Co-reporter:Kayleigh L. Arthur, Matthew A. Turner, James C. Reynolds, and Colin S. Creaser
Analytical Chemistry March 21, 2017 Volume 89(Issue 6) pp:3452-3452
Publication Date(Web):February 23, 2017
DOI:10.1021/acs.analchem.6b04315
Full scan field asymmetric waveform ion mobility spectrometry (FAIMS) combined with liquid chromatography and mass spectrometry (LC-FAIMS-MS) is shown to enhance peak capacity for omics applications. A miniaturized FAIMS device capable of rapid compensation field scanning has been incorporated into an ultrahigh performance liquid chromatography (UHPLC) and time-of-flight mass spectrometry analysis, allowing the acquisition of full scan FAIMS and MS nested data sets within the time scale of a UHPLC peak. Proof of principle for the potential of scanning LC-FAIMS-MS in omics applications is demonstrated for the nontargeted profiling of human urine using a HILIC column. The high level of orthogonality between FAIMS and MS provides additional unique compound identifiers with detection of features based on retention time, FAIMS dispersion field and compensation field (DF and CF), and mass-to-charge (m/z). Extracted FAIMS full scan data can be matched to standards to aid the identification of unknown analytes. The peak capacity for features detected in human urine using LC-FAIMS-MS was increased approximately threefold compared to LC-MS alone due to a combination of the reduction of chemical noise and separation of coeluting isobaric species across the entire analytical space. The use of FAIMS-selected in source collision induced dissociation (FISCID) yields fragmentation of ions, which reduces sample complexity associated with overlapping fragmentation patterns and provides structural information on the selected precursor ions.
Co-reporter:Kayleigh L. Arthur, Matthew A. Turner, Alan D. Brailsford, Andrew T. Kicman, David A. Cowan, James C. Reynolds, and Colin S. Creaser
Analytical Chemistry July 18, 2017 Volume 89(Issue 14) pp:7431-7431
Publication Date(Web):June 14, 2017
DOI:10.1021/acs.analchem.7b00940
The combination of field asymmetric waveform ion mobility spectrometry with liquid chromatography–mass spectrometry (LC-FAIMS-MS) has been developed for the analysis of glucuronide and sulfate metabolites of seven anabolic-androgenic steroids in urine. Separation by FAIMS-MS was investigated in positive ion mode for selected cationic adducts (H+, NH4+, Na+, K+, and Cs+). LC-FAIMS-MS analysis of the doubly sodiated adducts ([M + 2Na – H]+) of isobaric and coeluting steroid metabolites allowed their rapid (8 min) qualitative and quantitative determination in spiked urine using hydrophilic interaction liquid chromatography prior to FAIMS-MS separation, with discrimination >95% achieved between the steroids investigated. A quantitative evaluation of the LC-FAIMS-MS method was performed giving limits of detection in the range 1–6 ng mL–1, limits of quantification in the range 3–20 ng mL–1, with reproducibility (%RSD < 10%; n = 6) and linearity (R2 > 0.99). The LC-FAIMS-MS method demonstrates increases in signal-to-noise ratios for the doubly sodiated steroid metabolites in unspiked urine (>250%) by the reduction of isobaric interferences from the matrix. An alternative or additional tool for identification of the steroid metabolites is based on the observations of different patterns of sodium acetate clusters that are characteristic for each metabolite.
Co-reporter:Caitlyn Da Costa, Matthew Turner, James C. Reynolds, Samuel Whitmarsh, Tom Lynch, and Colin S. Creaser
Analytical Chemistry 2016 Volume 88(Issue 4) pp:2453
Publication Date(Web):January 18, 2016
DOI:10.1021/acs.analchem.5b04595
The analysis of corrosion inhibitors in the presence and absence of an oil matrix is reported using electrospray ionization (ESI) and desorption electrospray ionization (DESI), hyphenated with miniaturized high-field asymmetric waveform ion mobility spectrometry (FAIMS) and mass spectrometry (MS). The target analytes were successfully ionized in solution by ESI and directly from steel surfaces using DESI ambient ionization at levels ≥0.0004% w/w (4 ppm) in oil. Differences in the mass spectral profiles observed for the additive/oil mixture are attributed to differences between the ESI and DESI ionization processes. The use of FAIMS improved selectivity for ESI generated analyte ions through reduction in the chemical noise resulting from the oil matrix. DESI enabled the direct, rapid, native state interrogation of oil samples on steel surfaces without sample pretreatment, and the hyphenation of DESI with the miniaturized FAIMS enhanced the relative analyte responses of the surface-active corrosion inhibitors.
