Co-reporter:Wenwen Gong;Pingxiang Xu;Shanshan Guo;Xiaorong Li;Zengliang Jin;Yuming Zhao;Ming Fan
RSC Advances (2011-Present) 2017 vol. 7(Issue 41) pp:25414-25421
Publication Date(Web):2017/05/10
DOI:10.1039/C7RA03025H
The objective of this study was to compare the pharmacokinetics and metabolism of zaleplon (ZAL) in rats under hypoxic and normoxic condition and the effect of hypoxia on the protein expression and activities of the main metabolic enzyme CYP3A1/2. The LC-MS/MS method was successfully used for the determination of ZAL in rat plasma after an oral administration under normoxic and hypoxic exposure. The expressions of CYP3A1/2 proteins were determined by the Western blotting method. The activity of CYP3A1/2 in rat liver microsomes was determined by the LC-MS/MS method using testosterone as the probe drug. The metabolites of ZAL in rats were identified by LC-MSn by comparison of their ESI-MSn spectra and chromatographic retention times to those of the parent drug between the normoxic and the hypoxic group. The results indicated that the values of AUC and Cmax were significantly higher in the hypoxia exposure for 3 d (H3) group than that in the normoxic group, and the Vd and CL were markedly lower in the H3 group than those in the normoxic group. Hypoxia could markedly inhibit the protein expression and activities of CYP3A1/2, resulting in reduction of the metabolic rate of the drug and enhancement of the systematic exposure. Our data indicated that the expression and activity of CYP3A1/2 mainly affected the drug metabolism and pharmacokinetic characteristics of ZAL under the hypoxic condition.
Co-reporter:Lu Bai;Ying Han;Pingxiang Xu;Binbin Xia;Yuming Zhao;Xiaorong Li
RSC Advances (2011-Present) 2017 vol. 7(Issue 84) pp:53355-53361
Publication Date(Web):2017/11/16
DOI:10.1039/C7RA07369K
The objective of this paper was to investigate the plasma pharmacokinetics and brain distribution kinetics of lapachol in rats. A sensitive and specific high-performance liquid chromatography-tandem mass spectrometry method was developed and validated for quantification of the bioactive naphthoquinone lapachol in rat plasma and brain dialysates after oral administration. The analytes were determined using the negative electrospray ionization mode in multiple reaction monitoring (MRM). The chromatographic separation was on a ZORBAX SB-C18 column coupled with a C18 guard column using a mobile phase composed of acetonitrile–water containing 0.1% formic acid at a flow rate of 0.5 mL min−1. The methods were sensitive with good linearity and no endogenous material interferences. The inter- and intra-day precision and accuracy of lapachol in plasma and the brain were lower than 12%. The methods were successfully applied to the quantification and pharmacokinetic study of lapachol in rats. The results indicated that the disposition profile of lapachol fitted to first order elimination and the two-compartment open model. Lapachol could pass through the blood brain barrier and went through enterohepatic circulation in rats with extending in vivo exposure time after oral administration. In summary, these findings provide an important pharmacological foundation for developing a novel drug and the clinical use of lapachol.
Co-reporter:Shanshan Guo, Shousi Lu, Pingxiang Xu, Yi Ma, Liang Zhao, Yuming Zhao, Wei Gu and Ming Xue
Dalton Transactions 2016 vol. 45(Issue 18) pp:7665-7671
Publication Date(Web):25 Mar 2016
DOI:10.1039/C6DT00395H
Herein, we report a biomimetic method to synthesize needle-like calcium phosphate (CaP) with dimensions of ∼130 nm length and ∼30 nm width using carbon dots (CDs) and sodium carboxymethylcellulose as dual templates. In addition to acting as the template, the CDs enable the CaP/CDs hybrid composites to emit blue fluorescence under UV excitation. Moreover, the prepared CaP/CDs exhibited a negligible cytotoxicity towards HeLa cells. The potential of these CaP/CDs as a fluorescent probe for cell labeling was tested. In addition, it was demonstrated that the CaP/CDs were capable of selective detection of copper ions in drinking water.
