Co-reporter:Joseph A. Korn, Jan Urban, Andy Dang, Huong T. H. Nguyen, and František Tureček
The Journal of Physical Chemistry Letters September 7, 2017 Volume 8(Issue 17) pp:4100-4100
Publication Date(Web):August 15, 2017
DOI:10.1021/acs.jpclett.7b01856
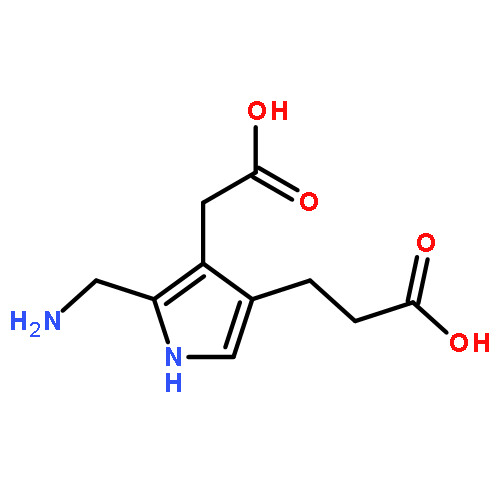
We report the generation of deoxyriboadenosine dinucleotide cation radicals by gas-phase electron transfer to dinucleotide dications and their noncovalent complexes with crown ether ligands. Stable dinucleotide cation radicals of a novel hydrogen-rich type were generated and characterized by tandem mass spectrometry and UV–vis photodissociation (UVPD) action spectroscopy. Electron structure theory analysis indicated that upon electron attachment the dinucleotide dications underwent a conformational collapse followed by intramolecular proton migrations between the nucleobases to give species whose calculated UV–vis absorption spectra matched the UVPD action spectra. Hydrogen-rich cation radicals generated from chimeric riboadenosine 5′-diesters gave UVPD action spectra that pointed to novel zwitterionic structures consisting of aromatic π-electron anion radicals intercalated between stacked positively charged adenine rings. Analogies with DNA ionization are discussed.
Co-reporter:Huong T. H. Nguyen, Prokopis C. Andrikopoulos, Daniel Bím, Lubomír Rulíšek, Andy Dang, and František Tureček
The Journal of Physical Chemistry B July 13, 2017 Volume 121(Issue 27) pp:6557-6557
Publication Date(Web):June 14, 2017
DOI:10.1021/acs.jpcb.7b04661
Peptide cation-radicals containing the threonine residue undergo radical-induced dissociations upon collisional activation and photon absorption in the 210–400 nm range. Peptide cation-radicals containing a radical defect at the N-terminal residue, [•Ala-Thr-Ala-Arg+H]+, were generated by electron transfer dissociation (ETD) of peptide dications and characterized by UV–vis photodissociation action spectroscopy combined with time-dependent density functional theory (TD-DFT) calculations of absorption spectra, including thermal vibronic band broadening. The action spectrum of [•Ala-Thr-Ala-Arg+H]+ ions was indicative of the canonical structure of an N-terminally deaminated radical whereas isomeric structures differing in the position of the radical defect and amide bond geometry were excluded. This indicated that exothermic electron transfer to threonine peptide ions did not induce radical isomerizations in the fragment cation-radicals. Several isomeric structures, ion–molecule complexes, and transition states for isomerizations and dissociations were generated and analyzed by DFT and Møller–Plesset perturbational ab initio calculations to aid interpretation of the major dissociations by loss of water, hydroxyl radical, C3H6NO•, C3H7NO, and backbone cleavages. Born–Oppenheimer molecular dynamics (BOMD) in combination with DFT gradient geometry optimizations and intrinsic reaction coordinate analysis were used to search for low-energy cation-radical conformers and transition states. BOMD was also employed to analyze the reaction trajectory for loss of water from ion–molecule complexes.
Co-reporter:Robert Pepin;Erik D. Layton;Yang Liu
Journal of The American Society for Mass Spectrometry 2017 Volume 28( Issue 1) pp:164-181
Publication Date(Web):2017 January
DOI:10.1007/s13361-016-1512-z
Electron transfer to doubly and triply charged heptapeptide ions containing polar residues Arg, Lys, and Asp in combination with nonpolar Gly, Ala, and Pro or Leu generates stable and metastable charge-reduced ions, (M + 2H)+●, in addition to standard electron-transfer dissociation (ETD) fragment ions. The metastable (M + 2H)+● ions spontaneously dissociate upon resonant ejection from the linear ion trap, giving irregularly shaped peaks with offset m/z values. The fractions of stable and metastable (M + 2H)+● ions and their mass shifts depend on the presence of Pro-4 and Leu-4 residues in the peptides, with the Pro-4 sequences giving larger fractions of the stable ions while showing smaller mass shifts for the metastables. Conversion of the Asp and C-terminal carboxyl groups to methyl esters further lowers the charge-reduced ion stability. Collisional activation and photodissociation at 355 nm of mass-selected (M + 2H)+● results in different dissociations that give sequence specific MS3 spectra. With a single exception of charge-reduced (LKGLADR + 2H)+●, the MS3 spectra do not produce ETD sequence fragments of the c and z type. Hence, these (M + 2H)+● ions are covalent radicals, not ion–molecule complexes, undergoing dramatically different dissociations in the ground and excited electronic states. The increased stability of the Pro-4 containing (M + 2H)+● ions is attributed to radicals formed by opening of the Pro ring and undergoing further stabilization by hydrogen atom migrations. UV–VIS photodissociation action spectroscopy and time-dependent density functional theory calculations are used in a case in point study of the stable (LKGPADR + 2H)+● ion produced by ETD. In contrast to singly-reduced peptide ions, doubly reduced (M + 3H)+ ions are stable only when formed from the Pro-4 precursors and show all characteristics of even electron ions regarding no photon absorption at 355 nm or ion-molecule reactions, and exhibiting proton driven collision induced dissociations.
Co-reporter:Christopher J. Shaffer;Jonathan Martens
Journal of The American Society for Mass Spectrometry 2016 Volume 27( Issue 7) pp:1176-1185
Publication Date(Web):2016 July
DOI:10.1007/s13361-016-1390-4
We report a combined experimental and computational study aimed at elucidating the structure of N-terminal fragment ions of the c type produced by electron transfer dissociation of photo-leucine (L*) peptide ions GL*GGKX. The c4 ion from GL*GGK is found to retain an intact diazirine ring that undergoes selective photodissociation at 355 nm, followed by backbone cleavage. Infrared multiphoton dissociation action spectra point to the absence in the c4 ion of a diazoalkane group that could be produced by thermal isomerization of vibrationally hot ions. The c4 ion from ETD of GL*GGK is assigned an amide structure by a close match of the IRMPD action spectrum and calculated IR absorption. The energetics and kinetics of c4 ion dissociations are discussed.
Co-reporter:Emilie Viglino;Dr. Christopher J. Shaffer ;Dr. Franti&x161;ek Ture&x10d;ek
Angewandte Chemie International Edition 2016 Volume 55( Issue 26) pp:7469-7473
Publication Date(Web):
DOI:10.1002/anie.201602604
Abstract
We report the first application of UV/Vis photodissociation action spectroscopy for the structure elucidation of tyrosine peptide cation radicals produced by oxidative intramolecular electron transfer in gas-phase metal complexes. Oxidation of Tyr-Ala-Ala-Ala-Arg (YAAAR) produces Tyr-O radicals by combined electron and proton transfer involving the phenol and carboxyl groups. Oxidation of Ala-Ala-Ala-Tyr-Arg (AAAYR) produces a mixture of cation radicals involving electron abstraction from the Tyr phenol ring and N-terminal amino group in combination with hydrogen-atom transfer from the Cα positions of the peptide backbone.
Co-reporter:Emilie Viglino;Dr. Christopher J. Shaffer ;Dr. Franti&x161;ek Ture&x10d;ek
Angewandte Chemie 2016 Volume 128( Issue 26) pp:7595-7599
Publication Date(Web):
DOI:10.1002/ange.201602604
Abstract
We report the first application of UV/Vis photodissociation action spectroscopy for the structure elucidation of tyrosine peptide cation radicals produced by oxidative intramolecular electron transfer in gas-phase metal complexes. Oxidation of Tyr-Ala-Ala-Ala-Arg (YAAAR) produces Tyr-O radicals by combined electron and proton transfer involving the phenol and carboxyl groups. Oxidation of Ala-Ala-Ala-Tyr-Arg (AAAYR) produces a mixture of cation radicals involving electron abstraction from the Tyr phenol ring and N-terminal amino group in combination with hydrogen-atom transfer from the Cα positions of the peptide backbone.
Co-reporter:Robert Pepin;Kenneth J. Laszlo;Aleš Marek
Journal of The American Society for Mass Spectrometry 2016 Volume 27( Issue 10) pp:1647-1660
Publication Date(Web):2016 October
DOI:10.1007/s13361-016-1437-6
Heptapeptide ions containing combinations of polar Lys, Arg, and Asp residues with non-polar Leu, Pro, Ala, and Gly residues were designed to study polar effects on gas-phase ion conformations. Doubly and triply charged ions were studied by ion mobility mass spectrometry and electron structure theory using correlated ab initio and density functional theory methods and found to exhibit tightly folded 3D structures in the gas phase. Manipulation of the basic residue positions in LKGPADR, LRGPADK, KLGPADR, and RLGPADK resulted in only minor changes in the ion collision cross sections in helium. Replacement of the Pro residue with Leu resulted in only marginally larger collision cross sections for the doubly and triply charged ions. Disruption of zwitterionic interactions in doubly charged ions was performed by converting the C-terminal and Asp carboxyl groups to methyl esters. This resulted in very minor changes in the collision cross sections of doubly charged ions and even slightly diminished collision cross sections in most triply charged ions. The experimental collision cross sections were related to those calculated for structures of lowest free energy ion conformers that were obtained by extensive search of the conformational space and fully optimized by density functional theory calculations. The predominant factors that affected ion structures and collision cross sections were due to attractive hydrogen bonding interactions and internal solvation of the charged groups that overcompensated their Coulomb repulsion. Structure features typically assigned to the Pro residue and zwitterionic COO-charged group interactions were only secondary in affecting the structures and collision cross sections of these gas-phase peptide ions.
Co-reporter:Emilie Viglino;Cheuk Kuen Lai;Xiaoyan Mu
Journal of The American Society for Mass Spectrometry 2016 Volume 27( Issue 9) pp:1454-1467
Publication Date(Web):2016 September
DOI:10.1007/s13361-016-1425-x
We report a comprehensive study of collision-induced dissociation (CID) and near-UV photodissociation (UVPD) of a series of tyrosine-containing peptide cation radicals of the hydrogen-rich and hydrogen-deficient types. Stable, long-lived, hydrogen-rich peptide cation radicals, such as [AAAYR + 2H]+● and several of its sequence and homology variants, were generated by electron transfer dissociation (ETD) of peptide-crown-ether complexes, and their CID-MS3 dissociations were found to be dramatically different from those upon ETD of the respective peptide dications. All of the hydrogen-rich peptide cation radicals contained major (77%–94%) fractions of species having radical chromophores created by ETD that underwent photodissociation at 355 nm. Analysis of the CID and UVPD spectra pointed to arginine guanidinium radicals as the major components of the hydrogen-rich peptide cation radical population. Hydrogen-deficient peptide cation radicals were generated by intramolecular electron transfer in CuII(2,2′:6′,2″-terpyridine) complexes and shown to contain chromophores absorbing at 355 nm and undergoing photodissociation. The CID and UVPD spectra showed major differences in fragmentation for [AAAYR]+● that diminished as the Tyr residue was moved along the peptide chain. UVPD was found to be superior to CID in localizing Cα-radical positions in peptide cation radical intermediates.
Co-reporter:Robert Pepin; Alessio Petrone; Kenneth J. Laszlo; Matthew F. Bush; Xiaosong Li;František Tureček
The Journal of Physical Chemistry Letters 2016 Volume 7(Issue 14) pp:2765-2771
Publication Date(Web):July 8, 2016
DOI:10.1021/acs.jpclett.6b01187
Ab initio molecular dynamics (AIMD) with density functional theory (DFT) was applied to explore conformational motions and collision cross sections (Ω) of folded (2) and extended (7) conformers of doubly charged peptide ions, (Ala–Ala–Leu–Arg + 2H)2+, in the gas phase at 300 and 473 K. The experimental Ω of (Ala–Ala–Leu–Arg +2H)2+ was measured as 149 ± 1.2 Å2 at 298 K. Thermally distributed mean values of Ω for 2 and 7 at 300 and 473 K were only 0.8–1.1% larger than for the equilibrium 0 K structures. Long (>10 ps) trajectory calculations indicated entropy-driven conformational change of 2 to 7 that occurred at random within a ∼ 4 ps time window. The experimental Ω was found to fit the calculated population averaged values for 2 and 7, indicating a rapid conformer interconversion. Overall, thermal breathing had only a minor effect on the peptide ion collision cross sections.
Co-reporter:Christopher J. Shaffer
Journal of The American Society for Mass Spectrometry 2016 Volume 27( Issue 4) pp:633-645
Publication Date(Web):2016 April
DOI:10.1007/s13361-016-1338-8
Noncovalent complexes of hydrophobic peptides GLLLG and GLLLK with photoleucine (L*) tagged peptides G(L*nLm)K (n = 1,3, m = 2,0) were generated as singly charged ions in the gas phase and probed by photodissociation at 355 nm. Carbene intermediates produced by photodissociative loss of N2 from the L* diazirine rings underwent insertion into X−H bonds of the target peptide moiety, forming covalent adducts with yields reaching 30%. Gas-phase sequencing of the covalent adducts revealed preferred bond formation at the C-terminal residue of the target peptide. Site-selective carbene insertion was achieved by placing the L* residue in different positions along the photopeptide chain, and the residues in the target peptide undergoing carbene insertion were identified by gas-phase ion sequencing that was aided by specific 13C labeling. Density functional theory calculations indicated that noncovalent binding to GL*L*L*K resulted in substantial changes of the (GLLLK + H)+ ground state conformation. The peptide moieties in [GL*L*LK + GLLLK + H]+ ion complexes were held together by hydrogen bonds, whereas dispersion interactions of the nonpolar groups were only secondary in ground-state 0 K structures. Born-Oppenheimer molecular dynamics for 100 ps trajectories of several different conformers at the 310 K laboratory temperature showed that noncovalent complexes developed multiple, residue-specific contacts between the diazirine carbons and GLLLK residues. The calculations pointed to the substantial fluidity of the nonpolar side chains in the complexes. Diazirine photochemistry in combination with Born-Oppenheimer molecular dynamics is a promising tool for investigations of peptide–peptide ion interactions in the gas phase.
Co-reporter:Huong T.H. Nguyen, Christopher J. Shaffer, Aaron R. Ledvina, Joshua J. Coon, František Tureček
International Journal of Mass Spectrometry 2015 Volume 378() pp:20-30
Publication Date(Web):15 February 2015
DOI:10.1016/j.ijms.2014.06.028
•Serine peptide cation-radicals of the z-type were studied.•Energy-resolved collision-induced dissociations are reported.•CID is compared with time-resolved infrared multiphoton dissociation.•z-type peptide cation-radicals undergo photodissociation at 355 nm.•Structures and energies of intermediates and transition states were calculated.The serine residue displays specific effects on the dissociations of peptide fragment cation-radicals of the z+●-type which are produced by electron transfer dissociation. Energy-resolved collision-induced dissociation (ER-CID), time-resolved infrared multiphoton dissociation (TR-IRMPD), and single-photon UV photodissociation at 355 nm revealed several competitive dissociation pathways consisting of loss of OH radical, water, and backbone cleavages occurring at N-terminal and C-terminal positions relative to the serine residue. The activation modes using slow-heating and UV photon absorption resulted in different relative intensities of fragment ions. This indicated that the dissociations proceeded through several channels with different energy-dependent kinetics. The experimental data were interpreted with the help of electron structure calculations that provided fully optimized structures and relative energies for cis and trans amide isomers of the z4+● ions as well as isomerization, dissociation, and transition state energies. UV photon absorption by the z4+● ions was due to Cα-radical amide groups created by ETD that provided a new chromophore absorbing at 355 nm.
Co-reporter:Aaron R. Ledvina, Joshua J. Coon, František Tureček
International Journal of Mass Spectrometry 2015 Volume 377() pp:44-53
Publication Date(Web):1 February 2015
DOI:10.1016/j.ijms.2014.02.015