Co-reporter:Aditya Malkar, Emma Wilson, Tim Harrrison, Dominick Shaw and Colin Creaser
Analytical Methods 2016 vol. 8(Issue 27) pp:5407-5413
Publication Date(Web):20 Jun 2016
DOI:10.1039/C6AY00938G
Current clinical tests employed to diagnose asthma are inaccurate and limited by their invasive nature. New metabolite profiling technologies offer an opportunity to improve asthma diagnosis using non-invasive sampling. A rapid analytical method for metabolite profiling of saliva is reported using ultra-high performance liquid chromatography combined with high resolution time-of-flight mass spectrometry (UHPLC-MS). The only sample pre-treatment required was protein precipitation with acetonitrile. The method has been applied to a pilot study of saliva samples obtained by passive drool from well phenotyped patients with asthma and healthy controls. Stepwise data reduction and multivariate statistical analysis was performed on the complex dataset obtained from the UHPLC-MS analysis to identify potential metabolomic biomarkers of asthma in saliva. Ten discriminant features were identified that distinguished between moderate asthma and healthy control samples with an overall recognition ability of 80% during training of the model and 97% for model cross-validation. The reported method demonstrates the potential for a non-invasive approach to the clinical diagnosis of asthma using mass spectrometry-based metabolic profiling of saliva.
Co-reporter:Caitlyn Da Costa, Samuel Whitmarsh, Tom Lynch, Colin S. Creaser
International Journal of Mass Spectrometry 2016 Volume 405() pp:24-31
Publication Date(Web):10 July 2016
DOI:10.1016/j.ijms.2016.05.011
•Direct analysis in real time of lubricant additives is reported.•Desorption of additives is demonstrated from filter paper, glass and steel surfaces.•Thermal fragmentation of quaternary amine corrosion inhibitors yields structurally diagnostic ions.•Quantitative determination of an antioxidant in oil is demonstrated.The application of direct analysis in real time combined with mass spectrometry (DART-MS) to the qualitative analysis of lubricant and oil additives, and the quantitative analysis of a lubricant antioxidant additive is reported. The additives were analysed alone and in the presence of a base oil, from filter paper, glass and steel surfaces, showing the potential of the DART-MS technique for the direct, rapid analysis of lubricant oil additives. The quantitative capabilities of the technique were evaluated for the antioxidant in an oil matrix at concentrations in the range 0.1–8 mg/mL in oil (1–80 μg antioxidant on spot), using a structural analogue of the antioxidant as an internal standard. The linearity (R2 = 0.997), precision (% RSD = 2.6%) and LOD (0.04 mg/mL in oil) of the method demonstrates that DART-MS is capable of the rapid determination of additives in oil without pre-extraction.Figure optionsDownload full-size imageDownload high-quality image (116 K)Download as PowerPoint slide
Co-reporter:Kayleigh L. Arthur;Gary A. Eiceman
Journal of The American Society for Mass Spectrometry 2016 Volume 27( Issue 5) pp:800-809
Publication Date(Web):2016 May
DOI:10.1007/s13361-016-1351-y
Miniaturised field asymmetric waveform ion mobility spectrometry (FAIMS), combined with mass spectrometry (MS), has been applied to the study of self-assembling, noncovalent supramolecular complexes of 3-methylxanthine (3-MX) in the gas phase. 3-MX forms stable tetrameric complexes around an alkali metal (Na+, K+) or ammonium cation, to generate a diverse array of complexes with single and multiple charge states. Complexes of (3-MX)n observed include: singly charged complexes where n = 1–8 and 12 and doubly charged complexes where n = 12–24. The most intense ions are those associated with multiples of tetrameric units, where n = 4, 8, 12, 16, 20, 24. The effect of dispersion field on the ion intensities of the self-assembled complexes indicates some fragmentation of higher order complexes within the FAIMS electrodes (in-FAIMS dissociation), as well as in-source collision induced dissociation within the mass spectrometer. FAIMS-MS enables charge state separation of supramolecular complexes of 3-MX and is shown to be capable of separating species with overlapping mass-to-charge ratios. FAIMS selected transmission also results in an improvement in signal-to-noise ratio for low intensity complexes and enables the visualization of species undetectable without FAIMS.
