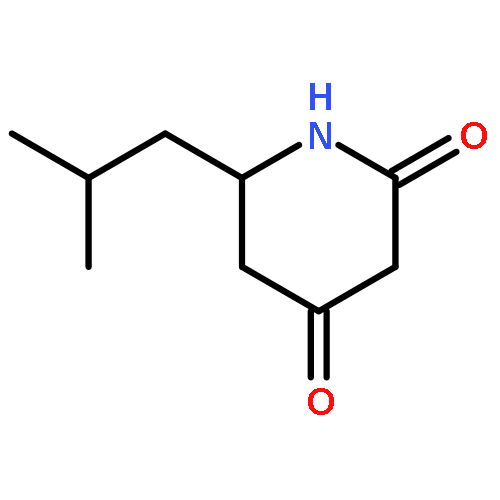


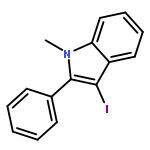
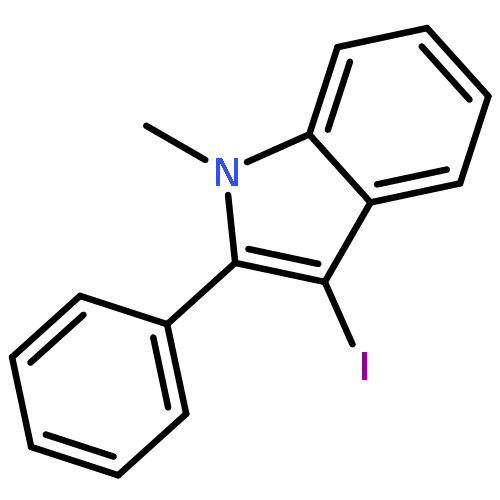
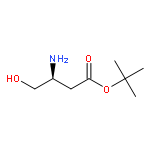
![tert-butyl [(1S)-1-(cyanomethyl)-3-methylbutyl]carbamate](http://img.cochemist.com/ccimg/172700/172695-24-8.png)
![tert-butyl [(1S)-1-(cyanomethyl)-3-methylbutyl]carbamate](http://img.cochemist.com/ccimg/172700/172695-24-8_b.png)
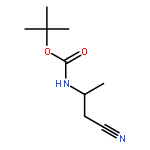
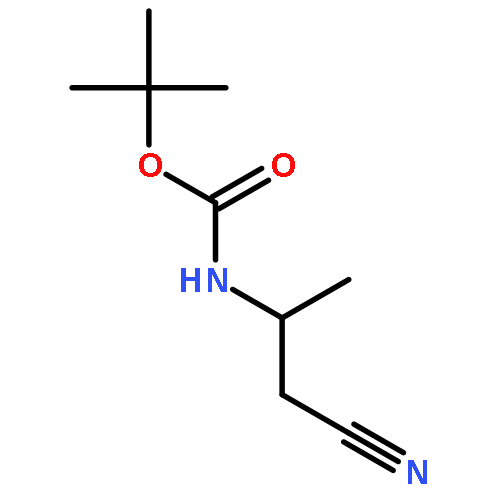
![Carbamic acid, [(1R)-2-cyano-1-methylethyl]-, 1,1-dimethylethyl ester (9CI)](http://img.cochemist.com/ccimg/170400/170367-68-7.png)
![Carbamic acid, [(1R)-2-cyano-1-methylethyl]-, 1,1-dimethylethyl ester (9CI)](http://img.cochemist.com/ccimg/170400/170367-68-7_b.png)










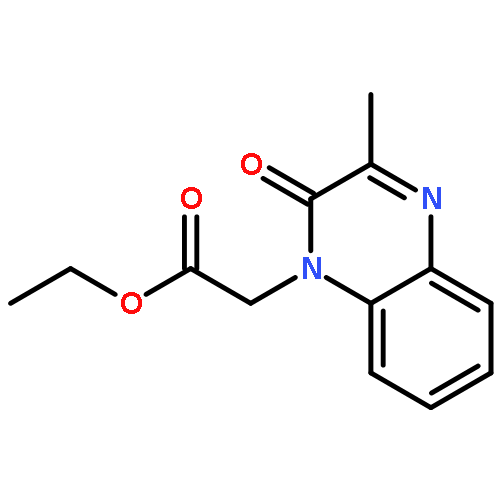
![ACETIC ACID, [(3-METHYL-2-QUINOXALINYL)OXY]-](http://img.cochemist.com/ccimg/110300/110281-70-4.png)
![ACETIC ACID, [(3-METHYL-2-QUINOXALINYL)OXY]-](http://img.cochemist.com/ccimg/110300/110281-70-4_b.png)
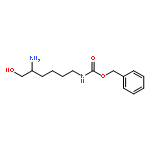
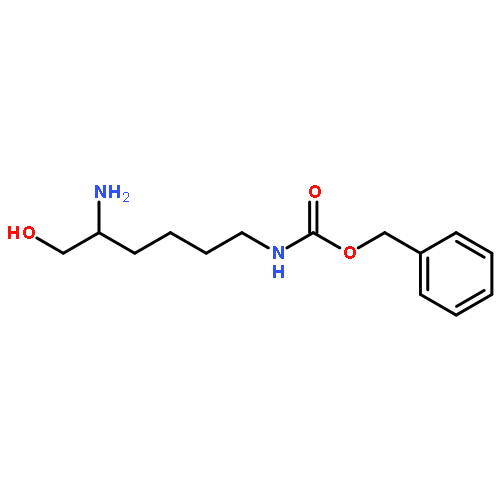
![1,4-DITHIA-7-AZASPIRO[4.4]NONANE-8-CARBOXYLIC ACID, (8S)-](http://img.cochemist.com/ccimg/83600/83552-44-7.png)
![1,4-DITHIA-7-AZASPIRO[4.4]NONANE-8-CARBOXYLIC ACID, (8S)-](http://img.cochemist.com/ccimg/83600/83552-44-7_b.png)