•Time-resolved IRMPD and energy-resolved CID spectra of peptide cation-radicals.•Ab initio theory and RRKM calculations of peptide cation-radical dissociations.•Combination of experiment and theory provides improved mechanistic insight.We report a combined experimental and computational study of energy-resolved collision-induced dissociation (ER-CID) and time-resolved infrared multiphoton dissociation (TR-IRMPD) of z4 ions prepared by electron transfer dissociation of peptide (Ala-Ala-Asn-Ala-Arg + 2H)2+ ions. The z4 cation-radicals, ANAR+, undergo competitive dissociations by backbone cleavage and loss of a CONH2 radical from the Asn side chain. The backbone cleavage proceeds by radical-assisted dissociation of the Asn Cα-CO bond, forming an x2 ion intermediate which rapidly dissociates by HNCO elimination to yield a stable z2 fragment ion, AR+. The ER-CID and TR-IRMPD data were consistent with the consecutive nature of the backbone dissociation but showed different branching ratios for the two major fragmentations. The ER-CID data showed branching ratios 0.6–1.0 for the side-chain and backbone cleavages whereas the TR-IRMPD data showed an earlier onset for the latter dissociation. Computational analysis of the potential energy surface with density functional theory and ab initio calculations was carried out to provide structures and energies for the reactant ions as well as several intermediates, products, and transition states. Dissociation pathways for cis and trans amide conformers were distinguished and their energies were evaluated. The threshold dissociation energies for the backbone and side-chain dissociations were similar in accordance with the experimental ER-CID branching ratio. The TR-IRMPD data were interpreted by different absorbances of intermediates produced by hydrogen atom migrations along the dissociation pathways.
Co-reporter:Christopher J. Shaffer;Aleš Marek
Journal of The American Society for Mass Spectrometry 2015 Volume 26( Issue 8) pp:1367-1381
Publication Date(Web):2015 August
DOI:10.1007/s13361-015-1139-5
Electron transfer dissociation of peptide ions with the diazirine-containing residue photomethionine (M*) results in side-chain dissociations by loss of C3H7N2 radicals in addition to standard backbone cleavages. The side-chain dissociations are particularly prominent upon activation of long-lived, charge-reduced, cation radicals (GM*GGR + 2H)+●. Investigation of these cation radicals by near-UV photodissociation and collisional activation revealed different fragmentation products and mechanisms resulting from these ion activation modes. The dissociations observed for photomethionine were dramatically different from those previously reported for the lower homologue photoleucine; here, a difference by a single methylene group in the side chain had a large effect on the chemistries of the cation radicals upon ETD and further activation. ETD intermediates and products were probed by tandem 355-nm UV photodissociation-collision induced dissociation and found to contain chromophores that resulted from electron attachment to the diazirine ring. The nature of the newly formed chromophores and ion energetics and kinetics were investigated by electron structure calculations combining ab initio and density functional theory methods and Rice-Ramsperger-Kassel-Marcus (RRKM) theory. The dramatic difference between the dissociations of L* and M* containing peptide cation radicals is explained by electronic effects that play a role in stabilizing critical reaction intermediates and steer the dissociations into kinetically favored reaction channels. In addition, a new alternating UVPD-ETD-UVPD MS4 experiment is introduced and utilized for ion structure elucidation.
Co-reporter:Aleš Marek;Christopher J. Shaffer
Journal of The American Society for Mass Spectrometry 2015 Volume 26( Issue 3) pp:415-431
Publication Date(Web):2015 March
DOI:10.1007/s13361-014-1047-0
Electron transfer to gas-phase peptide ions with diazirine-containing amino acid residue photoleucine (L*) triggers diazirine ring reduction followed by cascades of residue-specific radical reactions. Upon electron transfer, substantial fractions of (GL*GGR +2H)+● cation-radicals undergo elimination of [NH4O] radicals and N2H2 molecules from the side chain. The side-chain dissociations are particularly prominent on collisional activation of long-lived (GL*GGR +2H)+● cation-radicals formed by electron transfer dissociation of noncovalent peptide-18-crown-6-ether ion complexes. The ion dissociation products were characterized by multistage tandem mass spectrometry (MSn) and ion mobility measurements. The elimination of [NH4O] was elucidated with the help of 2H, 15 N, and 18O-labeled peptide ions and found to specifically involve the amide oxygen of the N-terminal residue. The structures, energies, and electronic states of the peptide radical species were elucidated by a combination of near-UV photodissociation experiments and electron structure calculations combining ab initio and density functional theory methods. Electron transfer reaching the ground electronic states of charge reduced (GL*GGR +2H)+● cation-radicals was found to reduce the diazirine ring. In contrast, backbone N − Cα bond dissociations that represent a 60%–75% majority of all dissociations because of electron transfer are predicted to occur from excited electronic states.
Co-reporter:Huong T. H. Nguyen; Christopher J. Shaffer; Robert Pepin;František Tureček
The Journal of Physical Chemistry Letters 2015 Volume 6(Issue 23) pp:4722-4727
Publication Date(Web):November 12, 2015
DOI:10.1021/acs.jpclett.5b02354
UV photodissociation (UVPD) action spectroscopy is reported to provide a sensitive tool for the detection of radical sites in gas-phase peptide ions. UVPD action spectra of peptide cation radicals of the z-type generated by electron-transfer dissociation point to the presence of multiple structures formed as a result of spontaneous isomerizations by hydrogen atom migration. N-terminal Cα radicals are identified as the dominant components, but the content of isomers differing in the radical defect position in the backbone or side chain depends on the nature of the aromatic residue with phenylalanine being more prone to isomerization than tryptophan. These results illustrate that spontaneous hydrogen atom migrations can occur in peptide cation-radicals upon electron-transfer dissociation.
Co-reporter:Robert Pepin and František Tureček
The Journal of Physical Chemistry B 2015 Volume 119(Issue 7) pp:2818-2826
Publication Date(Web):January 16, 2015
DOI:10.1021/jp510244d
Peptide fragment ions of the z-type were used as kinetic ion thermometers to gauge the internal energy of peptide cation-radicals produced by electron transfer in the gas-phase. Electron transfer dissociation (ETD)-produced z2 ions containing the leucine residue, z2(Leu-Lys) and z2(Leu-Arg), were found to undergo spontaneous dissociation by loss of C3H7 that was monitored by time-resolved kinetic measurements on the time scale of the linear ion trap mass spectrometer. Kinetic modeling of the dissociations, including collisional cooling and product loss by neutralization, provided unimolecular rate constants for dissociation that were converted to the z ion internal energies using RRKM theory. The internal energy of z2(Leu-Lys) and z2(Leu-Arg) fragment ions was found to decrease with the increasing size of the precursor peptide ion, indicating vibrational energy partitioning between the ion and neutral fragments and ergodic behavior. The experimentally determined excitation in the peptide cation-radicals upon electron transfer (285–327 kJ mol–1) was found to be lower than that theoretically calculated from the reaction exothermicity. The reasons for this missing energy are discussed.
Co-reporter:Huong T. H. Nguyen, Christopher J. Shaffer, and František Tureček
The Journal of Physical Chemistry B 2015 Volume 119(Issue 10) pp:3948-3961
Publication Date(Web):February 17, 2015
DOI:10.1021/jp511717c
Electron transfer reduction of gas-phase ions generated from histidine-containing peptides forms stable cation–radicals that absorb light at 355 nm, as studied for AAHAR, AAHAK, DSHAK, FHEK, HHGYK, and HHSHR. Laser photodissociation of mass-selected cation–radicals chiefly resulted in loss of H atoms, contrasting dissociations induced by slow collisional heating. The 355 nm absorption was due to new chromophores created by electron transfer and radical rearrangements in the cation–radicals. The chromophores were identified by time-dependent density functional theory calculations as 2H,3H-imidazoline and 2H-dihydrophenol radicals, formed by hydrogen atom transfer to the histidine and tyrosine side chain groups, respectively. These radicals undergo facile C–H bond dissociations upon photon absorption. In contrast, dissociations of histidine peptide cation–radicals containing the 1H,3H-imidazoline ring prefer loss of 4-methylimidazole via a multistep reaction pathway. The isomeric cation–radicals can be distinguished by a combination of collision-induced dissociation and near-UV photodissociation. The TD-DFT excitation energies in model imidazoline radicals were benchmarked on EOM-CCSD energies, and a satisfactory agreement was found for the M06-2X and ωB97XD functionals. The combination of electron transfer, photodissociation, collisional activation, and theory is presented as a powerful tool for studying structures and electronic properties of peptide cation–radicals in the gas phase.
Co-reporter:Michael Volný, Joelle Rolfs, Bejan Hakimi, Petr Fryčák, Thomas Schneider, Dingsheng Liu, Gloria Yen, Daniel T. Chiu, and František Tureček
Analytical Chemistry 2014 Volume 86(Issue 7) pp:3647
Publication Date(Web):March 13, 2014
DOI:10.1021/ac500365r
We report a microfluidic device, using segmented flow in a two-phase system of immiscible liquids, which delivers aqueous droplets into a modified commercial mass spectrometer. The interface coupling the microfluidics to the mass spectrometer achieves up to 96% sample transfer efficiency to the vacuum chamber. Sample ionization is assisted by multipass infrared laser beam in the interface. The system achieves low femtomole detection limits of several analytes ranging from drugs to proteins. Sample ionization in this segmented-flow sampling was found to be remarkably insensitive to the presence of buffer salts and other matrices.
Co-reporter:Mariana Barcenas, Chang Xue, Tatyana Marushchak-Vlaskin, C. Ronald Scott, Michael H. Gelb, and František Tureček
Analytical Chemistry 2014 Volume 86(Issue 15) pp:7962
Publication Date(Web):July 14, 2014
DOI:10.1021/ac501994b
We report new substrates for quantitative enzyme activity measurements of human palmitoyl protein thioesterase (PPT1) and tripeptidyl peptidase (TPP1) in dried blood spots from newborns using tandem mass spectrometry. Deficiencies in these enzyme activities due to inborn errors of metabolism cause neuronal ceroid lipofuscinoses. The assays use synthetic compounds that were designed to mimic the natural substrates. Incubation produces nanomole quantities of enzymatic products per a blood spot that are quantified by tandem mass spectrometry using synthetic internal standards and selected reaction monitoring. The assays utilize a minimum steps for sample workup and can be run in a duplex format for the detection of neuronal ceroid lipofuscinoses or potentially multiplexed with other mass spectrometry-based assays for newborn screening of lysosomal storage disorders.
Co-reporter:Sara Øvad Pedersen, Camilla Skinnerup Byskov, Frantisek Turecek, and Steen Brøndsted Nielsen
The Journal of Physical Chemistry A 2014 Volume 118(Issue 24) pp:4256-4265
Publication Date(Web):May 29, 2014
DOI:10.1021/jp504153p
The strong UV chromophores thymine (Thy) and uracil (Ura) have identical heteroaromatic rings that only differ by one methyl substituent. While their photophysics has been elucidated in detail, the effect on the excited states of base protonation and single water molecules is less explored. Here we report gas-phase absorption spectra of ThyH+ and UraH+ and monohydrated ions and demonstrate that the substituent is not only responsible for spectral shifts but also influences the tautomer distribution, being different for bare and monohydrated ions. Spectra interpretation is aided by calculations of geometrical structures and transition energies. The lowest free-energy tautomer (denoted 178, enol–enol form) accounts for 230–280 nm (ThyH+) and 225–270 nm (UraH+) bands. ThyH+ hardly absorbs above 300 nm, whereas a discernible band is measured for UraH+ (275–320 nm), ascribed to the second lowest free-energy tautomer (138, enol–keto form) comprising a few percent of the UraH+ population at room temperature. Band widths are similar to those measured of cold ions in support of very short excited-state lifetimes. Attachment of a single water increases the abundance of 138 relative to 178, 138 now clearly present for ThyH+. 138 resembles more the tautomer present in aqueous solution than 178 does, and 138 may indeed be a relevant transition structure. The band of ThyH+(178) is unchanged, that of UraH+(178) is nearly unchanged, and that of UraH+(138) blue-shifts by about 10 nm. In stark contrast to protonated adenine, more than one solvating water molecule is required to re-establish the absorption of ThyH+ and UraH+ in aqueous solution.
Co-reporter:Aleš Marek;František Tureček
Journal of The American Society for Mass Spectrometry 2014 Volume 25( Issue 5) pp:778-789
Publication Date(Web):2014 May
DOI:10.1007/s13361-014-0832-0
Gas-phase dissociations were investigated for several peptide ions containing the Gly-Leu* N-terminal motif where Leu* was a modified norleucine residue containing the photolabile diazirine ring. Collisional activation of gas-phase peptide cations resulted in facile N2 elimination that competed with backbone dissociations. A free lysine ammonium group can act as a Brønsted acid to facilitate N2 elimination. This dissociation was accompanied by insertion of a lysine proton in the side chain of the photoleucine residue, as established by deuterium labeling and gas-phase sequencing of the products. Electron structure calculations were used to provide structures and energies of reactants, intermediates, and transition states for Gly-Leu*-Gly-Gly-Lys amide ions that were combined with RRKM calculations of unimolecular rate constants. The calculations indicated that Brønsted acid-catalyzed eliminations were kinetically preferred over direct loss of N2 from the diazirine ring. Mechanisms are proposed to explain the proton-initiated reactions and discuss the reaction products. The non-catalyzed diazirine ring cleavage and N2 loss is proposed as a thermometer dissociation for peptide ion dissociations.
Co-reporter:František Tureček and Ryan R. Julian
Chemical Reviews 2013 Volume 113(Issue 8) pp:6691
Publication Date(Web):May 7, 2013
DOI:10.1021/cr400043s
Co-reporter:Dingsheng Liu, Bejan Hakimi, Michael Volny, Joelle Rolfs, Xudong Chen, Frantisek Turecek, and Daniel T. Chiu
Analytical Chemistry 2013 Volume 85(Issue 13) pp:6190
Publication Date(Web):June 14, 2013
DOI:10.1021/ac400844p
This Letter describes the controlled generation of double emulsions in the gas phase, which was carried out using an integrated emitter in a poly(dimethylsiloxane) (PDMS) microfluidic chip. The integrated emitter was formed using a molding approach, in which metal wires with desirable diameters were used as emitter molds. The generation of double emulsions in air was achieved with electrohydrodynamics actuation, which offers controllable force exerting on the double emulsions. We developed this capability for future integration of droplet microfluidics with mass spectrometry (MS), where each aqueous droplet in the microchannel is introduced into the gas phase as a double emulsion for subsequent ionization and MS analysis.
Co-reporter:Frantisek Turecek, Christopher L. Moss, Ioannis Pikalov, Robert Pepin, Kerim Gulyuz, Nicolas C. Polfer, Matthew F. Bush, Jeffery Brown, Jonathan Williams, Keith Richardson
International Journal of Mass Spectrometry 2013 Volumes 354–355() pp:249-256
Publication Date(Web):15 November 2013
DOI:10.1016/j.ijms.2013.06.021
•We elucidated structures of gas-phase phosphopeptide ions.•Ion mobility data were compatible with calculated structures.•Action IR spectroscopy was difficult to interpret.The goal of this paper was to investigate gas-phase structures of conformers of doubly charged phosphopeptide ions (ApSAAR + 2H)2+ and (AApSAR + 2H)2+ using traveling-wave and drift-tube ion mobility and IR action spectroscopy measurements in combination with molecular dynamics and electronic structure calculations. Lowest free-energy conformers were identified by extensive search of the conformational space with combined molecular dynamics, semi-empirical, density functional theory and perturbational Møller–Plesset ab initio calculations. The low-energy conformers had collisional cross sections that were compatible with the experimental data but did not allow the conformers to be distinguished. IR absorption spectra that were calculated by density functional theory for harmonic normal modes showed several intense bands that were absent in the experimental action spectrum. Agreement with experiment was improved upon implementing second-order anharmonic corrections to the calculated vibrational frequencies. Several factors affecting the action spectra are discussed such as effects of mode anharmonicity on the absorption band intensities, the magnitude of frequency shifts in anharmonic corrections, as well as non-linear effects in multiple-photon absorption that may suppress the detection of strongly H-bonded modes in these measurements.