Co-reporter:Robert W. Smith, Lisa B. Cox, Aswandi Yudin, James C. Reynolds, Mark Powell and Colin S. Creaser
Analytical Methods 2015 vol. 7(Issue 1) pp:34-39
Publication Date(Web):04 Nov 2014
DOI:10.1039/C4AY02026J
The determination of N-methyl pyrrolidine, a potential impurity in the cephalosporin antibiotic cefepime, by direct infusion ESI combined with field asymmetric waveform ion mobility spectrometry-mass spectrometry (ESI-FAIMS-MS) is demonstrated. The addition of a chip-based FAIMS separation prior to detection by time-of-flight mass spectrometry enables selective transmission of NMP in the presence of cefepime without interference from NMP formed by CID in the mass spectrometer interface. The limits of detection and quantification of NMP in cefepime were 0.011% (w/w) and 0.036% (w/w) NMP in cefepime respectively, well below the 0.3% (w/w) threshold concentration for NMP in cefepime. The % relative standard deviation was 3.9% with linearity for standard additions in the range 0.005–0.5 μg ml−1 NMP.
Co-reporter:Neil A. Devenport, Daniel J. Blenkhorn, Daniel J. Weston, James C. Reynolds, and Colin S. Creaser
Analytical Chemistry 2014 Volume 86(Issue 1) pp:357
Publication Date(Web):November 27, 2013
DOI:10.1021/ac403133t
A direct, ambient ionization method has been developed for the determination of creatinine in urine that combines derivatization and thermal desorption with extractive electrospray ionization and ion mobility-mass spectrometry. The volatility of creatinine was enhanced by a rapid on-probe aqueous acylation reaction, using a custom-made thermal desorption probe, allowing thermal desorption and ionization of the monoacylated derivative. The monoacyl creatinine [M + H]+ ion (m/z 156) was subjected to mass-to-charge selection and collision induced dissociation to remove the acyl group, generating the protonated creatinine [M + H]+ product ion at m/z 114 before an ion mobility separation was applied to reduce chemical noise. Stable isotope dilution using creatinine-d3 as internal standard was used for quantitative measurements. The direct on-probe derivatization allows high sample throughput with a typical cycle time of 1 min per sample. The method shows good linearity (R2 = 0.986) and repeatability (%RSD 8–10%) in the range of 0.25–2.0 mg/mL. The creatinine concentrations in diluted urine samples from a healthy individual were determined to contain a mean concentration of 1.44 mg/mL creatinine with a precision (%RSD) of 9.9%. The reactive ambient ionization approach demonstrated here has potential for the determination of involatile analytes in urine and other biofluids.
Co-reporter:Neil A. Devenport, Laura C. Sealey, Faisal H. Alruways, Daniel J. Weston, James C. Reynolds, and Colin S. Creaser
Analytical Chemistry 2013 Volume 85(Issue 13) pp:6224
Publication Date(Web):June 10, 2013
DOI:10.1021/ac401054n
A direct, ambient ionization method has been developed using atmospheric pressure thermal desorption–extractive electrospray–mass spectrometry (AP/TD-EESI-MS) for the detection of the genotoxic impurity (GTI) methyl p-toluenesulfonate (MTS) in a surrogate pharmaceutical matrix. A custom-made thermal desorption probe was used to the desorb and vaporize MTS from the solid state, by rapid heating to 200 °C then cooling to ambient temperature, with a cycle time of 6 min. The detection of MTS using EESI with a sodium acetate doped solvent to generate the [MTS+Na]+ adduct ion provided a significant sensitivity enhancement relative to the [M+H]+ ion generated using a 0.1% formic acid solvent modifier. The MTS detection limit is over an order of magnitude below the long-term daily threshold of toxicological concern (TTC) of 1.5 μg/g and the potential for quantitative analysis has been determined using starch as a surrogate active pharmaceutical ingredient (API).
Co-reporter:Robert W. Smith, Danielle E. Toutoungi, James C. Reynolds, Anthony W.T. Bristow, Andrew Ray, Ashley Sage, Ian D. Wilson, Daniel J. Weston, Billy Boyle, Colin S. Creaser
Journal of Chromatography A 2013 Volume 1278() pp:76-81
Publication Date(Web):22 February 2013
DOI:10.1016/j.chroma.2012.12.065
The incorporation of a chip-based high field asymmetric waveform ion mobility spectrometry (FAIMS) separation in the ultra (high)-performance liquid chromatography–high resolution mass spectrometry (UHPLC–HRMS) determination of the (R/S) ibuprofen 1-β-O-acyl glucuronide metabolite in urine is reported. UHPLC–FAIMS–HRMS reduced matrix chemical noise, improved the limit of quantitation approximately two-fold and increased the linear dynamic range compared to the determination of the metabolite without FAIMS separation. A quantitative evaluation of the prototype UHPLC–FAIMS–HRMS system showed better reproducibility for the drug metabolite (%RSD 2.7%) at biologically relevant concentrations in urine. In-source collision induced dissociation of the FAIMS-selected deprotonated metabolite was used to fragment the ion prior to mass analysis, enhancing selectivity by removing co-eluting species and aiding the qualitative identification of the metabolite by increasing the signal-to-noise ratio of the fragment ions.Highlights► A novel UHPLC–FAIMS–HRMS method for quantification of a drug metabolite in urine is presented. ► Chemical noise is reduced using FAIMS separation following UHPLC. ► FAIMS separation increases the linear dynamic range, reduces the limit of quantification and improves precision.