Co-reporter:Huanli Xu;Qunying Chen;Hong Wang;Ru Yuan;Pingxiang Xu;Xiaorong Li;Ming Xue;Lu Bai
Journal of Experimental & Clinical Cancer Research 2016 Volume 35( Issue 1) pp:
Publication Date(Web):2016/12/01
DOI:10.1186/s13046-016-0455-3
Lapachol is a natural naphthoquinone compound that possesses extensive biological activities. The aim of this study is to investigate the inhibitory effects of lapachol on rat C6 glioma both in vitro and in vivo, as well as the potential mechanisms.The antitumor effect of lapachol was firstly evaluated in the C6 glioma model in Wistar rats. The effects of lapachol on C6 cell proliferation, apoptosis and DNA damage were detected by 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS)/ phenazinemethosulfate (PMS) assay, hoechst 33358 staining, annexin V-FITC/PI staining, and comet assay. Effects of lapachol on topoisomerase I (TOP I) and topoisomerase II (TOP II) activities were detected by TOP I and TOP II mediated supercoiled pBR322 DNA relaxation assays and molecular docking. TOP I and TOP II expression levels in C6 cells were also determined.High dose lapachol showed significant inhibitory effect on the C6 glioma in Wistar rats (P < 0.05). It was showed that lapachol could inhibit proliferation, induce apoptosis and DNA damage of C6 cells in dose dependent manners. Lapachol could inhibit the activities of both TOP I and II. Lapachol-TOP I showed relatively stronger interaction than that of lapachol-TOP II in molecular docking study. Also, lapachol could inhibit TOP II expression levels, but not TOP I expression levels.These results showed that lapachol could significantly inhibit C6 glioma both in vivo and in vitro, which might be related with inhibiting TOP I and TOP II activities, as well as TOP II expression.
Co-reporter:Su Su, Yanan Wang, Lu Bai, Binbin Xia, Xiaorong Li, Yu Tang, Pingxiang Xu, Ming Xue
Journal of Pharmaceutical and Biomedical Analysis 2015 Volume 104() pp:38-46
Publication Date(Web):10 February 2015
DOI:10.1016/j.jpba.2014.11.010
•A novel LC–ESI–MSn method is used to investigate the in vivo metabolism of isobavachalcone.•The fragmentation pathways of isobavachalcone were elucidated using ESI-MSn in both positive and negative ion mode.•Five phase I metabolites are biotransformed via hydroxylation, reduction, cyclization and oxidative cleavage.•Ten phase II metabolites are mainly identified as the glucuronide conjugates and sulfated conjugate.•These findings are reported for the first time and contributed further understanding of the intermediate processes and metabolic mechanism.Isobavachalcone (IBC) is a prenylated chalcone and belongs to the class of flavonoids, which is an active component isolated from Psoralea corylifolia L. IBC showed a range of significant pharmacological activities, including antibacterial, anticancer, anti-reverse transcriptase and antioxidant actions. In this research, the mass spectral fragmentation pattern of IBC was investigated to predict the in vivo metabolites, and five phase I metabolites and ten phase II metabolites of IBC in rat bile were elucidated and identified after oral administration using novel LC–ESI-MSn and LC–NMR method. The molecular structures of these metabolites were proposed on the basis of the characteristics of their precursor ions, product ions, MS/MS fragment behaviors and chromatographic retention time. The phase I metabolites were mainly biotransformed via the hydroxylation, reduction, cyclization and oxidative cleavage reactions. The phase II metabolites were mainly identified as the glucuronide conjugates and sulfated conjugates. All these findings were reported for the first time and would contribute to a further understanding of the in vivo intermediate processes and metabolic mechanism of isobavachalcone and its analogs.The proposed metabolic pathways of isobavachalcone in rat.
Co-reporter:Jin-Feng Yao, Ning Zhou, Lu Bai, Ping-Xiang Xu, Ke-Liang Liu, Ming Xue
Journal of Chromatography B 2014 Volume 962() pp:94-101
Publication Date(Web):1 July 2014
DOI:10.1016/j.jchromb.2014.05.035
•Five novel LHRH antagonists consisted of 10 amino acids were administrated to rats via cassette dosing.•LC–MS/MS was developed for simultaneous determination of these LHRH antagonists in rat plasma.•Pharmacokinetic profiles of the LHRH antagonists were examined in rats.•The results provide useful methods and abundant metabolic information for the peptides.Long acting luteinizing hormone-releasing hormone (LHRH) antagonists designed to be protease-resistant were a series of novel decapeptides structurally similar to LHRH. In the present work, a high-throughput method based on a LC–MS/MS has been developed for the simultaneous determination of pharmacokinetics of five LHRH antagonists in rat via cassette dosing. The method was performed under selected reaction monitoring (SRM) in positive ion mode. The analytes were extracted from rat plasma by liquid–liquid extraction with acetonitrile. Chromatographic separation of the analytes was successfully achieved on a Hypersil Gold (100 mm × 2.1 mm, 3 μm) using a mobile phase composed of acetonitrile–water (30:70) containing 0.05% (v/v) formic acid. The result showed good linearity and selectivity were obtained for all antagonists. The limits of quantification of the five LHRH antagonists were from 5 to 10 ng/mL. The average extract recoveries in the rat plasma were all over 72%. The intra-day and inter-day precisions (R.S.D. %) were all within 10% and the accuracy was ranged from 92.54 to 109.05%. This method has been successfully applied to the pharmacokinetic studies of the five LHRH antagonists. The results indicated that the plasma drug concentrations versus time curves after intravenous injection of five antagonists via cassette dosing were all fitted to a two-compartment model. The pharmacokinetic parameters of five LHRH antagonists suggested that LY616 could be the more stable candidate drugs and optimized as the candidate drug for further study. Our studies enabled high-throughput rapid screening for pharmacokinetic assessment of new peptide candidates, and provided abundant information on the metabolic properties of these LHRH antagonists.