Co-reporter:Magdalena Zimnicka, Thomas W. Chung, Christopher L. Moss, and František Tureček
The Journal of Physical Chemistry A 2013 Volume 117(Issue 6) pp:1265-1275
Publication Date(Web):July 5, 2012
DOI:10.1021/jp305865q
Thioxodipeptides Gly-thio-Lys (GtK), Ala-thio-Lys (AtK), and Ala-thio-Arg (AtR) in which the amide group has been modified to a thioxoamide were made into dications by electrospray ionization and converted to cation-radicals, (GtK + 2H)+•, (AtK + 2H)+•, and (AtR + 2H)+•, by electron transfer dissociation (ETD) tandem mass spectrometry using fluoranthene anion-radical as an electron donor. The common and dominant dissociation of these cation-radicals was the loss of a hydrogen atom. The dissociation products were characterized by collision-induced dissociation (CID) multistage tandem mass spectrometry up to CID-MS5. The ground electronic states of several (GtK + 2H)+•, (AtK + 2H)+•, and (AtR + 2H)+• conformers were explored by extensive ab initio and density functional theory calculations of the potential energy surface. In silico electron transfer to the precursor dications, (GtK + 2H)2+, (AtK + 2H)2+, and (AtR + 2H)2+, formed zwitterionic intermediates containing thioenol anion-radical and ammonium cation groups that were local energy minima on the potential energy surface of the ground electronic state. The zwitterions underwent facile isomerization by N-terminal ammonium proton migration to the thioenol anion-radical group forming aminothioketyl intermediates. Combined potential energy mapping and RRKM calculations of dissociation rate constants identified N–Cα bond cleavages as the most favorable dissociation pathways, in a stark contrast to the experimental results. This discrepancy is interpreted as being due to the population upon electron transfer of low-lying excited electronic states that promote loss of hydrogen atoms. For (GtK + 2H)+•, these excited states were characterized by time-dependent density functional theory as A–C states that had large components of Rydberg-like 3s molecular orbitals at the N-terminal and lysine ammonium groups that are conducive to hydrogen atom loss.
Co-reporter:Aleš Marek;Robert Pepin;Bo Peng
Journal of The American Society for Mass Spectrometry 2013 Volume 24( Issue 11) pp:1641-1653
Publication Date(Web):2013 November
DOI:10.1007/s13361-013-0630-0
Gas-phase conformations and electron transfer dissociations of pentapeptide ions containing the photo-Leu residue (L*) were studied. Exhaustive conformational search including molecular dynamics force-field, semi-empirical, ab initio, and density functional theory calculations established that the photo-Leu residue did not alter the gas-phase conformations of (GL*GGK + 2H)2+ and (GL*GGK-NH2 + H)+ ions, which showed the same conformer energy ranking as the unmodified Leu-containing ions. This finding is significant in that it simplifies conformational analysis of photo-labeled peptide ions. Electron transfer dissociation mass spectra of (GL*GGK + 2H)2+, (GL*GGK-NH2 + 2H)2+,(GL*GGKK + 2H)2+, (GL*GLK + 2H)2+, and (GL*LGK + 2H)2+ showed 16 %–21 % fragment ions originating by radical rearrangements and cleavages in the diazirine ring. These side-chain dissociations resulted in eliminations of N2H3, N2H4, [N2H5], and [NH4O] neutral fragments and were particularly abundant in long-lived charge-reduced cation-radicals. Deuterium labeling established that the neutral hydrazine molecules mainly contained two exchangeable and two nonexchangeable hydrogen atoms from the peptide and underwent further H/D exchange in an ion–molecule complex. Electron structure calculations on the charge-reduced ions indicated that the unpaired electron was delocalized between the diazirine and amide π* electronic systems in the low electronic states of the cation-radicals. The diazirine moiety in GL*GGK-NH2was calculated to have an intrinsic electron affinity of 1.5 eV, which was further increased by the Coulomb effect of the peptide positive charge. Mechanisms are proposed for the unusual elimination of hydrazine from the photo-labeled peptide ions.
Co-reporter:Brian J. Wolfe, Farideh Ghomashchi, Tim Kim, Cynthia A. Abam, Martin Sadilek, Rhona Jack, Jerry N. Thompson, C. Ronald Scott, Michael H. Gelb, and Frantisek Turecek
Bioconjugate Chemistry 2012 Volume 23(Issue 3) pp:557
Publication Date(Web):February 28, 2012
DOI:10.1021/bc200609x
The clinical phenotype of Sanfilippo Syndrome is caused by one of four enzyme deficiencies that are associated with a defect in mucopolysaccharide metabolism. The four subtypes (A, B, C, and D) are each caused by an enzyme deficiency involved in the degradation of heparan sulfate. We have developed a highly efficient synthesis of the substrates and internal standards required for the enzymatic assay of each of the four enzymes. The synthesis of the substrates involves chemical modification of a common intermediate. The substrates and internal standards allow the measurement of the enzymes relevant to heparan N-sulfatase (type A); N-acetyl-α-glucosaminidase (type B); acetyl-CoA:α-glucosamide N-acetyltransferase (type C); and N-acetylglucosamine 6-sulfatase (type D). The internal standards are similar to the substrates and allow for the accurate quantification of the enzyme assays using tandem mass spectrometry. The synthetic substrates incorporate a coumarin moiety and can also be used in fluorometric enzyme assays. We confirm that all four substrates can detect the appropriate Sanfilippo Syndrome in fibroblast lysates, and the measured enzyme activities are distinctly lower by a factor of 10 when compared to fibroblast lysates from unaffected persons.
Co-reporter:Christopher L. Moss, František Tureček
International Journal of Mass Spectrometry 2012 Volumes 316–318() pp:57-67
Publication Date(Web):15 April 2012
DOI:10.1016/j.ijms.2011.11.017
Protonation sites in a model pentapeptide Ala-Ala-βAb-Ala-Ala, where βAb was β-aminobutyric acid, were studied by ab initio and density functional theory calculations. Gas-phase dication tautomers protonated at the N-terminus and amide oxygens were found to be substantially more stable than tautomers protonated at amide nitrogens. This order of ion stability did not change upon solvation with methanol. Conformational analysis of dication tautomers indicated similar degrees of internal solvation by hydrogen bonding in amide O- and N-protonated ions in the gas phase. Because of the low stability of amide N-protonated tautomers, their formation by electrospray of non-basic peptides is highly unlikely. Computational analysis of Ala-Ala-βAb-Ala-Ala cation-radicals indicated substantially lower transition-state energies for NCα bond dissociations at Ala residues than for the NCβ bond dissociation at βAb. The formation of β-radicals as z fragments was found to require a high threshold energy. Cleavage of the CONH2 bond leading to b and y fragments was hampered by a high-energy transition state for the formation of an N-protonated cation-radical intermediate as well as by a high threshold energy for the fragment formation. The calculated energies for transition states and dissociation thresholds explain the less efficient NCβ bond dissociation upon electron capture or transfer in peptide ions containing β-amino acid residues.Graphical abstractHighlights► Protonation in peptides containing β-amino acid residues occurs at amide O-atoms. ► NCα bond dissociations outcompete NCβ bond cleavages at β-amino acid residues. ► Hydrogen atom migrations between amide groups compete with NCα bond cleavage.
Co-reporter:Magdalena Zimnicka;Christopher L. Moss
Journal of The American Society for Mass Spectrometry 2012 Volume 23( Issue 4) pp:608-620
Publication Date(Web):2012 April
DOI:10.1007/s13361-011-0184-y
Charge tags using basic auxiliary functional groups 6-aminoquinolinylcarboxamido, 4-aminopyrimidyl-1-methylcarboxamido, 2-aminobenzoimidazolyl-1-methylcarboxamido, and the fixed-charge 4-(dimethylamino)pyridyl-1-carboxamido moiety are evaluated as to their properties in electron transfer dissociation mass spectra of arginine C-terminated peptides. The neutral tags have proton affinities that are competitive with those of amino acid residues in peptides. Charge reduction by electron transfer from fluoranthene anion-radicals results in peptide backbone dissociations that improve sequence coverage by providing extensive series of N-terminal c-type fragments without impeding the formation of C-terminal z fragments. Comparison of ETD mass spectra of free and tagged peptides allows one to resolve ambiguities in fragment ion assignment through mass shifts of c ions. Simple chemical procedures are reported for N-terminal tagging of Arg-containing tryptic peptides.
Co-reporter:Christopher L. Moss;Wenkel Liang
Journal of The American Society for Mass Spectrometry 2012 Volume 23( Issue 3) pp:446-459
Publication Date(Web):2012 March
DOI:10.1007/s13361-011-0283-9
We report a new approach to investigating the mechanisms of fast peptide cation-radical dissociations based on an analysis of time-resolved reaction progress by Ehrenfest dynamics, as applied to an Ala-Arg cation-radical model system. Calculations of stationary points on the ground electronic state that were carried out with effective CCSD(T)/6-311++G(3df,2p) could not explain the experimental branching ratios for loss of a hydrogen atom, ammonia, and N–Cα bond dissociation in (AR + 2H)+●. The Ehrenfest dynamics results indicate that the ground and low-lying excited electronic states of (AR + 2H)+● follow different reaction courses in the first 330 femtoseconds after electron attachment. The ground (X) state undergoes competing loss of N-terminal ammonia and isomerization to an aminoketyl radical intermediate that depend on the vibrational energy of the charge-reduced ion. The A and B excited states involve electron capture in the Arg guanidine and carboxyl groups and are non-reactive on the short time scale. The C state is dissociative and progresses to a fast loss of an H atom from the Arg guanidine group. Analogous results were obtained by using the B3LYP and CAM-B3LYP density functionals for the excited state dynamics and including the universal M06-2X functional for ground electronic state calculations. The results of this Ehrenfest dynamics study indicate that reaction pathway branching into the various dissociation channels occurs in the early stages of electron attachment and is primarily determined by the electronic states being accessed. This represents a new paradigm for the discussion of peptide dissociations in electron based methods of mass spectrometry.
Co-reporter:František Tureček, Christopher L. Moss, Thomas W. Chung
International Journal of Mass Spectrometry 2012 s 330–332() pp: 207-219
Publication Date(Web):
DOI:10.1016/j.ijms.2012.08.001
Co-reporter:Aaron R. Ledvina;Thomas W. Chung
Journal of The American Society for Mass Spectrometry 2012 Volume 23( Issue 8) pp:1351-1363
Publication Date(Web):2012 August
DOI:10.1007/s13361-012-0409-8
Dissociations of z4 ions from pentapeptides AAXAR where X = H, Y, F, W, and V produce dominant z2 ions that account for >50 % of the fragment ion intensity. The dissociation has been studied in detail by experiment and theory and found to involve several isomerization and bond-breaking steps. Isomerizations in z4 ions proceed by amide trans→cis rotations followed by radical-induced transfer of a β-hydrogen atom from the side chain, forming stable Cβ radical intermediates. These undergo rate-determining cleavage of the Cα–CO bond at the X residue followed by loss of the neutral AX fragment, forming x2 intermediates. The latter were detected by energy-resolved resonant excitation collision-induced dissociation (CID) and infrared multiphoton dissociation (IRMPD) experiments. The x2 intermediates undergo facile loss of HNCO to form z2 fragment ions, as also confirmed by energy-resolved CID and IRMPD MS4 experiments. The loss of HNCO from the x2 ion from AAHWR is kinetically hampered by the Trp residue that traps the OCNH radical group in a cyclic intermediate.
Co-reporter:Thomas W. Chung;Renjie Hui;Aaron Ledvina
Journal of The American Society for Mass Spectrometry 2012 Volume 23( Issue 8) pp:1336-1350
Publication Date(Web):2012 August
DOI:10.1007/s13361-012-0408-9
Amino acid residue-specific backbone and side-chain dissociations of peptide z ions in MS3 spectra were elucidated for over 40 pentapeptides with arginine C-terminated sequences of the AAXAR and AAHXR type, nonapeptides of the AAHAAXX"AR and AAHAXAX"AR type, and AAHAAXX"AAR decapeptides. Peptide zn ions containing amino acid residues with readily transferrable benzylic or tertiary β-hydrogen atoms (Phe, Tyr, His, Trp, Val) underwent facile backbone cleavages to form dominant zn-2 or zn-3 ions. These backbone cleavages are thought to be triggered by a side-chain β-hydrogen atom transfer to the z ion Cα radical site followed by homolytic dissociation of the adjacent Cα–CO bond, forming xn-2 cation-radicals that spontaneously dissociate by loss of HNCO. Amino acid residues that do not have readily transferrable β-hydrogen atoms (Gly, Ala) do not undergo the zn → zn-2 dissociations. The backbone cleavages compete with side-chain dissociations in z ions containing Asp and Asn residues. Side-chain dissociations are thought to be triggered by α-hydrogen atom transfers that activate the Cβ–Cγ or Cβ–heteroatom bonds for dissociations that dominate the MS3 spectra of z ions from peptides containing Leu, Cys, Lys, Met, Ser, Arg, Glu, and Gln residues. The Lys, Arg, Gln, and Glu residues also participate in γ-hydrogen atom transfers that trigger other side-chain dissociations.
Co-reporter:Magdalena Zimnicka ; Joshua A. Gregersen ;František Tureček
Journal of the American Chemical Society 2011 Volume 133(Issue 26) pp:10290-10301
Publication Date(Web):May 26, 2011
DOI:10.1021/ja203611x
We report the first preparation of a stable aminothioketyl radical, CH3C•(SH)NHCH3 (1), by fast electron transfer to protonated thioacetamide in the gas phase. The radical was characterized by neutralization–reionization mass spectrometry and ab initio calculations at high levels of theory. The unimolecular dissociations of 1 were elucidated with deuterium-labeled radicals CH3C•(SD)NHCH3 (1a), CH3C•(SH)NDCH3 (1b), CH3C•(SH)NHCD3 (1c), and CD3C•(SH)NHCH3 (1d). The main dissociations of 1 were a highly specific loss of the thiol H atom and a specific loss of the N-methyl group, which were competitive on the potential energy surface of the ground electronic state of the radical. RRKM calculations on the CCSD(T)/aug-cc-pVTZ potential energy surface indicated that the cleavage of the S–H bond in 1 dominated at low internal energies, Eint < 232 kJ mol–1. The cleavage of the N–CH3 bond was calculated to prevail at higher internal energies. Loss of the thiol hydrogen atom can be further enhanced by dissociations originating from the B excited state of 1 when accessed by vertical electron transfer. Hydrogen atom addition to the thioamide sulfur atom is calculated to have an extremely low activation energy that may enable the thioamide group to function as a hydrogen atom trap in peptide radicals. The electronic properties and reactivity of the simple aminothioketyl radical reported here may be extrapolated and applied to elucidate the chemistry of thioxopeptide radicals and cation radicals of interest to protein structure studies.
Co-reporter:Brian J. Wolfe, Sophie Blanchard, Martin Sadilek, C. Ronald Scott, Frantisek Turecek, and Michael H. Gelb
Analytical Chemistry 2011 Volume 83(Issue 3) pp:1152
Publication Date(Web):December 30, 2010
DOI:10.1021/ac102777s
We have developed a tandem mass spectrometry based assay of iduronate-2-sulfatase (IdS) activity for the neonatal detection of mucopolysaccharidosis II (MPS-II, Hunter Syndrome). The assay uses a newly designed synthetic substrate (IdS-S) consisting of α-l-iduronate-2-sulfate, which is glycosidically conjugated to a coumarin and a linker containing a tert-butyloxycarbamido group. A short synthesis of the substrate has been developed that has the potential of being scaled to multigram quantities. Sulfate hydrolysis of IdS-S by IdS found within a 3 mm dried blood spot specifically produces a nonsulfated product (IdS-P) which is detected by electrospray tandem mass spectrometry and quantified using a deuterium-labeled internal standard, both carried out in positive ion mode. Analysis of DBS from 75 random human newborns showed IdS activities in the range of 4.8−16.2 (mean 9.1) μmol/(h L of blood), which were clearly distinguished from the activities measured for 14 MPS-II patients at 0.17−0.52 (mean 0.29) μmol/(h L of blood). The assay shows low blank activity, 0.15 ± 0.03 μmol/(h L of blood). The within-assay coefficient of variation (CV) was 3.1% while the interassay CV was 15%.