Co-reporter:Robert W. Smith, James C. Reynolds, Sze-Ling Lee and Colin S. Creaser
Analytical Methods 2013 vol. 5(Issue 16) pp:3799-3802
Publication Date(Web):11 Jul 2013
DOI:10.1039/C3AY40676H
Thermal desorption has been combined with field asymmetric waveform ion mobility spectrometry and mass spectrometry for the rapid, direct analysis of isobaric potentially genotoxic impurities (PGIs) in a surrogate active pharmaceutical ingredient. FAIMS-selected PGIs were detected with limits of quantification <0.2 ppm, below the threshold of toxicological concern, with %RSD <8.4%, at the 1 ppm level.
Co-reporter:Aditya Malkar;Neil A. Devenport;Helen J. Martin;Pareen Patel
Metabolomics 2013 Volume 9( Issue 6) pp:1192-1201
Publication Date(Web):2013 December
DOI:10.1007/s11306-013-0541-x
A method has been developed for metabolite profiling of the salivary metabolome based on protein precipitation and ultra-high performance liquid chromatography coupled with ion mobility-mass spectrometry (UHPLC–IM–MS). The developed method requires 0.5 mL of human saliva, which is easily obtainable by passive drool. Standard protocols have been established for the collection, storage and pre-treatment of saliva. The use of UHPLC allows rapid global metabolic profiling for biomarker discovery with a cycle time of 15 min. Mass spectrometry imparts the ability to analyse a diverse number of species reproducibly over a wide dynamic range, which is essential for profiling of biofluids. The combination of UHPLC with IM–MS provides an added dimension enabling complex metabolic samples to be separated on the basis of retention time, ion mobility and mass-to-charge ratio in a single chromatographic run. The developed method has been applied to targeted metabolite identification and untargeted metabolite profiling of saliva samples collected before and after exercise-induced physiological stress. δ-Valerolactam has been identified as a potential biomarker on the basis of retention time, MS/MS spectrum and ion mobility drift time.
Co-reporter:Victoria E. Wright;Fernando Castro-Gómez
International Journal for Ion Mobility Spectrometry 2013 Volume 16( Issue 1) pp:61-67
Publication Date(Web):2013 March
DOI:10.1007/s12127-013-0122-8
Collision cross sections (CCS) have been measured for three salen ligands, and their complexes with copper and zinc using travelling-wave ion mobility-mass spectrometry (TWIMS) and drift tube ion mobility-mass spectrometry (DTIMS), allowing a comparative size evaluation of the ligands and complexes. CCS measurements using TWIMS were determined using peptide and TAAH calibration standards. TWIMS measurements gave significantly larger CCS than DTIMS in helium, by 9 % for TAAH standards and 3 % for peptide standards, indicating that the choice of calibration standards is important in ensuring the accuracy of TWIMS-derived CCS measurements. Repeatability data for TWIMS was obtained for inter- and intra-day studies with mean RSDs of 1.1 % and 0.7 %, respectively. The CCS data obtained from IM-MS measurements are compared to CCS values obtained via the projection approximation, the exact hard spheres method and the trajectory method from X-ray coordinates and modelled structures using density functional theory (DFT) based methods.
Co-reporter:Lauren J. Brown, Robert W. Smith, Danielle E. Toutoungi, James C. Reynolds, Anthony W. T. Bristow, Andrew Ray, Ashley Sage, Ian D. Wilson, Daniel J. Weston, Billy Boyle, and Colin S. Creaser
Analytical Chemistry 2012 Volume 84(Issue 9) pp:4095
Publication Date(Web):March 28, 2012
DOI:10.1021/ac300212r
Miniaturized ultra high field asymmetric waveform ion mobility spectrometry (FAIMS) is used for the selective transmission of differential mobility-selected ions prior to in-source collision-induced dissociation (CID) and time-of-flight mass spectrometry (TOFMS) analysis. The FAIMS-in-source collision induced dissociation-TOFMS (FISCID-MS) method requires only minor modification of the ion source region of the mass spectrometer and is shown to significantly enhance analyte detection in complex mixtures. Improved mass measurement accuracy and simplified product ion mass spectra were observed following FAIMS preselection and subsequent in-source CID of ions derived from pharmaceutical excipients, sufficiently close in m/z (17.7 ppm mass difference) that they could not be resolved by TOFMS alone. The FISCID-MS approach is also demonstrated for the qualitative and quantitative analysis of mixtures of peptides with FAIMS used to filter out unrelated precursor ions thereby simplifying the resulting product ion mass spectra. Liquid chromatography combined with FISCID-MS was applied to the analysis of coeluting model peptides and tryptic peptides derived from human plasma proteins, allowing precursor ion selection and CID to yield product ion data suitable for peptide identification via database searching. The potential of FISCID-MS for the quantitative determination of a model peptide spiked into human plasma in the range of 0.45–9.0 μg/mL is demonstrated, showing good reproducibility (%RSD < 14.6%) and linearity (R2 > 0.99).