Co-reporter:Hai-Ting Cheng, Xiao-li Li, Xiao-rong Li, Yu-hang Li, Li-juan Wang, Ming Xue
Food Chemistry 2012 130(4) pp: 1031-1035
Publication Date(Web):
DOI:10.1016/j.foodchem.2011.07.126
Co-reporter:Jin-Feng Yao;Ning Zhou;Yu-Jian Lv;Ruifeng Zhang;Ke-Liang Liu
Amino Acids 2012 Volume 43( Issue 4) pp:1557-1566
Publication Date(Web):2012 October
DOI:10.1007/s00726-012-1231-0
Long-acting luteinizing hormone-releasing hormone (LHRH) antagonists designed to be protease resistant consisted of a series of novel decapeptides structurally similar to LHRH. The aim of this study was to evaluate the in vitro metabolic stability of the LHRH decapeptides using pancreatin and homogenates models and identify the metabolites in rat liver homogenate for the purpose of illustrating the metabolic features of the decapeptides. The major metabolites in rat liver homogenate were identified by LC–ESI-MSn. The half-lives of the 11 LHRH decapeptides were from 44 to 330 min in the pancreatin model. The half-lives of the five decapeptides in rat liver, kidney and lung homogenates were between 8 and 462 min. The most stable decapeptides were the LY616 and LY608 peptides with half-lives of 36 min in liver homogenate. Two major cleavage sites were found by analysing the metabolites of the LY618 peptide in rat liver homogenate, between the Pal3-Ser4 and the Leu7-Ilys8 peptide bonds. The major metabolites were produced via cleavages of peptide bonds at these sites, and further metabolic reactions such as hydroxylation, oxidative dechlorination, alcohol dehydration and isopropyl dealkylation were also observed.
Co-reporter:Yun Liu, Xiaorong Li, Yuhang Li, Lijuan Wang, Ming Xue
Journal of Pharmaceutical and Biomedical Analysis 2010 53(3) pp: 698-704
Publication Date(Web):
DOI:10.1016/j.jpba.2010.03.041
Co-reporter:Ying Liu, Yuyin Kou, Ming Xue, Yanxia Xu, Liu He, Jinxiu Ruan, Keliang Liu
Talanta 2010 Volume 82(Issue 4) pp:1200-1211
Publication Date(Web):15 September 2010
DOI:10.1016/j.talanta.2010.06.032
The structural elucidation of the metabolites of phencynonate and its analogue thiencynonate in rats was performed by liquid chromatography–electrospray ionization tandem mass spectrometry (LC–ESI-MSn) in positive ion mode, by comparing their changes in molecular masses (ΔM), retention times and spectral patterns with those of the parent drug. Phencynonate and thiencynonate were easily biotransformed in vivo by the pathways of N-demethylated, oxidative, hydroxylated and methoxylated to form seventeen metabolites that retained the some features of the two parent molecules. These metabolites included ten phencynonate metabolites (N-demethyphencynonate monoxide, N-demethyhydroxy phencynonate, phencynonate monoxide, hydroxyphencynonate, phencynonate dioxide, methoxyphencynonate, dihydroxyphencynonate, dihydroxyphencynonate, hydroxymethoxy phencynonate, trihydroxyphencynonate) and seven thiencynonate metabolites (N-demethy thiencynonate, N-demethythiencynonate monoxide, N-demethyhydroxythiencynonate, thiencynonate monoxide, hydroxythiencynonate, hydroxythiencynonate monoxide, dihydroxy thiencynonate). The described method has wide applicability to rapidly screen and provide structural information of these metabolites. The identifications of precise structures of these metabolites need to be confirmed by other techniques such as the 1H and 13C NMR.