Co-reporter:Zdeněk Spáčil, Susan Elliott, Steven L. Reeber, Michael H. Gelb, C. Ronald Scott, and František Tureček
Analytical Chemistry 2011 Volume 83(Issue 12) pp:4822
Publication Date(Web):May 6, 2011
DOI:10.1021/ac200417u
We report a comparative study of triplex tandem mass spectrometry (MS/MS) based assays of lysosomal enzymes in dried blood spots for the early detection of Pompe, Fabry, and Hurler diseases in newborns. Four methods have been evaluated that differed in sample handling and the equipment used. A newly developed method uses assay quenching with acetonitrile to precipitate blood proteins followed by analysis on an LC–electrospray/MS/MS system capable of multiple consecutive sample injections on two parallel chromatographic columns. This method requires 1.5 min per a triplex analysis of enzyme products and internal standards, which matches the throughput of the previously reported flow injection method. LC separation reduces matrix effects and allows for more facile sample workup. The new LC-based method showed figures of merit that were superior to those of the currently used method based on liquid–liquid extraction into ethyl acetate and flow injection into the mass spectrometer. The other methods we investigated for comprehensive comparison involved liquid–liquid extraction into ethyl acetate followed by LC–ESI-MS/MS and acetonitrile quenching followed by direct flow injection. Both methods using acetonitrile quenching were found to be robust and provide good quality data while requiring fewer liquid transfer steps and less disposable material and labor than did the extraction methods. The individual merits of the new methods are discussed to present an evaluated alternative approach to high-throughput analysis in newborn screening laboratories.
Co-reporter:Thomas W. Chung, František Tureček
International Journal of Mass Spectrometry 2011 Volume 301(1–3) pp:55-61
Publication Date(Web):30 March 2011
DOI:10.1016/j.ijms.2010.06.025
Two types of aminoketyl radicals and cation-radicals are distinguished by each's structure and reactivity. As proper aminoketyl radicals we denote the presumed intermediates of NCα bond dissociations induced by electron attachment to protonated peptides. These radicals have pyramidized aminoketyl groups, CαC(OH)NH, high spin density on the central carbon atom, and undergo facile NCα bond dissociations. The critical energies for NCα bond cleavages in proper aminoketyl radicals are summarized here and typically do not exceed 60 kJ mol−1 in peptide cation-radicals. In contrast, a different type of intermediates, which we call improper aminoketyl radicals, is formed by collisional dissociation of peptide cation-radicals, such as decarboxylation of zn fragments. Improper aminoketyl radicals have near planar CαC(OH)NH groups, and the spin density is delocalized over several atoms adjacent to the aminoketyl moiety. Improper aminoketyl radicals show higher transition energies for NCα bond cleavage and undergo H-atom transfers resulting in side-chain losses.Proper and improper aminoketyl radicals and cation-radicals are two types of intermediates that are distinguished by structure and reactivity.
Co-reporter:Thomas W. Chung, František Tureček
International Journal of Mass Spectrometry 2011 Volume 306(2–3) pp:99-107
Publication Date(Web):15 September 2011
DOI:10.1016/j.ijms.2010.08.021
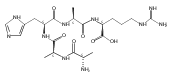
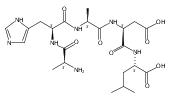
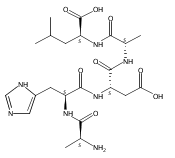
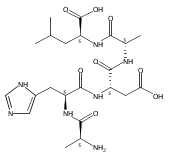
We report electron-transfer dissociation (ETD) mass spectra of histidine-containing peptides DSHAK, FHEK, HHGYK, and HHSHR from trypsinolysis of histatin 5. ETD of both doubly and triply protonated peptides provided sequence ions of the c and z type. In addition, electron transfer to doubly protonated peptides produced abundant long-lived cation-radicals, (M+2H)+, whose relative intensities depended on the peptide sequence and number of histidine residues. CID-MS3 spectra of (M+2H)+ cation-radicals were entirely different from the ETD spectra of the doubly charged ions and involved radical-driven losses of C4H6N2 neutral fragments from the histidine residues and charge-driven backbone cleavages forming b and y ions. Product ions from CID of (M+2H)+ were further characterized by CID-MS4 spectra to distinguish the histidine residues undergoing loss of C4H6N2. The ETD-CID-MSn mass spectra are interpreted by considering radical-induced rearrangements of histidine side chains in the long-lived charge-reduced ions.Graphical abstractResearch highlights▶ ETD of histidine-rich peptides provides complete sequence coverage. ▶ Charge-reduced survivor ions undergo specific rearrangements. ▶ Multi-stage CID reveals the histidine rearrangement sites.
Co-reporter:Thomas W. Chung;Christopher L. Moss
Journal of The American Society for Mass Spectrometry 2011 Volume 22( Issue 1) pp:13-30
Publication Date(Web):2011/01/01
DOI:10.1007/s13361-010-0012-9
Pyridiniummethylcarbonyl moieties that were previously designed on the basis of electronic structure analysis are now utilized as fixed-charge tags with tunable electronic properties to be used for N-terminal peptide derivatization and sequencing by electron-transfer dissociation. Dipeptides AK and KA were derivatized at the peptide N-terminus with 4-dimethylaminopyridinium-N-acetyl (DMAP-ac) and pyridinium-N-acetyl (pyrid-ac) tags of increasing intrinsic recombination energies. Upon the capture of a free electron or electron transfer from fluoranthene anions, (DMAP-ac-AK+H)2+, (DMAP-ac-KA+H)2+, (pyrid-ac-AK+H)2+ and (pyrid-ac-KA+H)2+ ions, as well as underivatized (AK+2H)2+, completely dissociated. The fixed-charge tags steered the dissociation upon electron transfer to form abundant backbone N–Cα bond cleavages, whereas the underivatized peptide mainly underwent H-atom and side-chain losses. Precursor ion structures for the tagged peptides were analyzed by an exhaustive conformational search combined with B3LYP/6-31+G(d,p) geometry optimization and single-point energy calculations in order to select the global energy minima. Structures, relative energies, transition states, ion–molecule complexes, and dissociation products were identified for several charge-reduced species from the tagged peptides. The electronic properties of the charge tags and their interactions with the peptide moieties are discussed. Electrospray ionization and electron-transfer dissociation of larger peptides are illustrated with a DMAP-tagged pentapeptide.
Co-reporter:Christopher L. Moss;Thomas W. Chung
Journal of The American Society for Mass Spectrometry 2011 Volume 22( Issue 4) pp:731-751
Publication Date(Web):2011 April
DOI:10.1007/s13361-011-0083-2
Electron transfer and capture mass spectra of a series of doubly charged ions that were phosphorylated pentapeptides of a tryptic type (pS,A,A,A,R) showed conspicuous differences in dissociations of charge-reduced ions. Electron transfer from both gaseous cesium atoms at 100 keV kinetic energies and fluoranthene anion radicals in an ion trap resulted in the loss of a hydrogen atom, ammonia, and backbone cleavages forming complete series of sequence z ions. Elimination of phosphoric acid was negligible. In contrast, capture of low-energy electrons by doubly charged ions in a Penning ion trap induced loss of a hydrogen atom followed by elimination of phosphoric acid as the dominant dissociation channel. Backbone dissociations of charge-reduced ions also occurred but were accompanied by extensive fragmentation of the primary products. z-Ions that were terminated with a deaminated phosphoserine radical competitively eliminated phosphoric acid and H2PO4 radicals. A mechanism is proposed for this novel dissociation on the basis of a computational analysis of reaction pathways and transition states. Electronic structure theory calculations in combination with extensive molecular dynamics mapping of the potential energy surface provided structures for the precursor phosphopeptide dications. Electron attachment produces a multitude of low lying electronic states in charge-reduced ions that determine their reactivity in backbone dissociations and H- atom loss. The predominant loss of H atoms in ECD is explained by a distortion of the Rydberg orbital space by the strong dipolar field of the peptide dication framework. The dipolar field steers the incoming electron to preferentially attach to the positively charged arginine side chain to form guanidinium radicals and trigger their dissociations.
Co-reporter:František Tureček ; Thomas W. Chung ; Christopher L. Moss ; Jean A. Wyer ; Anneli Ehlerding ; Anne I. S. Holm ; Henning Zettergren ; Steen Brøndsted Nielsen ; Preben Hvelplund ; Julia Chamot-Rooke ; Benjamin Bythell ;Béla Paizs
Journal of the American Chemical Society 2010 Volume 132(Issue 31) pp:10728-10740
Publication Date(Web):July 15, 2010
DOI:10.1021/ja907808h
Electron-transfer and -capture dissociations of doubly protonated peptides gave dramatically different product ions for a series of histidine-containing pentapeptides of both non-tryptic (AAHAL, AHAAL, AHADL, AHDAL) and tryptic (AAAHK, AAHAK, AHAAK, HAAAK, AAAHR, AAHAR, AHAAR, HAAAR) type. Electron transfer from gaseous Cs atoms and fluoranthene anions triggered backbone dissociations of all four N−Cα bonds in the peptide ions in addition to loss of H and NH3. Substantial fractions of charge-reduced cation-radicals did not dissociate on an experimental time scale ranging from 10−6 to 10−1 s. Multistage tandem mass spectrometric (MSn) experiments indicated that the non-dissociating cation-radicals had undergone rearrangements. These were explained as being due to proton migrations from N-terminal ammonium and COOH groups to the C-2′ position of the reduced His ring, resulting in substantial radical stabilization. Ab initio calculations revealed that the charge-reduced cation-radicals can exist as low-energy zwitterionic amide π* states which were local energy minima. These states underwent facile exothermic proton migrations to form aminoketyl radical intermediates, whereas direct N−Cα bond cleavage in zwitterions was disfavored. RRKM analysis indicated that backbone N−Cα bond cleavages did not occur competitively from a single charge-reduced precursor. Rather, these bond cleavages proceeded from distinct intermediates which originated from different electronic states accessed by electron transfer. In stark contrast to electron transfer, capture of a free electron by the peptide ions mainly induced radical dissociations of the charge-carrying side chains and loss of a hydrogen atom followed by standard backbone dissociations of even-electron ions. The differences in dissociation are explained by different electronic states being accessed upon electron transfer and capture.
Co-reporter:Trisha A. Duffey, Martin Sadilek, C. Ronald Scott, Frantisek Turecek, and Michael H. Gelb
Analytical Chemistry 2010 Volume 82(Issue 22) pp:9587
Publication Date(Web):October 20, 2010
DOI:10.1021/ac102090v
We report a new assay of N-acetylgalactosamine-4-sulfatase (aryl sulfatase B) activity in dried blood spots (DBS) for the early detection of mucopolysaccharidosis VI (Maroteaux−Lamy syndrome) in newborn screening. The assay uses a synthetic substrate consisting of N-acetylgalactosamine-4-sulfate moiety glycosidically linked to a hydrophobic residue and furnished with a tert-butyloxycarbamido group as a marker for specific mass spectrometric fragmentation. Incubation with aryl sulfatase B present in DBS converts the substrate to a desulfated product which is detected by electrospray tandem mass spectrometry and quantified using a homologous internal standard. Assay and workup procedures were optimized to be compatible with the work flow in newborn screening laboratories. Analysis of DBS from human newborns showed clear distinction of aryl sulfatase B activity from 89 healthy individuals where it ranged between 1.4 and 16.9 μmol/(h L of blood), with an average activity of 7.4 μmol/(h L of blood), and an MPS-VI patient that had an activity of 0.12 μmol/(h L of blood). Results are also reported for the aryl sulfatase B assay in DBS from groups of normal felines and felines affected with MPS-VI.
Co-reporter:Thomas W. Chung;František Tureček
Journal of The American Society for Mass Spectrometry 2010 Volume 21( Issue 8) pp:1279-1295
Publication Date(Web):2010 August
DOI:10.1016/j.jasms.2010.02.018
Backbone z-type fragment ions formed by electron-transfer dissociation (ETD) of doubly protonated peptides AAHAL, AHDAL, and AHADL were subjected to collisional activation and their dissociation products were studied by ETD-CID-MS3 and MS4. Electron structure theory calculations were performed to elucidate ion structures and reaction mechanisms. All z ions showed competitive eliminations of C3H7 and C4H8 from the C-terminal Leu side chain. The energetics and kinetics of these dissociations were studied computationally for the z4 ion from AAHAL, and optimized structures are reported for several intermediates and transition states. RRKM calculations on the combined B3LYP and PMP2/6-311++G(2d,p) potential energy surface provided unimolecular rate constants that closely reproduced the experimental branching ratios for C3H7 and C4H8 eliminations. Mechanisms were also studied for the loss of CO2 from z ions generated by ETD of AHDAL and AHADL and for a specific radical-induced Asp-Cα-CO backbone cleavage. CID of the z ions under study did not produce any fragment ions that would indicate cascade backbone dissociations triggered by the radical sites. In contrast, the majority of backbone dissociations occurred at bonds that were remote from the radical sites (spin-remote dissociations) and were triggered by proton migrations that were analogous to those considered for standard peptide ion fragmentations.
Co-reporter:František Tureček, Chunxiang Yao, Y. M. Eva Fung, Shigeo Hayakawa, Mami Hashimoto and Hiroshi Matsubara
The Journal of Physical Chemistry B 2009 Volume 113(Issue 20) pp:7347-7366
Publication Date(Web):April 23, 2009
DOI:10.1021/jp900719n
Radicals containing the histidine residue have been generated in the gas phase by femtosecond electron transfer to protonated histidine-N-methylamide (1H+), Nα-acetylhistidine-N-methylamide (2H+), Nα-glycylhistidine (3H+), and Nα-histidylglycine (4H+). Radicals generated by collisional electron transfer from dimethyldisulfide to ions 1H+ and 2H+ at 7 keV collision energies were found to dissociate completely on the microsecond time scale, as probed by reionization to cations. The main dissociations produced fragments from the imidazole side chain and the cleavage of the Cα—CO bond, whereas products of N—Cα bond cleavage were not observed. Electron transfer from gaseous potassium atoms to ions 3H+ and 4H+ at 2.97 keV collision energies not only caused backbone N—Cα bond dissociations but also furnished fractions of stable radicals that were detected after conversion to anions. Ion structures, ion−electron recombination energies, radical structures, electron affinities, and dissociation and transition-state energies were obtained by combined density functional theory and Møller−Plesset perturbational calculations (B3-PMP2) and basis sets ranging from 6-311+G(2d,p) to aug-cc-pVTZ. The Rice−Ramsperger−Kassel−Marcus theory was used to calculate rate constants on the B3-PMP2 potential energy surfaces to aid interpretation of the mass spectrometric data. The stability of Nα-histidylglycine-derived radicals is attributed to an exothermic isomerization in the imidazole ring, which is internally catalyzed by reversible proton transfer from the carboxyl group. The isomerization depends on the steric accessibility of the histidine side chain and the carboxyl group and involves a novel cation radical−COO salt-bridge intermediate.
Co-reporter:Grady R. Blacken;Michael Volný
Journal of The American Society for Mass Spectrometry 2009 Volume 20( Issue 6) pp:915-926
Publication Date(Web):2009 June
DOI:10.1016/j.jasms.2009.01.006
Zirconium, titanium, and hafnium oxide-coated stainless steel surfaces are fabricated by reactive landing of gas-phase ions produced by electrospray ionization of group IVB metal alkoxides. The surfaces are used for in situ enrichment of phosphopeptides before analysis by matrix-assisted laser desorption ionization (MALDI) mass spectrometry. To evaluate this method we characterized ZrO2 (zirconia) surfaces by (1) comparison with the other group IVB metal oxides of TiO2 (titania) and HfO2 (hafnia), (2) morphological characterization by SEM image analysis, and (3) dependence of phosphopeptide enrichment on the metal oxide layer thickness. Furthermore, we evaluated the necessity of the reactive landing process for the construction of useful metal oxide surfaces by preparing surfaces by electrospray deposition of Zr, Ti, and Hf alkoxides directly onto polished metal surfaces at atmospheric pressure. Although all three metal oxide surfaces evaluated were capable of phosphopeptide enrichment from complex peptide mixtures, zirconia performed better than hafnia or titania as a result of morphological characteristics illustrated by the SEM analysis. Metal oxide coatings that were fabricated by atmospheric pressure deposition were still capable of in situ phosphopeptide enrichment, although with inferior efficiency and surface durability. We show that zirconia surfaces prepared by reactive landing of gas-phase ions can be a useful tool for high throughput screening of novel phosphorylation sites and quantitation of phosphorylation kinetics.