Co-reporter:Neil A. Devenport, James C. Reynolds, Daniel J. Weston, Ian D. Wilson and Colin S. Creaser
Analyst 2012 vol. 137(Issue 15) pp:3510-3513
Publication Date(Web):22 Jun 2012
DOI:10.1039/C2AN35495K
The direct extraction of urinary analytes deposited on reversed-phase thin-layer chromatography (RP-TLC) plates is demonstrated using a solvent gradient extraction procedure without prior chromatographic development. The surface sample probe TLC-MS interface used for the gradient extraction is compared to direct loop injection into the electrospray ion source for biofluid profiling. The gradient elution is shown to enhance ion intensities, as urinary salts are eluted in aqueous formic acid in the early part of the gradient reducing ion suppression. The retention of urinary components on the C18 RP-TLC plate was confirmed by monitoring analyte responses with, and without, an aqueous wash phase prior to the solvent gradient extraction. The use of gradient elution allows fractionation of the complex biological matrix as a result of differential retention of urine components on the undeveloped RP-TLC plate. The direct gradient analysis of TLC plates has also been combined with ion mobility-mass spectrometry to further resolve the complex urinary profile and identify co-eluting compounds.
Co-reporter:Emma L. Harry, Anthony W. T. Bristow, Ian D. Wilson and Colin S. Creaser
Analyst 2011 vol. 136(Issue 8) pp:1728-1732
Publication Date(Web):25 Feb 2011
DOI:10.1039/C0AN00700E
The potential of ion mobility (IM) spectrometry in combination with mass spectrometry (MS) for real-time reaction monitoring is reported. The combined IM-MS approach using electrospray ionization affords gas-phase analyte characterization based on both mass-to-charge (m/z) ratio and gas-phase ion mobility (drift time). The use of IM-MS analysis is demonstrated for the monitoring of the reaction products formed when 7-fluoro-6-hydroxy-2-methylindole is deprotonated by aqueous sodium hydroxide. Real-time reaction monitoring was carried out over a period of several hours, with the reaction mixture sampled and analysed at intervals of several minutes. Product ion relative intensity is enhanced selectively in the ion mobility-selected mass spectrum, compared to mass spectrometry alone. The combined IM-MS approach has potential as a rapid and selective technique to aid pharmaceutical process control and for the elucidation of reaction mechanism.
Co-reporter:Gushinder Kaur-Atwal, James C. Reynolds, Christopher Mussell, Elodie Champarnaud, Tom W. Knapman, Alison E. Ashcroft, Gavin O'Connor, Steven D. R. Christie and Colin S. Creaser
Analyst 2011 vol. 136(Issue 19) pp:3911-3916
Publication Date(Web):12 Aug 2011
DOI:10.1039/C1AN15450H
UPLC-ion mobility spectrometry separations combined with mass spectrometry (UPLC-IM-MS) and tandem mass spectrometry (UPLC-IM-MS/MS) have been investigated for the simultaneous determination of testosterone and epitestosterone glucuronides in urine. The glucuronide epimers of testosterone and epitestosterone were separated by ion mobility spectrometry prior to mass analysis on the basis of differences in their collision cross sections, which have been measured in nitrogen. Combining ion mobility separation with UPLC/MS enhances the analysis of these low-abundance steroids in urine by selective interrogation of specific retention time, mass-to-charge and mobility regions. Detection limits for the UPLC-IM-MS/MS analysis of TG and ETG were 9.9 ng mL−1 and 98 ng mL−1 respectively, equivalent to 0.7 ng mL−1 and 7.4 ng mL−1 in urine, with linear dynamic ranges corresponding to 0.7–108 ng mL−1 and 7.4–147 ng mL−1 in urine. Repeatability (%RSD) for urine extracts was 0.64% and 2.31% for TG and ETG respectively.