Co-reporter:Mingming Wang, Haixue Dai, Xiaorong Li, Yuhang Li, Lijuan Wang, Ming Xue
Journal of Chromatography B 2010 Volume 878(13–14) pp:915-924
Publication Date(Web):15 April 2010
DOI:10.1016/j.jchromb.2010.02.014
Tanshinone I and its analogue dihydrotanshinone I are the major active components isolated from Salvia miltiorrhiza Bunge and Salvia Przewalskii Maxim. These compounds have been found to possess significant antibacterial, anti-dermatophytic, antioxidant, anti-inflammatory and anticancer activities. Fifteen phase I metabolites and two phase II metabolites of tanshinone I and dihydrotanshinone I in rat bile were elucidated and identified by a sensitive HPLC–ESI-MSn method. The molecular structures of the metabolites are presented on the basis of the characteristics of their precursor ions, product ions and chromatographic retention times. The results indicate that the phase I metabolites are biotransformed through four main pathways: dehydrogenation, hydroxylation, furan ring cleavage and oxidation metabolism. Phase II metabolites were mainly identified as the sulfated conjugates which showed a characteristic neutral loss of 80 Da. The biotransformed pathways of tanshinone I and dihydrotanshinone I were proposed on the basis of the investigation.
Co-reporter:Ying Liu, Mingming Wang, Ming Xue, Yuhang Li, Xiaorong Li, Jinxiu Ruan, Keliang Liu
Journal of Chromatography B 2008 Volume 873(Issue 1) pp:41-50
Publication Date(Web):15 September 2008
DOI:10.1016/j.jchromb.2008.07.049
The structural elucidation of metabolites of penehyclidine in rats, a novel anti-cholinergic drug, by the method of liquid chromatography–electrospray ionization mass spectrometry, gas chromatography–mass spectrometry with electron impact ion source and stable isotope ion cluster was described. Identification and elucidation of the phase I and phase II metabolites were performed by comparing the daughter ion pairs of stable isotope cluster, changes of the protonated molecular masses, full scan MSn spectra and retention times with those of the parent drug, penehyclidine and penehyclidine deuterium-labeled. Penehyclidine was easily biotransformed by the pathways of oxidative, hydroxylated, methoxylated and phase II conjugated reactions to form several metabolites that retained the some features of the parent molecules. Twelve metabolites (penehyclidine monoxide, hydroxypenehyclidine, penehyclidine dioxide, hydroxypenehyclidine monoxide, dihydroxypenehyclidine, dihydroxypenehyclidine (position isomer), dihydroxypenehyclidine monoxide, trihydroxypenehyclidine, methoxypenehyclidine dioxide, dimethoxypenehyclidine, trihydroxymethoxypenehyclidine and glucuronide conjugated hydroxypenehyclidine) were identified. The results from electrospray ion and electron impact ion data with the stable isotope cluster showed the qualitative differences in the mass spectral patterns, suggesting that these technologies should be used in parallel to ensure comprehensive metabolites detection and characterization. The described method has wide applicability to rapidly screen and provide structural information of metabolites.
Co-reporter:Haixue Dai, Xiaorong Li, Xiaoli Li, Lu Bai, Yuhang Li, Ming Xue
Phytomedicine (15 November 2012) Volume 19(Issue 14) pp:1256-1262
Publication Date(Web):15 November 2012
DOI:10.1016/j.phymed.2012.08.007
Cryptotanshinone, derived from the roots of Salvia miltiorrhiza Bge and Salvia przewalskii Maxim, is the major active component and possesses significant antibacterial, antidermatophytic, antioxidant, anti-inflammatory and anticancer activities. The objective of this study was to investigate the intestinal absorptive characteristics of cryptotanshinone as well as the absorptive behavior influenced by co-administration of the diterpenoid tanshinones and danxingfang using an in vitro everted rat gut sac model. The results showed a good linear correlation between cryptotanshinone of absorption and the incubation time from 10 to 70 min. The concentration dependence showed that a non-linear correlation existed between the cryptotanshinone absorption and the concentration at 100 μg/ml. Coexisting diterpenoid tanshinones and danxingfang could significantly enhance the absorption of cryptotanshinone. Coexisting diterpenoid tanshinones and danxingfang, which influenced cryptotanshinone's absorption, manifested as similar to that of the P-glycoprotein inhibitor. The underlying mechanism of the improvement of oral bioavailability was proposed that coexisting diterpenoid tanshinones and danxingfang could decrease the efflux transport of cryptotanshinone by P-glycoprotein.