Co-reporter:Changtong Hao;František Tureček
Journal of The American Society for Mass Spectrometry 2009 Volume 20( Issue 4) pp:639-651
Publication Date(Web):2009 April
DOI:10.1016/j.jasms.2008.12.001
1,n-Alkanediammonium cations in noncovalent complexes with two dibenzo-18-crown-6-ether (DBCE) ligands undergo an unusual intramolecular tandem hydrogen atom and proton transfer to the crown ether ligand upon charge reduction by electron capture. Deuterium labeling established that both migrating hydrogens originated from the ammonium groups. The double hydrogen transfer was found to depend on the length of the alkane chain connecting the ammonium groups. Ab initio calculations provided structures for select alkanediammonium·dibenzo-18-crown-6-ether complexes and dissociation products. This first observation of an intra-complex hydrogen transfer is explained by the unusual electronic properties of the complexes and the substantial hydrogen atom affinity of the aromatic rings in the crown ligand.
Co-reporter:Anne I.S. Holm, Mikkel K. Larsen, Subhasis Panja, Preben Hvelplund, Steen Brøndsted Nielsen, Ryan D. Leib, William A. Donald, Evan R. Williams, Changtong Hao, František Tureček
International Journal of Mass Spectrometry 2008 Volume 276(2–3) pp:116-126
Publication Date(Web):1 October 2008
DOI:10.1016/j.ijms.2008.04.021
Complexes of doubly protonated 1,n-diaminoalkanes with one or two molecules of 18-crown-6-ether undergo consecutive and competitive dissociations upon electron capture from a free thermal electron and femtosecond collisional electron transfer from Na and Cs atoms. The electron capture dissociation (ECD) and electron capture-induced dissociation (ECID) mass spectra show very different products and product ion intensities. In ECD, the reduced precursor ions dissociate primarily by loss of an ammonium hydrogen and the crown ether ligand. In ECID, ions from many more dissociation channels are observed and depend on whether collisions occur with Na or Cs atoms. ECID induces highly endothermic CC bond cleavages along the diaminoalkane chain, which are not observed with ECD. Adduction of one or two crown ethers to diaminoalkanes results in different electron capture cross-sections that follow different trends for ECD and ECID. Electron structure calculations at the B3-PMP2/6-311++G(2d,p) level of theory were used to determine structures of ions and ion radicals and the energetics for protonation, electron transfer, and ion dissociations for most species studied experimentally. The calculations revealed that the crown ether ligand substantially affected the recombination energy of the diaminoalkane ion and the electronic states accessed by electron attachment.
Co-reporter:Subhasis Panja;Steen Brøndsted Nielsen
Journal of The American Society for Mass Spectrometry 2008 Volume 19( Issue 12) pp:1726-1742
Publication Date(Web):2008 December
DOI:10.1016/j.jasms.2008.08.001
Collisional electron transfer from gaseous Cs atoms was studied for singly and doubly protonated peptides Gly-Arg (GR) and Ala-Arg (AR) at 50- and 100-keV kinetic energies. Singly protonated GR and AR were discharged to radicals that in part rearranged by migration of a Cα hydrogen atom onto the guanidine group. The Cα-radical isomers formed were detected as stable anions following transfer of a second electron. In addition to the stabilizing rearrangements, the radicals underwent side-chain and backbone dissociations. The latter formed z fragments that were detected as the corresponding anions. Analysis of the (GR+H)· radical potential energy surface using electronic structure theory in combination with Rice-Ramsperger-Kassel-Marcus calculations of rate constants indicated that the arginine Cα hydrogen atom was likely to be transferred to the arginine side-chain on the experimental timescale of ≤200 ns. Transfer of the Gly Cα-H was calculated to have a higher transition-state energy and was not kinetically competitive. Collisional electron transfer to doubly protonated GR and AR resulted in complete dissociation of (GR+2H)+· and (AR+2H)+· ions by loss of H, ammonia, and N-Cα bond cleavage. Electronic structure theory analysis of (GR+2H)+· indicated the presence of multiple conformers and electronic states that differed in reactivity and steered the dissociations to distinct channels. Electron attachment to (GR+2H)2+ resulted in the formation of closely spaced electronic states of (GR+2H)+· in which the electron density was delocalized over the guanidinium, ammonium, amide, and carboxyl groups. The different behavior of (GR+H)· and (GR+2H)+· is explained by the different timescales for dissociation and different internal energies acquired upon electron transfer.
Co-reporter:Jace W. Jones;Scott A. Shaffer;Robert K. Ernst;David R. Goodlett;František Tureček;
Proceedings of the National Academy of Sciences 2008 105(35) pp:12742-12747
Publication Date(Web):August 27, 2008
DOI:10.1073/pnas.0800445105
Lipid A isolated from several bacteria (Escherichia coli, Pseudomonas aeruginosa, Salmonella enterica, and various strains of Yersinia) showed abundant formation of pyrophosphate anions upon ion dissociation. Pyrophosphate [H3P2O7]− and/or [HP2O6]− anions were observed as dominant fragments from diphosphorylated lipid A anions regardless of the ionization mode (matrix-assisted
laser desorption ionization or electrospray ionization), excitation mode (collisional activation or infrared photoexcitation),
or mass analyzer (time-of-flight/time-of-flight, tandem quadrupole, Fourier transform–ion cyclotron resonance mass spectrometry).
Dissociations of anions from model lipid phosphate, pyrophosphate, and hexose diphosphates confirmed that pyrophosphate fragments
were formed abundantly only in the presence of an intact pyrophosphate group in the analyte molecule and were not due to intramolecular
rearrangement upon ionization, ion-molecule reactions, or rearrangement following activation. This indicated that pyrophosphate
groups are present in diphosphorylated lipid A from a variety of Gram-negative bacteria.
Co-reporter:Jill K. Wolken, Chunxiang Yao, František Tureček, Michael J. Polce, Chrys Wesdemiotis
International Journal of Mass Spectrometry 2007 Volume 267(1–3) pp:30-42
Publication Date(Web):1 November 2007
DOI:10.1016/j.ijms.2007.02.016
Gas-phase cytosine molecules and cation–radicals represent a complex system of several nearly isoenergetic tautomers within each group. Computational methods differ in ordering the relative enthalpies of neutral cytosine tautomers. At our highest level of theory, CCSD(T)/aug-cc-pVTZ calculations find an enol form, anti-2-hydroxy-4-aminopyrimidine (2), to be the most stable neutral tautomer in the gas-phase, followed by its rotamer, syn-2-hydroxy-4-aminopyrimidine (3), the canonical oxo-form, 4-amino-1,2-dihydropyrimidin-2(1H)-one (1), imino-forms, 2-oxo-4-iminodihydro(1H,3H)pyrimidine (4 and 5), and another oxo-form, 4-amino-dihydropyrimidin-2(3H)-one (6). Other tautomers, such as anti-anti, syn-syn and syn-anti-2-hydroxy-4-iminodihydro(3H,4H)pyrimidines (7–9), are less stable. The adiabatic ionization energies of the major cytosine tautomers have been calculated to be 8.71, 8.64, 8.62, 8.58, 8.64, and 8.31 eV for 1, 2, 3, 4, 5, and 6, respectively. Cytosine cation–radicals show very close relative energies that increase in the order of 6+ (most stable) <2+ ≈ 3+ < 4+ ≈ 7+ ≈ 1+ < 5+. In addition, distonic ions having radical centers at C-5 (10+) and C-6 (11+ are found as low-energy isomers of 1+–7+. Metastable cytosine cation–radicals undergo ring-cleavage dissociations by eliminations of CO (major) and HNCO (minor). The energetics of these and other higher-energy dissociations, including the pertinent transition states, have been established by high-level ab initio and density functional theory calculations and plausible mechanisms have been proposed. Collisional neutralization of cytosine cation–radicals with trimethylamine and dimethyldisulfide as electron donors forms stable molecules that are detected as cation–radicals following collisional reionization. The dissociations observed upon neutralization–reionization mainly include ring-cleavages followed by loss of NCO, HNCO, and formation of C2H3N, C2H2N, and CO neutral fragments that are assigned to ion dissociations following reionization.
Co-reporter:Chunxiang Yao, František Tureček, Michael J. Polce, Chrys Wesdemiotis
International Journal of Mass Spectrometry 2007 Volume 265(2–3) pp:106-123
Publication Date(Web):1 September 2007
DOI:10.1016/j.ijms.2007.01.013
Cytosine cations were generated by chemical ionization and fast-atom bombardment of cytosine, and their dissociations in the gas phase were studied by tandem mass spectrometry and quantum chemistry calculations at levels of theory up to CCSD(T)/aug-cc-pVTZ. Metastable cytosine cations undergo loss of NH3, H2O, and [CHNO] molecules as major dissociations. Mechanisms for these dissociations were established by a combination of MS3, deuterium labeling, and calculations. Several tautomers of protonated cytosine were identified to exist as local energy minima. The tautomers are predicted to undergo facile prototropic isomerizations at internal energies below the lowest ion-dissociation thresholds. The lowest-energy elimination of ammonia proceeds from open-ring structures and is accompanied by exchange of the N-3 and N-7 positions in cytosine, so that either nitrogen atom can be lost in the ammonia molecule. Cytosine radicals corresponding to hydrogen atom adducts are stable when formed by femtosecond electron transfer to the cations. A fraction of the radicals dissociate by loss of hydrogen atom to form cytosine tautomers. Ring-cleavage dissociations leading to the loss of CO and HNCO are less abundant, and are predicted to proceed from excited electronic states accessed by vertical electron transfer.
Co-reporter:W. Timothy Elam;Buddy D. Ratner;František Turec̆ek;Michael Volný
Journal of Biomedical Materials Research Part B: Applied Biomaterials 2007 Volume 80B(Issue 2) pp:505-510
Publication Date(Web):12 JUL 2006
DOI:10.1002/jbm.b.30624
A novel dry process for immobilization of hyaluronan on stainless steel surfaces is presented. This process that we call reactive landing is based on an interaction of hyperthermal gas-phase hyaluronan ions with plasma-cleaned and activated stainless steel surfaces. Reactive landing is performed on a unique instrument that combines an in-situ plasma reactor with an electrospray ion source and ion transfer optics. Gas-phase hyaluronan anions are obtained by electrospray ionization of sodium hyaluronan solutions and immobilized by reactive landing on large-area stainless steel surfaces. The immobilized hyaluronan withstands extensive washing with polar solvents and solutions, and the washed surfaces maintain the protective properties against blood platelet activation. The mechanism of hyaluronan discharge and immobilization is discussed. © 2006 Wiley Periodicals, Inc. J Biomed Mater Res Part B: Appl Biomater, 2007
Co-reporter:Julia Chamot-Rooke;Christian Malosse
Journal of The American Society for Mass Spectrometry 2007 Volume 18( Issue 12) pp:2146-2161
Publication Date(Web):2007 December
DOI:10.1016/j.jasms.2007.09.009
Electron capture dissociation (ECD) was studied with doubly charged dipeptide ions that were tagged with fixed-charge tris-(2,4,6-trimethoxyphenyl)phosphonium-methylenecarboxamido (TMPP-ac) groups. Dipeptides GK, KG, AK, KA, and GR were each selectively tagged with one TMPP-ac group at the N-terminal amino group while the other charge was introduced by protonation at the lysine or arginine side-chain groups to give (TMPP-ac-peptide + H)2+ ions by electrospray ionization. Doubly tagged peptide derivatives were also prepared from GK, KG, AK, and KA in which the fixed-charge TMPP-ac groups were attached to the N-terminal and lysine side-chain amino groups to give (TMPP-ac-peptide-ac-TMPP)2+ dications by electrospray. ECD of (TMPP-ac-peptide + H)2+ resulted in 72% to 84% conversion to singly charged dissociation products while no intact charge-reduced (TMPP-ac-dipeptide + H)+· ions were detected. The dissociations involved loss of H, formation of (TMPP + H)+, and N-cα bond cleavages giving TMPP-CH2CONH2+ (c0) and c1 fragments. In contrast, ECD of (TMPP-ac-peptide-ac-TMPP)2+ resulted in 31% to 40% conversion to dissociation products due to loss of neutral TMPP molecules and 2,4,6-trimethoxyphenyl radicals. No peptide backbone cleavages were observed for the doubly tagged peptide ions. Ab initio and density functional theory calculations for (Ph3P-ac-GK + H)2+ and (H3P-ac-GK + H)2+ analogs indicated that the doubly charged ions contained the lysine side-chain NH3+ group internally solvated by the COOH group. The distance between the charge-carrying phosphonium and ammonium atoms was calculated to be 13.1–13.2 Å in the most stable dication conformers. The intrinsic recombination energies of the TMPP+-ac and (GK + H)+ moieties, 2. 7 and 3. 15 eV, respectively, indicated that upon electron capture the ground electronic states of the (TMPP-ac-peptide + H)+· ions retained the charge in the TMPP group. Ground electronic state (TMPP-ac-GK + H)+· ions were calculated to spontaneously isomerize by lysine H-atom transfer to the COOH group to form dihydroxycarbinyl radical intermediates with the retention of the charged TMPP group. These can trigger cleavages of the adjacent N-Cα bonds to give rise to the c1 fragment ions. However, the calculated transition-state energies for GK and GGK models suggested that the ground-state potential energy surface was not favorable for the formation of the abundant c0 fragment ions. This pointed to the involvement of excited electronic states according to the Utah-Washington mechanism of ECD.
Co-reporter:Jace W. Jones;Tomikazu Sasaki
Journal of The American Society for Mass Spectrometry 2007 Volume 18( Issue 3) pp:432-444
Publication Date(Web):2007 March
DOI:10.1016/j.jasms.2006.10.005
Electron capture dissociation was studied with tetradecapeptides and pentadecapeptides that were capped at N-termini with a 2-(4′-carboxypyrid-2′-yl)-4-carboxamide group (pepy), e. g., pepy-AEQLLQEEQLLQEL-NH2, pepy-AQEFGEQGQKALKQL-NH2, and pepy-AQEGSEQAQKFFKQL-NH2. Doubly and triply protonated peptide cations underwent efficient electron capture in the ion-cyclotron resonance cell to yield charge-reduced species. However, the electron capture was not accompanied by backbone dissociations. When the peptide ions were preheated by absorption of infrared photons close to the dissociation threshold, subsequent electron capture triggered ion dissociations near the remote C-terminus forming mainly (b11–14+1)+· fragment ions that were analogous to those produced by infrared multiphoton dissociation alone. Ab initio calculations indicated that the N-1 and N-1′ positions in the pepy moiety had topical gas-phase basicities (GB=923 kJ mol−1) that were greater than those of backbone amide groups. Hence, pepy was a likely protonation site in the doubly and triply charged ions. Electron capture in the protonated pepy moiety produced the ground electronic state of the charge-reduced cation-radical with a topical recombination energy, RE=5.43–5.46 eV, which was greater than that of protonated peptide residues. The hydrogen atom in the charge-reduced pepy moiety was bound by >160 kJ mol−1, which exceeded the hydrogen atom affinity of the backbone amide groups (21–41 kJ mol−1). Thus, the pepy moiety functioned as a stable electron and hydrogen atom trap that did not trigger radical-type dissociations in the peptide backbone that are typical of ECD. Instead, the internal energy gained by electron capture was redistributed over the peptide moiety, and when combined with additional IR excitation, induced proton-driven ion dissociations which occurred at sites that were remote from the site of electron capture. This example of a spin-remote fragmentation provided the first clear-cut experimental example of an ergodic dissociation upon ECD.
Co-reporter:Chunxiang Yao and František Tureček
Physical Chemistry Chemical Physics 2005 vol. 7(Issue 5) pp:912-920
Publication Date(Web):31 Jan 2005
DOI:10.1039/B414764B
The title hypervalent ammonium radicals were investigated by neutralization–reionization mass spectrometry and quantum chemical calculations. Methyl ammonium (1) forms a small fraction of metastable radicals from isotopomers CH3ND3