Co-reporter:Neil A. Devenport, James C. Reynolds, Ved Parkash, Jason Cook, Daniel J. Weston, Colin S. Creaser
Journal of Chromatography B 2011 Volume 879(Issue 32) pp:3797-3801
Publication Date(Web):15 December 2011
DOI:10.1016/j.jchromb.2011.10.016
The elastin degradation products, desmosine (DES) and isodesmosine (IDES) are highly stable, cross-linking amino-acids that are unique to mature elastin. The excretion of DES/IDES in urine, in the free form and with associated peptide fragments, provides an indicator of lung damage in chronic obstructive pulmonary disease (COPD). A quantitative ion mobility-mass spectrometry (IM-MS) method has been developed for the analysis of free DES/IDES in urine with deuterated IDES as an internal standard. Resolution of DES/IDES isomers was achieved in less than five minutes using ultra performance liquid chromatography (UPLC) combined with ion pairing. The optimized UPLC–IM-MS method provided a linear dynamic range of 10–300 ng/mL and a limit of quantitation of 0.028 ng/mL for IDES and 0.03 ng/mL for DES (0.55 ng and 0.61 ng on column respectively). The method reproducibility (%RSD) was <4% for DES and IDES. The UPLC–IM-MS method was applied to the analysis of urine samples obtained from healthy volunteers and COPD patients. The DES/IDES concentrations in healthy and COPD urine showed an increase in DES (79%) and IDES (74%) in the COPD samples, relative to healthy controls. The incorporation of an IM separation prior to m/z measurement by MS was shown to reduce non-target ion responses from the bio-fluid matrix.Highlights► We have analyzed free DES/IDES levels in urine (Healthy vs. COPD). ► Targeting Free DES/IDES provides a significant reduction in sample preparation. ► Resolution of the free DES/IDES isomers is achieved in 6 min. ► Free DES/IDES levels have been quantified using ion mobility-mass spectrometry. ► Free DES/IDES levels in urine are significantly elevated in COPD patients.
Co-reporter:L. J. Brown, D. E. Toutoungi, N. A. Devenport, J. C. Reynolds, G. Kaur-Atwal, P. Boyle, and C. S. Creaser
Analytical Chemistry 2010 Volume 82(Issue 23) pp:9827
Publication Date(Web):November 4, 2010
DOI:10.1021/ac102125u
Miniaturized ultra high field asymmetric waveform ion mobility spectrometry (ultra-FAIMS) combined with mass spectrometry (MS) has been applied to the analysis of standard and tryptic peptides, derived from α-1-acid glycoprotein, using electrospray and nanoelectrospray ion sources. Singly and multiply charged peptide ions were separated in the gas phase using ultra-FAIMS and detected by ion trap and time-of-flight MS. The small compensation voltage (CV) window for the transmission of singly charged ions demonstrates the ability of ultra-FAIMS-MS to generate pseudo-peptide mass fingerprints that may be used to simplify spectra and identify proteins by database searching. Multiply charged ions required a higher CV for transmission, and ions with different amino acid sequences may be separated on the basis of their differential ion mobility. A partial separation of conformers was also observed for the doubly charged ion of bradykinin. Selection on the basis of charge state and differential mobility prior to tandem mass spectrometry facilitates peptide and protein identification by allowing precursor ions to be identified with greater selectivity, thus reducing spectral complexity and enhancing MS detection.
Co-reporter:Mark D. Howdle, Christine Eckers, Alice M.-F. Laures, Colin S. Creaser
International Journal of Mass Spectrometry 2010 Volume 298(1–3) pp:72-77
Publication Date(Web):1 December 2010
DOI:10.1016/j.ijms.2009.08.007
The effect of drift gas and binary gas mixtures on the separation of active pharmaceutical ingredients and related compounds has been investigated for the first time using tri-wave ion mobility–mass spectrometry. The drift times for [M+H]+ ions were measured in argon, carbon dioxide, nitrogen, helium and binary mixtures of these gases as a function of pressure and drift gas composition. At the same drift cell pressure the drift times of the protonated ions decreased in order; carbon dioxide > argon > nitrogen > helium. The drift times of compounds in the different drift gases were dependent on the polarizability of the drift gas, the size of the drift gas and the reduced mass of the analyte ion/drift gas pair. Resolution of the active pharmaceutical ingredients Lamotrigine and Rosiglitazone increased with drift cell pressure for argon and nitrogen, but showed no significant increase in carbon dioxide. Enhanced selectivity is demonstrated for the separation of pharmaceutical components using binary drift gas mixtures in tri-wave IM–MS.The effect of drift gas and binary gas mixtures on the separation of active pharmaceutical ingredients and related compounds has been investigated using ion mobility–mass spectrometry.