Co-reporter:Xiaorong Li, Lijuan Wang, Yuhang Li, Lu Bai, Ming Xue
Regulatory Toxicology and Pharmacology (1 June 2011) Volume 60(Issue 1) pp:106-111
Publication Date(Web):1 June 2011
DOI:10.1016/j.yrtph.2011.02.013
Acute and subacute investigations were carried out to evaluate the safety of scutellarin, an active flavone glycoside that has been used to treating cardiocerebral vascular diseases and cerebral infarction in rodents. For the acute study, scutellarin was administered to mice by gavage at different dose levels. Scutellarin caused dose-dependent general behavior adverse effects, but the LD50 values could not be detected, and the maximum tolerated dose was more than 10 g/kg. In the subacute study, scutellarin was administered orally at doses of 100 and 500 mg/kg daily for 30 days to rats. Body weight, heart rate, blood pressure, biochemical, hematological and urine parameters were determined at the end of the experimental day. Daily oral administration for up to 30 days did not result in death or significant changes in hematology, blood chemistries or urinalysis. However, a 30 day regimen of scutellarin at doses of 100 or 500 mg/kg led to non-dose related decreases in BUN and triglyceride levels. Scutellarin was found to be minimally toxic or non-toxic in rodents. In view of the doses of the components used, the results from acute and subacute toxicity studies suggest that this component has a sufficient margin of safety for therapeutic use.
Co-reporter:Shanshan Guo, Shousi Lu, Pingxiang Xu, Yi Ma, Liang Zhao, Yuming Zhao, Wei Gu and Ming Xue
Dalton Transactions 2016 - vol. 45(Issue 18) pp:NaN7671-7671
Publication Date(Web):2016/03/25
DOI:10.1039/C6DT00395H
Herein, we report a biomimetic method to synthesize needle-like calcium phosphate (CaP) with dimensions of ∼130 nm length and ∼30 nm width using carbon dots (CDs) and sodium carboxymethylcellulose as dual templates. In addition to acting as the template, the CDs enable the CaP/CDs hybrid composites to emit blue fluorescence under UV excitation. Moreover, the prepared CaP/CDs exhibited a negligible cytotoxicity towards HeLa cells. The potential of these CaP/CDs as a fluorescent probe for cell labeling was tested. In addition, it was demonstrated that the CaP/CDs were capable of selective detection of copper ions in drinking water.









![2H-Naphtho[1,2-b]pyran-5,6-dione, 2,2-dimethyl-](http://img.cochemist.com/ccimg/15300/15297-93-5.png)
![2H-Naphtho[1,2-b]pyran-5,6-dione, 2,2-dimethyl-](http://img.cochemist.com/ccimg/15300/15297-93-5_b.png)
![2H-Naphtho[2,3-b]pyran-5,10-dione,2,2-dimethyl-](http://img.cochemist.com/ccimg/15300/15297-92-4.png)
![2H-Naphtho[2,3-b]pyran-5,10-dione,2,2-dimethyl-](http://img.cochemist.com/ccimg/15300/15297-92-4_b.png)
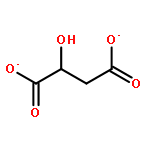
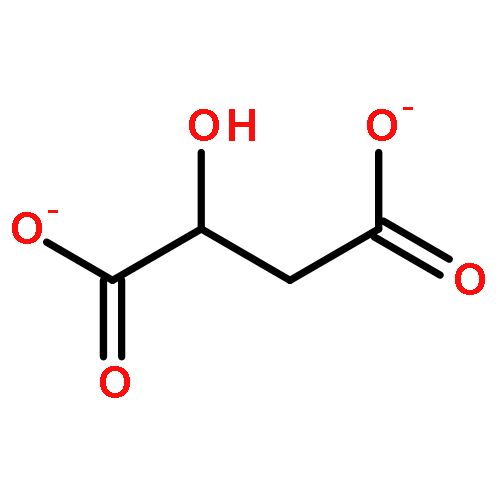








![3-(10,11-Dihydro-5H-dibenzo[b,f]azepin-5-yl)-N-methylpropan-1-amine](http://img.cochemist.com/ccimg/100/50-47-5.png)
![3-(10,11-Dihydro-5H-dibenzo[b,f]azepin-5-yl)-N-methylpropan-1-amine](http://img.cochemist.com/ccimg/100/50-47-5_b.png)


![(e)-1-[2,4-dihydroxy-3-(3-methylbut-2-enyl)phenyl]-3-(4-hydroxyphenyl)prop-2-en-1-one](http://img.cochemist.com/ccimg/20800/20784-50-3.png)
![(e)-1-[2,4-dihydroxy-3-(3-methylbut-2-enyl)phenyl]-3-(4-hydroxyphenyl)prop-2-en-1-one](http://img.cochemist.com/ccimg/20800/20784-50-3_b.png)