(1a) and CD3NH3
(1b) when these are produced by femtosecond electron transfer to vibrationally excited precursor cations. The branching ratios for dissociations of the N–C and N–(H,D) bonds in 1 favor the latter, kN–C/kN–H
= 0.39. The experimental results are in accord with ab initio/RRKM calculations that quantitatively reproduce the branching ratios for dissociations of 1. A small fraction of high-energy 1 dissociates to form ammonium methylide, −CH2NH3+. Ethyl ammonium (2) and its CH3CH2ND3 isotopomer (2a) dissociate completely on the microsecond time scale. The branching ratios for dissociations of the N–C and N–(H,D) bonds favor the former, kN–C/kN–H
= 2.04. This result is incompatible with the calculated potential energy surface of the ground doublet electronic state in 2 and is attributed to the formation and dissociations of excited electronic states.
Co-reporter:František Tureček, Xiaohong Chen
Journal of the American Society for Mass Spectrometry 2005 Volume 16(Issue 10) pp:1713-1726
Publication Date(Web):October 2005
DOI:10.1016/j.jasms.2005.06.010
Ab initio and density functional theory calculations at the B3-MP2 and CCSD(T)/6-311 + G(3df,2p) levels of theory are reported that address the protonation of adenine in the gas phase, water clusters, and bulk aqueous solution. The calculations point to N-1-protonated adenine (1+) as the thermodynamically most stable cationic tautomer in the gas phase, water clusters, and bulk solution. This strongly indicates that electrospray ionization of adenine solutions produces tautomer 1+ with a specificity calculated as 97–90% in the 298–473 K temperature range. The mechanisms for elimination of hydrogen atoms and ammonia from 1+ have also been studied computationally. Ion 1+ is calculated to undergo fast migrations of protons among positions N-1, C-2, N-3, N-10, N-7, and C-8 that result in an exchange of five hydrogens before loss of a hydrogen atom forming adenine cation radical at 415 kJ mol−1 dissociation threshold energy. The elimination of ammonia is found to be substantially endothermic requiring 376–380 kJ mol−1 at the dissociation threshold and depending on the dissociation pathway. The overall dissociation is slowed by the involvement of ion-molecule complexes along the dissociation pathways. The competing isomerization of 1+ proceeds by a sequence of ring opening, internal rotations, imine flipping, ring closures, and proton migrations to effectively exchange the N-1 and N-10 atoms in 1+, so that either can be eliminated as ammonia. This mechanism explains the previous N-1/N-10 exchange upon collision-induced dissociation of protonated adenine.
Co-reporter:Erik A. Syrstad, František Tureček
Journal of the American Society for Mass Spectrometry 2005 Volume 16(Issue 2) pp:208-224
Publication Date(Web):February 2005
DOI:10.1016/j.jasms.2004.11.001
The effects of positive charge on the properties of ammonium and amide radicals were investigated by ab initio and density functional theory calculations with the goal of elucidating the energetics of electron capture dissociation (ECD) of multiply charged peptide ions. The electronic properties of the amide group in N-methylacetamide (NMA) are greatly affected by the presence of a remote charge in the form of a point charge, methylammonium, or guanidinium cations. The common effect of the remote charge is an increase of the electron affinity of the amide group, resulting in exothermic electron capture. The NCα bond dissociation and transition state energies in charge-stabilized NMA anions are 20–50 kJ mol−1 greater than in the hydrogen atom adduct. The zwitterions formed by electron capture have proton affinities that were calculated as 1030–1350 kJ mol−1, and are sufficiently basic for the amide carbonyl to exothermically abstract a proton from the ammonium, guanidinium and imidazolium groups in protonated lysine, arginine, and histidine residues, respectively. A new mechanism is proposed for ECD of multiply charged peptide and protein cations in which the electron enters a charge-stabilized electronic state delocalized over the amide group, which is a superbase that abstracts a proton from a sterically proximate amino acid residue to form a labile aminoketyl radical that dissociates by NCα bond cleavage. This mechanism explains the low selectivity of NCα bond dissociations induced by electron capture, and is applicable to dissociations of peptide ions in which the charge carriers are metal ions or quaternary ammonium groups. The new amide superbase and the previously proposed mechanisms of ECD can be uniformly viewed as being triggered by intramolecular proton transfer in charge-reduced amide cation-radicals. In contrast, remote charge affects NH bond dissociation in weakly bound ground electronic states of hypervalent ammonium radicals, as represented by methylammonium, CH3NH3·, but has a negligible effect on the NH bond dissociation in the strongly bound excited electronic states. This refutes previous speculations that loss of “hot hydrogen” can occur from an excited state of an ammonium radical.
Co-reporter:Chunxiang Yao;Maria L. Cuadrado-Peinado;Miroslav Polášek Dr.;František Tureček Dr.
Angewandte Chemie 2005 Volume 117(Issue 41) pp:
Publication Date(Web):27 SEP 2005
DOI:10.1002/ange.200502363
Welch ein Radikalziel! Zwei zentrale Wasserstoffatomaddukte von 1-Methylcytosin wurden durch die gezielte chemische Erzeugung von Cytosinradikalen hergestellt. Der Femtosekundenstoßtransfer von Elektronen aus molekularen Elektronendonoren (Dimethyldisulfid und Trimethylamin) auf Gasphasentautomere von protoniertem 1-Methylcytosin liefert stabile Radikaladdukte, die die Wasserstoffatome entweder an N3 oder an C5 des Cytosinrings tragen (siehe Schema).
Co-reporter:Chunxiang Yao, Maria L. Cuadrado-Peinado, Miroslav Polá&x161;ek,Franti&x161;ek Ture&x10d;ek
Angewandte Chemie International Edition 2005 44(41) pp:6708-6711
Publication Date(Web):
DOI:10.1002/anie.200502363
Co-reporter:Yuko Ogata, Louis Scampavia, Tyan L. Carter, Erkang Fan, František Tureček
Analytical Biochemistry 2004 Volume 331(Issue 1) pp:161-168
Publication Date(Web):1 August 2004
DOI:10.1016/j.ab.2004.04.028
We report a fully automated affinity chromatography system using a lab-on-valve (LOV) apparatus coupled to an electrospray ionization ion-trap mass spectrometer (ESI-MS). The system allows simultaneous measurements of multiple ligand affinities to proteins immobilized on beads. Bead regeneration, column repacking, and repetitive measurements are achieved on the time scale of several minutes. In this study, the system was used to screen the binding of a peptide mixture to human and Trypanosoma brucei (T. brucei) truncated Pex5 (tPex5) proteins. Equilibrium dissociation constants (Kd) were measured for T. brucei tPex5 and compared to the values obtained by a fluorescence-based competition assay. The three peptides that showed affinity toward tPex5 had Kd values that were comparable in magnitude (within a factor of 5) and showed the same ranking order as those from manual fluorescence measurements. With 12 min of sample infusion, the entire sample-to-sample cycle takes about 15 min and can be repeated without any preparation between runs. For T. brucei tPex5 affinity measurements, 1 mg of protein was sufficient for 35 repetitive analyses in the automated LOV-ESI-MS apparatus. The system allows rapid determination of Kd in the range of 10−5–10−7 M for sample mixtures and is suitable for screening a large number of compounds against multiple proteins.
Co-reporter:R Srikanth, R Srinivas, K Bhanuprakash, S Vivekananda, E.A Syrstad, F Turec̆ek
Journal of the American Society for Mass Spectrometry 2002 Volume 13(Issue 3) pp:250-264
Publication Date(Web):March 2002
DOI:10.1016/S1044-0305(01)00360-9
The bicoordinated dihydroxyphosphenium ion P(OH)2+ (1+) was generated specifically by charge-exchange dissociative ionization of triethylphosphite and its connectivity was confirmed by collision induced dissociation and neutralization-reionization mass spectra. The major dissociation of 1+ forming PO+ ions at m/z 47 involved another isomer, OPOH2+ (2+), for which the optimized geometry showed a long POH2 bond. Dissociative 70-eV electron ionization of diethyl phosphite produced mostly 1+ together with a less stable isomer, HP(O)OH+ (3+). Ion 2+ is possibly co-formed with 1+ upon dissociative 70-eV electron ionization of methylphosphonic acid. Neutralization-reionization of 1+ confirmed that P(OH)2· (1) was a stable species. Dissociations of neutral 1, as identified by variable-time measurements, involved rate-determining isomerization to 2 followed by fast loss of water. A competitive loss of H occurs from long-lived excited states of 1 produced by vertical electron transfer. The A and B states undergo rate-determining internal conversion to vibrationally highly excited ground state that loses an H atom via two competing mechanisms. The first of these is the direct cleavage of one of the OH bonds in 1. The other is an isomerization to 3 followed by cleavage of the PH bond to form OPOH as a stable product. The relative, dissociation, and transition state energies for the ions and neutrals were studied by ab initio and density functional theory calculations up to the QCISD(T)/6–311+G(3df,2p) and CCSD(T)/aug-cc-pVTZ levels of theory. RRKM calculations were performed to investigate unimolecular dissociation kinetics of 1. Excited state geometries and energies were investigated by a combination of configuration interaction singles and time-dependent density functional theory calculations.
Co-reporter:Miroslav Polášek, František Tureček
Journal of the American Society for Mass Spectrometry 2000 Volume 11(Issue 5) pp:380-392
Publication Date(Web):May 2000
DOI:10.1016/S1044-0305(00)00106-9
Protonation sites in methyl nitrate (1) were evaluated computationally at the Gaussian 2(MP2) level of ab initio theory. The methoxy oxygen was the most basic site that had a calculated proton affinity of PA = 728–738 kJ mol−1 depending on the optimization method used to calculate the equilibrium geometry of the CH3O(H)–NO2+ ion (2+). Protonation at the terminal oxygen atoms in methyl nitrate was less exothermic; the calculated proton affinities were 725, 722, and 712 kJ mol−1 for the formation of the syn–syn, anti–syn, and syn–anti ion rotamers 3a+, 3b+, and 3c+, respectively. Ion 2+ was prepared by an ion–molecule reaction of NO2+ with methanol and used to generate the transient CH3O(H)–NO2· radical (2) by femtosecond collisional electron transfer. Exothermic protonation of 1 produced a mixture of 3a+–3c+ with 2+ that was used to generate transient radicals 3a–3c. Radical 2 was found to be unbound and dissociated without barrier to methanol and NO2. Radicals 3a–3c were calculated to be weakly bound. When formed by vertical neutralization, 3a–3c dissociated completely on the 4.2 μs time scale of the experiment. The main dissociations of 3a–3c were formations of CH3O· + HONO and CH3ONO + OH·. The gas-phase chemistry of radicals 3a–3c and their dissociation products, as studied by neutralization–reionization mass spectrometry, was dominated by Franck–Condon effects on collisional neutralization and reionization. The adiabatic ionization energies of 3a–3c were calculated as 7.54, 7.57, and 7.66 eV, respectively.
Co-reporter:Miroslav Polášek, Martin Sadı́lek, František Tureček
International Journal of Mass Spectrometry 2000 Volumes 195–196() pp:101-114
Publication Date(Web):21 January 2000
DOI:10.1016/S1387-3806(99)00139-6
[C,H3,N,O] cation radicals and molecules corresponding to the less stable isomers were investigated by a combination of ab initio and density functional calculations, and collision-induced dissociation and neutralization-reionization mass spectrometric measurements. The ions formed by loss of oxygen from a nitromethane cation radical and loss of hydroxyl radical from protonated nitromethane were identified as having the nitrosomethane structure (2+·). A novel rearrangement in the ethyl nitrite cation radical was found to result in an elimination of formaldehyde to produce 2+· but not isonitrosomethane, CH3ON+· (3+·). Dissociations of 2+· and 2 were investigated by variable-time neutralization-reionization mass spectrometry. Cleavage of the C–N bond was found to occur in both the neutral and ion with comparable rate constants. Ab initio calculations provided C–N bond dissociation energies that were very similar in 2 and 2+·, 161 and 164 kJ mol−1, respectively. The calculated heats of formation, adiabatic and vertical ionization energies, and recombination energies are reported for six [C,H3,N,O] ion and neutral isomers, formamide, nitrosomethane, isonitrosomethane, formaldoxime, formaldonitrone, and oxaziridine.
Co-reporter:Martin Sadı́lek, František Tureček
International Journal of Mass Spectrometry 1999 Volumes 185–187() pp:639-649
Publication Date(Web):29 April 1999
DOI:10.1016/S1387-3806(98)14147-7
Variable-time neutralization–reionization mass spectrometry was used to generate and detect the hypervalent radical dimethylsulfonium, (CH3)2SH·(1H·), and its isotopomers (CH3)2SD·(1HD·), (CD3)2SH·(1DH·), and (CD3)2SD·(1DD·). The successful detection of 1H·–1DD·was achieved in spite of severe interferences due to isobaric overlaps by the 13C, 33S, and 34S isotope satellites of residual dimethylsulfide cation radicals. Radicals 1H·–1DD·dissociated by cleavages of the S–(H,D) and S–C bonds in a ∼3:1 branching ratio. Ab initio calculations with large basis sets failed to find a local energy minimum for the (X)2A′ ground electronic state of 1H·, which dissociated without barrier to (CH3)2S and H·. Several excited states of 1H·were found by configuration interaction singles CIS/6-311++G(3df,2p) calculations that were within 2 eV of the repulsive ground state potential energy surface. The microsecond metastability and dissociations of 1H·–1DD· were ascribed to the properties of excited electronic states.
Co-reporter:Thomas W. Chung, František Tureček
Journal of the American Society for Mass Spectrometry (August 2010) Volume 21(Issue 8) pp:1279-1295
Publication Date(Web):1 August 2010
DOI:10.1016/j.jasms.2010.02.018
Backbone z-type fragment ions formed by electron-transfer dissociation (ETD) of doubly protonated peptides AAHAL, AHDAL, and AHADL were subjected to collisional activation and their dissociation products were studied by ETD-CID-MS3 and MS4. Electron structure theory calculations were performed to elucidate ion structures and reaction mechanisms. All z ions showed competitive eliminations of C3H7 and C4H8 from the C-terminal Leu side chain. The energetics and kinetics of these dissociations were studied computationally for the z4 ion from AAHAL, and optimized structures are reported for several intermediates and transition states. RRKM calculations on the combined B3LYP and PMP2/6-311++G(2d,p) potential energy surface provided unimolecular rate constants that closely reproduced the experimental branching ratios for C3H7 and C4H8 eliminations. Mechanisms were also studied for the loss of CO2 from z ions generated by ETD of AHDAL and AHADL and for a specific radical-induced Asp-Cα–CO backbone cleavage. CID of the z ions under study did not produce any fragment ions that would indicate cascade backbone dissociations triggered by the radical sites. In contrast, the majority of backbone dissociations occurred at bonds that were remote from the radical sites (spin-remote dissociations) and were triggered by proton migrations that were analogous to those considered for standard peptide ion fragmentations.ETD-formed zn ions undergo competitive dissociations involving hydrogen atom migrations.Download high-res image (146KB)Download full-size image
Co-reporter:Changtong Hao, František Tureček
Journal of the American Society for Mass Spectrometry (April 2009) Volume 20(Issue 4) pp:639-651
Publication Date(Web):1 April 2009
DOI:10.1016/j.jasms.2008.12.001
1,n-Alkanediammonium cations in noncovalent complexes with two dibenzo-18-crown-6-ether (DBCE) ligands undergo an unusual intramolecular tandem hydrogen atom and proton transfer to the crown ether ligand upon charge reduction by electron capture. Deuterium labeling established that both migrating hydrogens originated from the ammonium groups. The double hydrogen transfer was found to depend on the length of the alkane chain connecting the ammonium groups. Ab initio calculations provided structures for select alkanediammonium·dibenzo-18-crown-6-ether complexes and dissociation products. This first observation of an intra-complex hydrogen transfer is explained by the unusual electronic properties of the complexes and the substantial hydrogen atom affinity of the aromatic rings in the crown ligand.Electron capture in dibenzo-18-crown-6-ether-alkanediammmonium complexes triggers an unusual transfer of ammonium proton and hydrogen atom forming a crown-ether cation radical.Download high-res image (98KB)Download full-size image
.jpg)