Co-reporter:Mark D. Howdle;Christine Eckers
Journal of The American Society for Mass Spectrometry 2009 Volume 20( Issue 1) pp:1-9
Publication Date(Web):2009 January
DOI:10.1016/j.jasms.2008.10.002
Gas-phase ion mobility studies of mixtures containing polyethylene glycols (PEG) and an active pharmaceutical ingredient (API), lamivudine, have been carried out using electrospray ionization-ion mobility spectrometry-quadrupole-time-of-flight mass spectrometry (ESI-IMS-Q-TOF). In addition to protonated and cationized PEG oligomers, a series of high molecular weight ions were observed and identified as noncovalent complexes formed between lamivudine and PEG oligomers. The noncovalent complex ions were dissociated using collision induced dissociation (CID) after separation in the ion mobility drift tube to recover the protonated lamivudine free from interfering matrix ions and with a drift time associated with the precursor complex. The potential of PEG excipients to act as “shift reagents,” which enhance selectivity by moving the mass/mobility locus to an area of the spectrum away from interferences, is demonstrated for the analysis of lamivudine in a Combivir formulation containing PEG and lamivudine.
Co-reporter:Gushinder Kaur-Atwal;Gavin O’Connor
International Journal for Ion Mobility Spectrometry 2009 Volume 12( Issue 1) pp:1-14
Publication Date(Web):2009 March
DOI:10.1007/s12127-009-0021-1
Ion mobility spectrometry (IMS), using stand-alone instrumentation and hyphenated with mass spectrometry (IM-MS), has recently undergone significant expansion in the numbers of users and applications, particularly in sectors outside its established user base; predominantly military and security applications. Although several IMS reference standards have been proposed, there are no currently universally recognised reference standards for the calibration and evaluation of mobility spectrometers. This review describes current practices and the literature on chemical standards for validating IMS systems in positive and negative ion modes. The key qualities and requirements an ‘ideal’ reference standard must possess are defined, together with the instrumental and environmental factors such as temperature, electric field, humidity and drift gas composition that may need to be considered. Important challenges that have yet to be resolved are also identified and proposals for future development presented.
Co-reporter:Emma L. Harry, Daniel J. Weston, Anthony W.T. Bristow, Ian D. Wilson, Colin S. Creaser
Journal of Chromatography B 2008 Volume 871(Issue 2) pp:357-361
Publication Date(Web):15 August 2008
DOI:10.1016/j.jchromb.2008.04.043
The potential of drift tube ion mobility (IM) spectrometry in combination with high performance liquid chromatography (LC) and mass spectrometry (MS) for the metabonomic analysis of rat urine is reported. The combined LC–IM–MS approach using quadrupole/time-of-flight mass spectrometry with electrospray ionisation, uses gas-phase analyte characterisation based on both mass-to-charge (m/z) ratio and relative gas-phase mobility (drift time) following LC separation. The technique allowed the acquisition of nested data sets, with mass spectra acquired at regular intervals (65 μs) during each IMS separation (∼13 ms) and several IMS spectra acquired during the elution of a single LC peak, without increasing the overall analysis time compared to LC–MS. Preliminary results indicate that spectral quality is improved when using LC–IM–MS, compared to direct injection IM–MS, for which significant ion suppression effects were observed in the electrospray ion source. The use of reversed-phase LC employing fast gradient elution reduced sample preparation to a minimum, whilst maintaining the potential for high throughput analysis. Data mining allowed information on specific analytes to be extracted from the complex metabonomic data set. LC–IM–MS based approaches may have a useful role in metabonomic analyses by introducing an additional discriminatory dimension of ion mobility (drift time).
Co-reporter:Gushinder Kaur-Atwal, Daniel J. Weston, Philip S. Green, Susan Crosland, Philip L.R. Bonner, Colin S. Creaser
Journal of Chromatography B 2007 Volume 857(Issue 2) pp:240-245
Publication Date(Web):1 October 2007
DOI:10.1016/j.jchromb.2007.07.025
Capillary column immobilised metal affinity chromatography (IMAC) has been combined on-line with electrospray ionisation/quadrupole time-of-flight mass spectrometry for the fractionation of histidine-containing peptides. IMAC beads (Poros 20MC, 20 μm) containing imidodiacetate chelating groups on a cross-linked poly(styrene-divinylbenzene) support were packed into a fused silica column (250 μm i.d.), which was interfaced to the electrospray ion source of the spectrometer. A Cu(II) activated column was used to isolate histidine-containing peptides from tryptic and other peptide mixtures with an average breakthrough of 9.1%, to reduce the complexity of the mass spectral analysis. The analysis cycle time was reduced to less than 15 min, at an optimum flow rate of 7.5 μL/min, without sacrificing peptide selectivity. Direct coupling of capillary IMAC with MS allows on-line separation, using MS compatible loading and elution buffers, and detection in a high-throughput fashion when compared to off-line strategies.