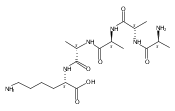






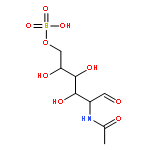
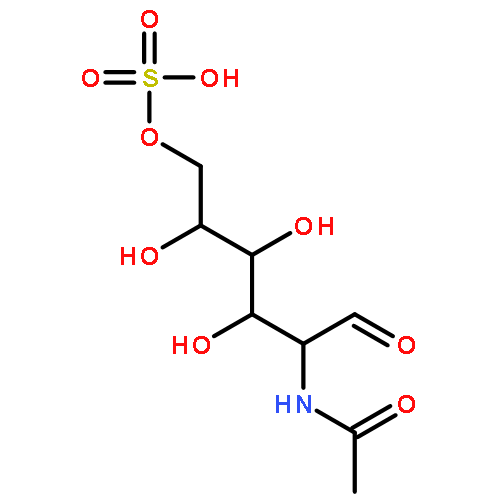

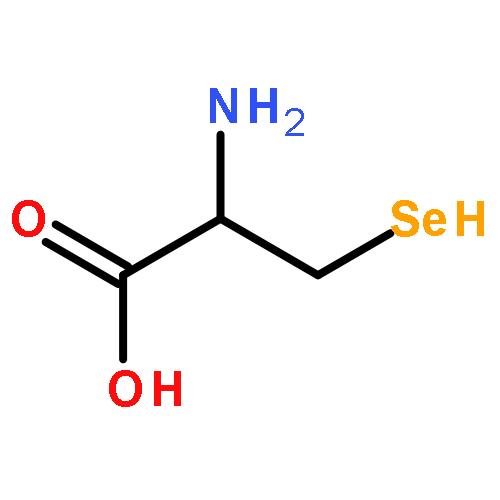









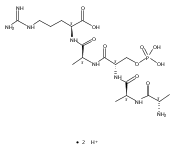

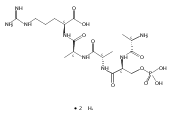
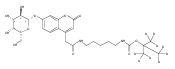

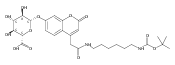
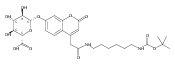
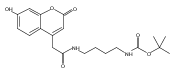
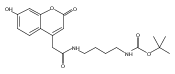

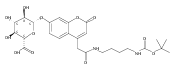
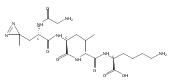


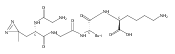
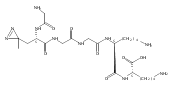

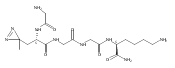
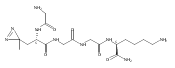
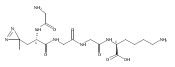


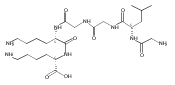

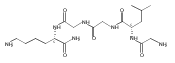
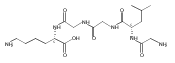

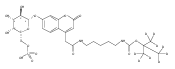
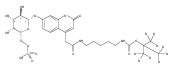






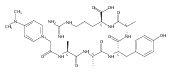
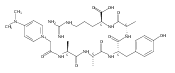
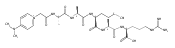
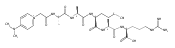

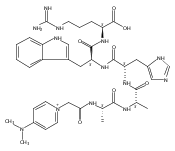




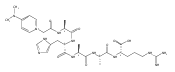
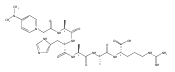

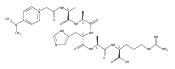

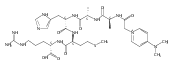
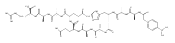

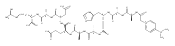
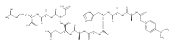
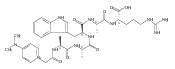

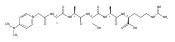

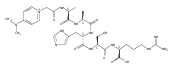
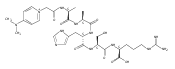


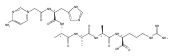
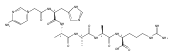


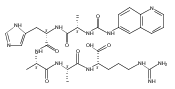

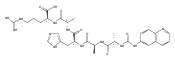
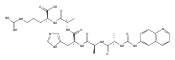

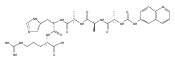

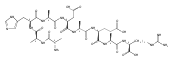
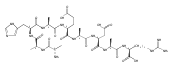
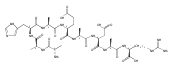







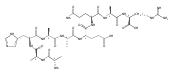
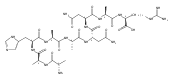

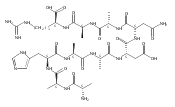
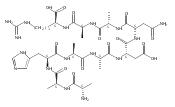
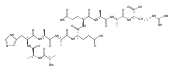


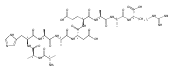

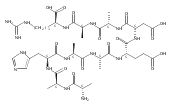
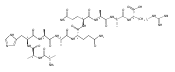
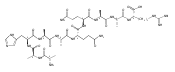

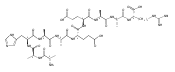
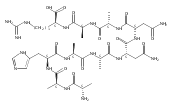
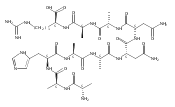
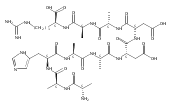

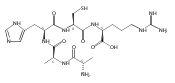
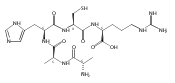
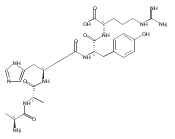
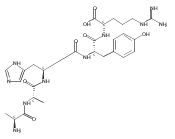
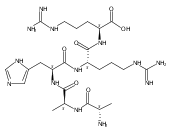
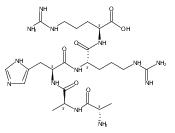


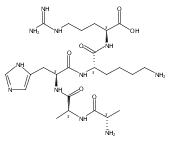
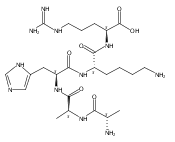
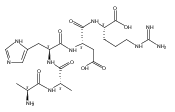
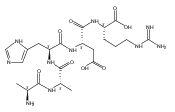
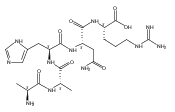
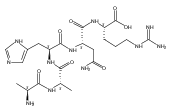
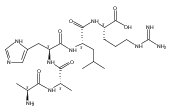
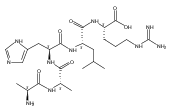
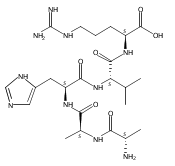
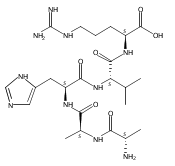



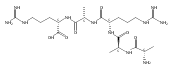
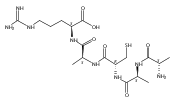

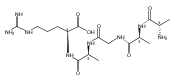

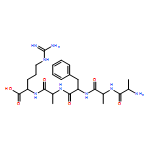
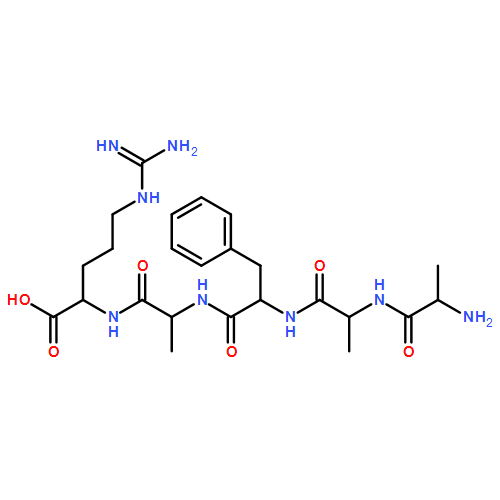

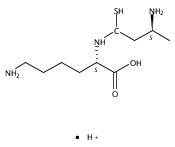


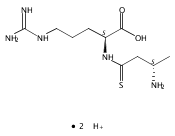


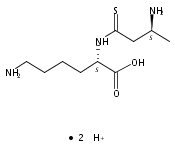
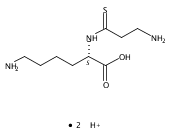












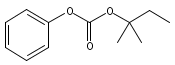
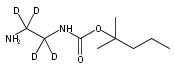
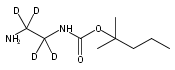

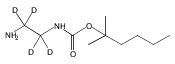
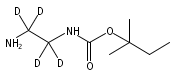

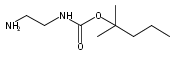
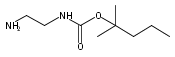
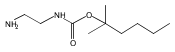

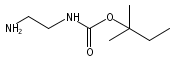
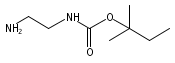

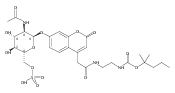
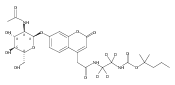
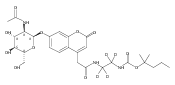

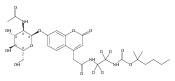

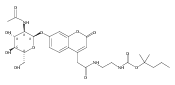
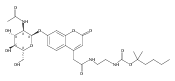





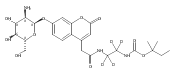
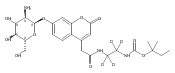



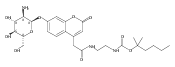
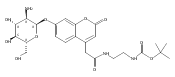

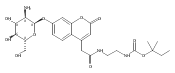
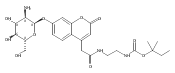
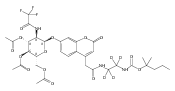

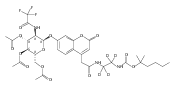

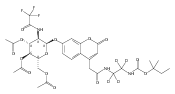
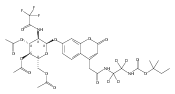
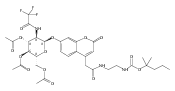



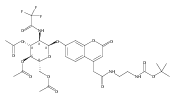







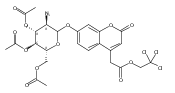
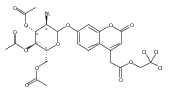

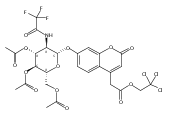
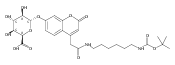
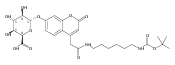
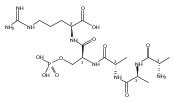
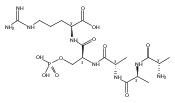
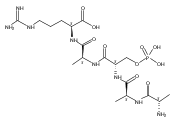
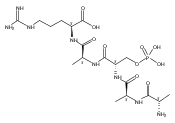
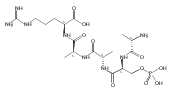
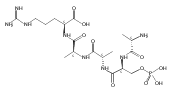
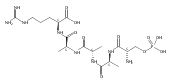
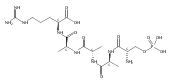
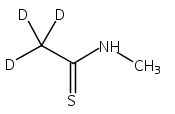
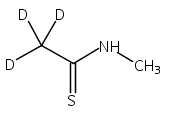



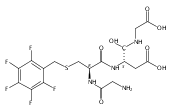
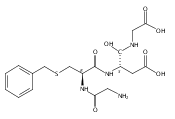






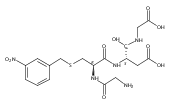
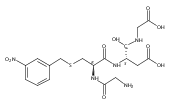
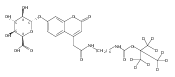
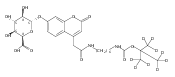
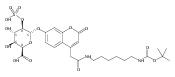

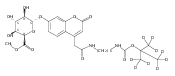
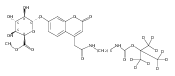

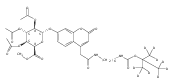
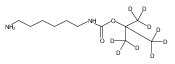
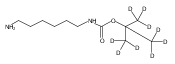
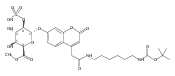
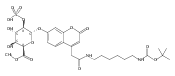

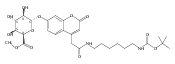
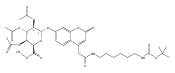



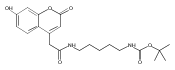
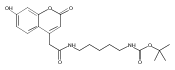



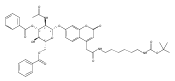
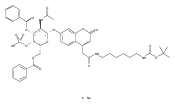



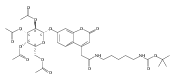
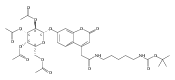

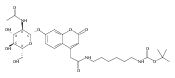

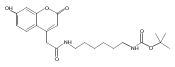

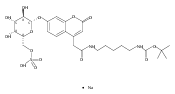

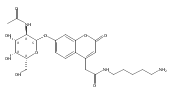
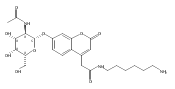

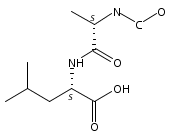
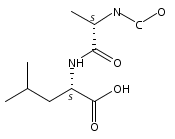
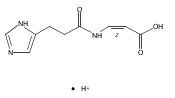
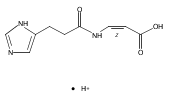
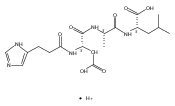
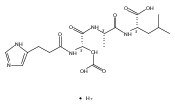
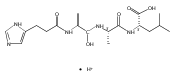
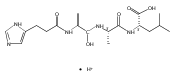
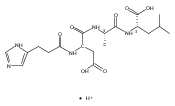



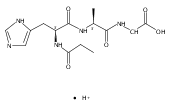
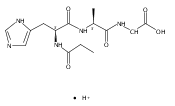
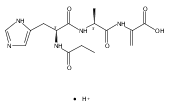

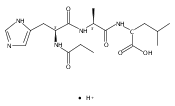
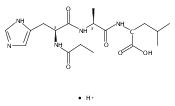
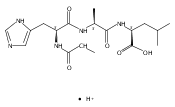










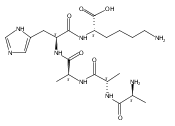

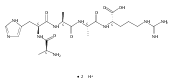






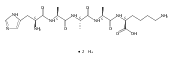
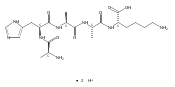

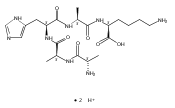
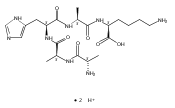
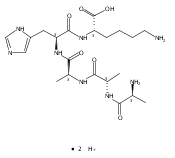


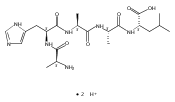
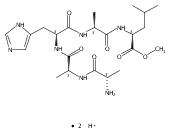







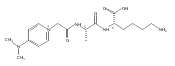
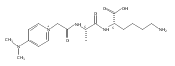
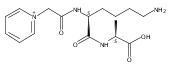


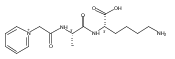
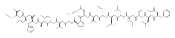
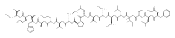


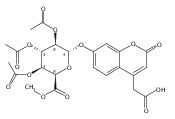



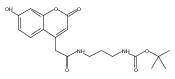
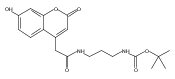
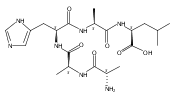


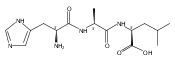
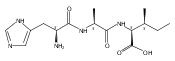
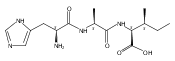

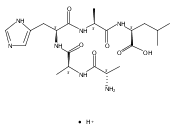

















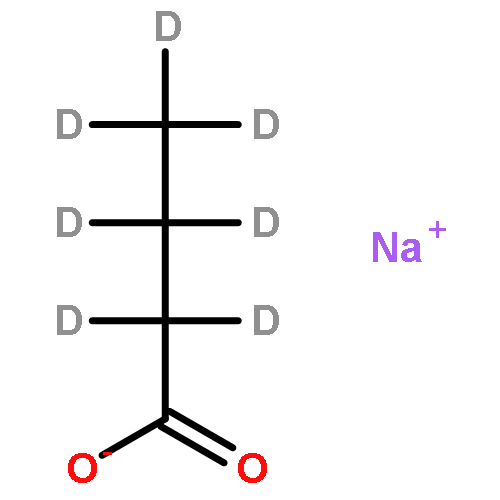













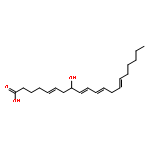
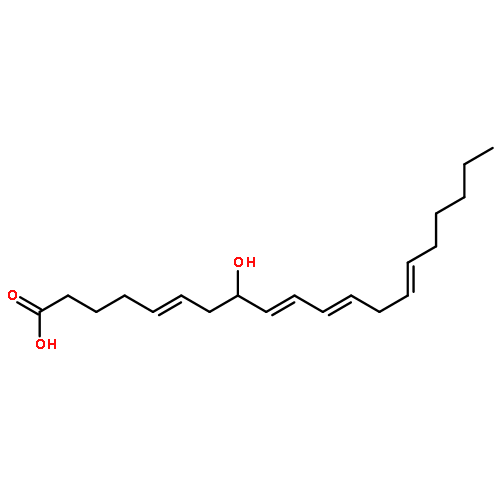
![Pyridinium, 1-[(4-methoxyphenyl)methyl]-, chloride](http://img.cochemist.com/ccimg/98400/98349-72-5.png)
![Pyridinium, 1-[(4-methoxyphenyl)methyl]-, chloride](http://img.cochemist.com/ccimg/98400/98349-72-5_b.png)
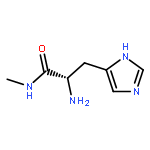
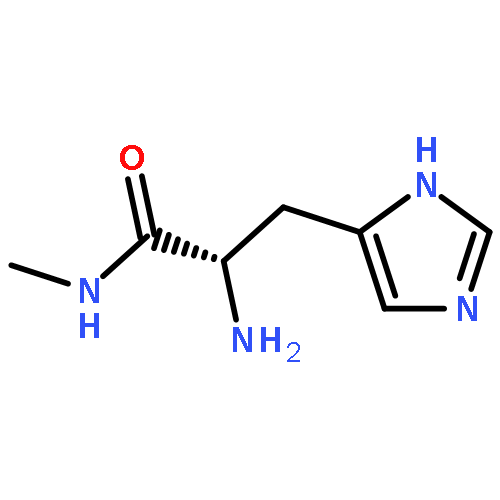









![N-[(E,2S,3R)-3-hydroxy-1-[(2R,5R)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxyoctadec-4-en-2-yl]octanamide](http://img.cochemist.com/ccimg/41700/41613-16-5.png)
![N-[(E,2S,3R)-3-hydroxy-1-[(2R,5R)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxyoctadec-4-en-2-yl]octanamide](http://img.cochemist.com/ccimg/41700/41613-16-5_b.png)




![1-[(4-METHYLPHENYL)METHYL]PYRIDIN-1-IUM;CHLORIDE](http://img.cochemist.com/ccimg/30100/30004-39-8.png)
![1-[(4-METHYLPHENYL)METHYL]PYRIDIN-1-IUM;CHLORIDE](http://img.cochemist.com/ccimg/30100/30004-39-8_b.png)

![Pyridinium, 1-[(4-chlorophenyl)methyl]-, chloride](http://img.cochemist.com/ccimg/19400/19340-03-5.png)
![Pyridinium, 1-[(4-chlorophenyl)methyl]-, chloride](http://img.cochemist.com/ccimg/19400/19340-03-5_b.png)
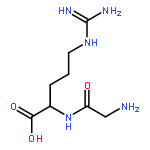
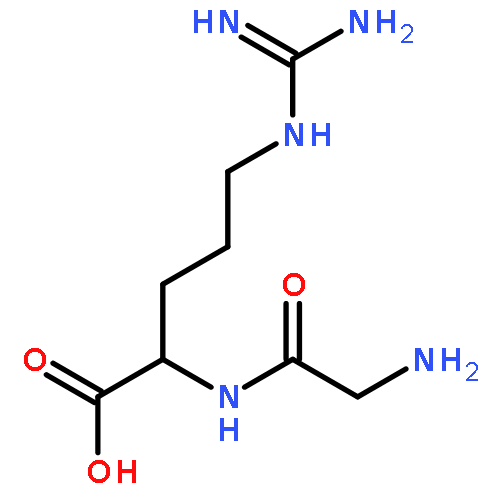


![Ferrate(2-), [7,12-diethenyl-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(4-)-κN21,κN22,κN23,κN24]-, hydrogen (1:2), (SP-4-2)-](/data/chemimg/522800/14875-96-8.png)
![Ferrate(2-), [7,12-diethenyl-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(4-)-κN21,κN22,κN23,κN24]-, hydrogen (1:2), (SP-4-2)-](/data/chemimg/522800/14875-96-8_b.png)
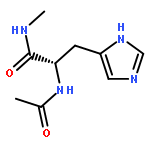
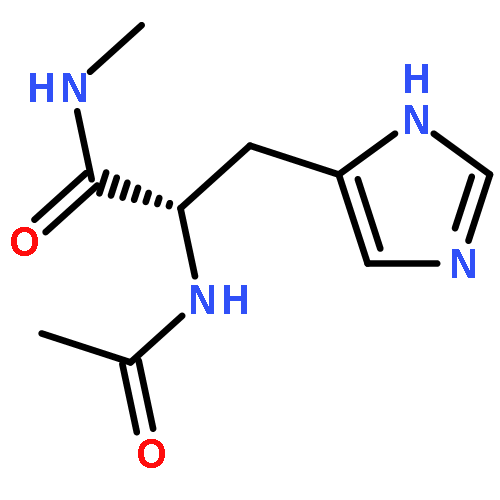
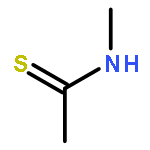
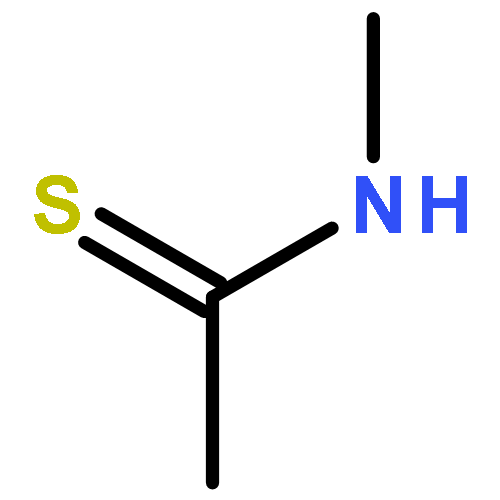
![1-[(4-nitrophenyl)methyl]pyridin-1-ium;chloride](/data/chemimg/50300/4329-72-0.png)
![1-[(4-nitrophenyl)methyl]pyridin-1-ium;chloride](/data/chemimg/50300/4329-72-0_b.png)
![Pentanamide, 2-amino-5-[(aminoiminomethyl)amino]-, (S)-](http://img.cochemist.com/ccimg/2800/2788-83-2.png)
![Pentanamide, 2-amino-5-[(aminoiminomethyl)amino]-, (S)-](http://img.cochemist.com/ccimg/2800/2788-83-2_b.png)





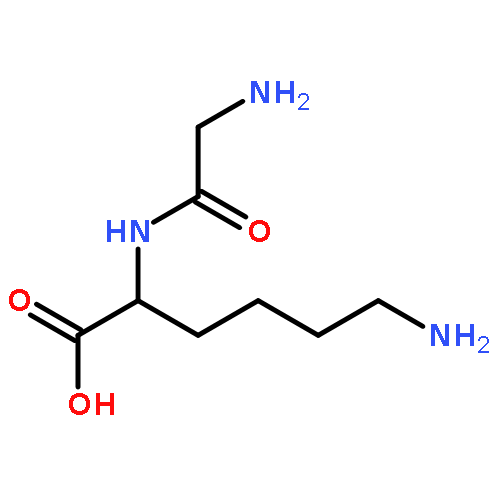






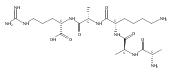



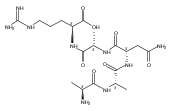
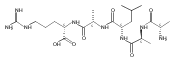
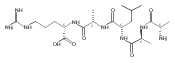
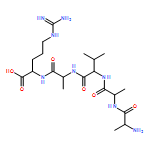
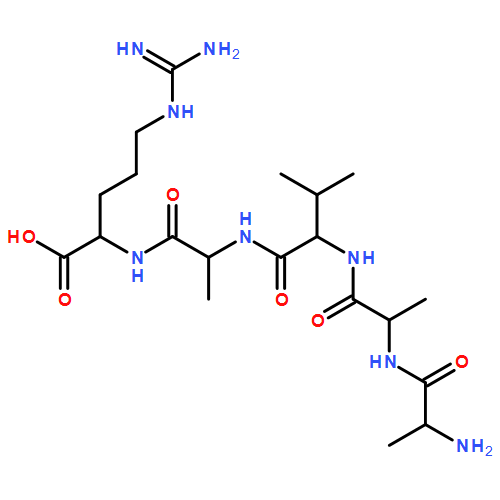

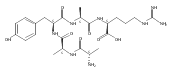
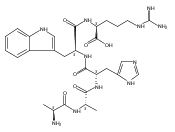
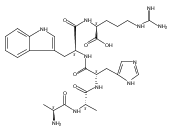

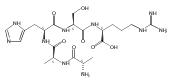
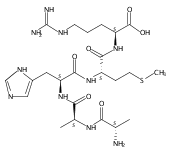
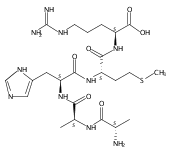



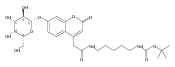
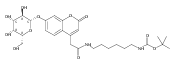
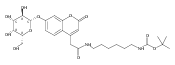
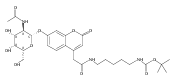
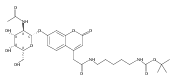



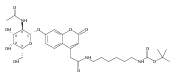

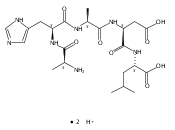

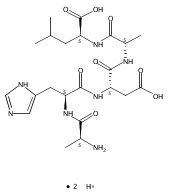

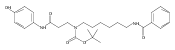


![5-Heptenoic acid,7-[(2R,3S,4S)-tetrahydro-4,6-dihydroxy-2-[(1E,3S)-3-hydroxy-1-octen-1-yl]-2H-pyran-3-yl]-,(5Z)-](http://img.cochemist.com/ccimg/54400/54397-85-2.png)
![5-Heptenoic acid,7-[(2R,3S,4S)-tetrahydro-4,6-dihydroxy-2-[(1E,3S)-3-hydroxy-1-octen-1-yl]-2H-pyran-3-yl]-,(5Z)-](http://img.cochemist.com/ccimg/54400/54397-85-2_b.png)