Co-reporter:Robert W. Smith, James C. Reynolds, Sze-Ling Lee and Colin S. Creaser
Analytical Methods (2009-Present) 2013 - vol. 5(Issue 16) pp:NaN3802-3802
Publication Date(Web):2013/07/11
DOI:10.1039/C3AY40676H
Thermal desorption has been combined with field asymmetric waveform ion mobility spectrometry and mass spectrometry for the rapid, direct analysis of isobaric potentially genotoxic impurities (PGIs) in a surrogate active pharmaceutical ingredient. FAIMS-selected PGIs were detected with limits of quantification <0.2 ppm, below the threshold of toxicological concern, with %RSD <8.4%, at the 1 ppm level.

![Pyridinium,2-[(4S)-4-amino-4-carboxybutyl]-1-[(5S)-5-amino-5-carboxypentyl]-3,5-bis[(3S)-3-amino-3-carboxypropyl]-](http://img.cochemist.com/ccimg/1000/991-01-5.png)
![Pyridinium,2-[(4S)-4-amino-4-carboxybutyl]-1-[(5S)-5-amino-5-carboxypentyl]-3,5-bis[(3S)-3-amino-3-carboxypropyl]-](http://img.cochemist.com/ccimg/1000/991-01-5_b.png)
![3'-azido-3'-deoxythymidine - 4-amino-1-[(2R,5S)-2-(hydroxymethyl)-1,3-oxathiolan-5-yl]pyrimidin-2(1H)-one (1:1)](http://img.cochemist.com/ccimg/165500/165456-81-5.png)
![3'-azido-3'-deoxythymidine - 4-amino-1-[(2R,5S)-2-(hydroxymethyl)-1,3-oxathiolan-5-yl]pyrimidin-2(1H)-one (1:1)](http://img.cochemist.com/ccimg/165500/165456-81-5_b.png)







![Phenol, 2-[(dimethylamino)methyl]-3-methoxy-](http://img.cochemist.com/ccimg/172000/171907-76-9.png)
![Phenol, 2-[(dimethylamino)methyl]-3-methoxy-](http://img.cochemist.com/ccimg/172000/171907-76-9_b.png)

![Pyridinium,4-[(4S)-4-amino-4-carboxybutyl]-1-[(5S)-5-amino-5-carboxypentyl]-3,5-bis[(3S)-3-amino-3-carboxypropyl]-](http://img.cochemist.com/ccimg/11100/11003-57-9.png)
![Pyridinium,4-[(4S)-4-amino-4-carboxybutyl]-1-[(5S)-5-amino-5-carboxypentyl]-3,5-bis[(3S)-3-amino-3-carboxypropyl]-](http://img.cochemist.com/ccimg/11100/11003-57-9_b.png)



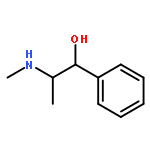
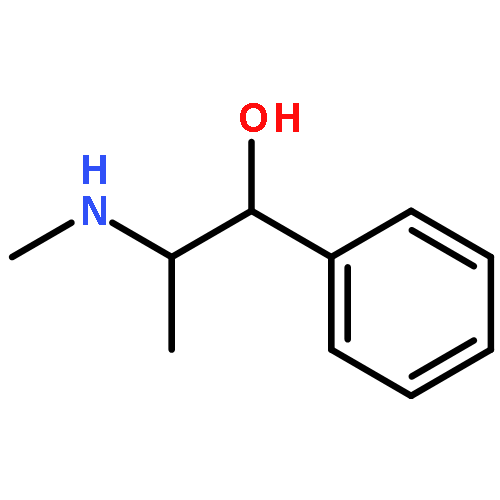
![b-D-Glucopyranuronic acid, 1-[a-methyl-4-(2-methylpropyl)benzeneacetate]](http://img.cochemist.com/ccimg/115100/115075-59-7.png)
![b-D-Glucopyranuronic acid, 1-[a-methyl-4-(2-methylpropyl)benzeneacetate]](http://img.cochemist.com/ccimg/115100/115075-59-7_b.png)
![Piperidine,3-[(1,3-benzodioxol-5-yloxy)methyl]-4-phenyl-, (3S,4R)-](http://img.cochemist.com/ccimg/324100/324024-00-2.png)
![Piperidine,3-[(1,3-benzodioxol-5-yloxy)methyl]-4-phenyl-, (3S,4R)-](http://img.cochemist.com/ccimg/324100/324024-00-2_b.png)