Co-reporter:Ferdinand Ndikuryayo, Behrooz Moosavi, Wen-Chao Yang, and Guang-Fu Yang
Journal of Agricultural and Food Chemistry October 4, 2017 Volume 65(Issue 39) pp:8523-8523
Publication Date(Web):September 14, 2017
DOI:10.1021/acs.jafc.7b03851
The development of new herbicides is receiving considerable attention to control weed biotypes resistant to current herbicides. Consequently, new enzymes are always desired as targets for herbicide discovery. 4-Hydroxyphenylpyruvate dioxygenase (HPPD, EC 1.13.11.27) is an enzyme engaged in photosynthetic activity and catalyzes the transformation of 4-hydroxyphenylpyruvic acid (HPPA) into homogentisic acid (HGA). HPPD inhibitors constitute a promising area of discovery and development of innovative herbicides with some advantages, including excellent crop selectivity, low application rates, and broad-spectrum weed control. HPPD inhibitors have been investigated for agrochemical interests, and some of them have already been commercialized as herbicides. In this review, we mainly focus on the chemical biology of HPPD, discovery of new potential inhibitors, and strategies for engineering transgenic crops resistant to current HPPD-inhibiting herbicides. The conclusion raises some relevant gaps for future research directions.Keywords: 4-hydroxyphenylpyruvate dioxygenase; agrochemical; chemical biology; herbicide; inhibitor;
Co-reporter:Ge-Fei Hao, Yang Zuo, Sheng-Gang Yang, Qian Chen, Yue Zhang, Chun-Yan Yin, Cong-Wei Niu, Zhen Xi, and Guang-Fu Yang
Journal of Agricultural and Food Chemistry July 19, 2017 Volume 65(Issue 28) pp:5581-5581
Publication Date(Web):June 27, 2017
DOI:10.1021/acs.jafc.7b01557
Tuning the binding selectivity through appropriate ways is a primary goal in the design and optimization of a lead toward agrochemical discovery. However, how to achieve rational design of selectivity is still a big challenge. Herein, we developed a novel computational fragment generation and coupling (CFGC) strategy that led to a series of highly potent and bioselective inhibitors targeting protoporphyrinogen IX oxidase. This enzyme plays a vital role in heme and chlorophyll biosynthesis, which has been proven to be associated with many drugs and agrochemicals. However, existing agrochemicals are nonbioselective, resulting in a great threat to nontargeted organisms. To the best of our knowledge, this is the first bioselective inhibitor targeting the tetrapyrrole biosynthesis pathway. In addition, the candidate showed excellent in vivo bioactivity and much better safety toward humans.Keywords: bioselectivity; enzyme inhibitors; fragment; herbicides; PPO;
Co-reporter:Li Xiong;Hua Li;Li-Na Jiang;Jing-Ming Ge;Wen-Chao Yang;Xiao Lei Zhu
Journal of Agricultural and Food Chemistry February 8, 2017 Volume 65(Issue 5) pp:1021-1029
Publication Date(Web):January 22, 2017
DOI:10.1021/acs.jafc.6b05134
A series of diphenyl ether-containing pyrazole-carboxamide derivatives was designed and synthesized as new succinate ubiquinone oxidoreductase (SQR) inhibitors. This highly potent molecular scaffold was developed from a moderately activie hit 3, obtained from our previous pharmacophore-linked fragment virtual screening (PFVS) method. The results of greenhouse tests indicated that some analogues showed good SQR inhibitory activity, with promising fungicidal activity against Rhizoctonia solani and Sphaerotheca fuliginea at a dosage of 200 mg/L. Most surprisingly, compound 62 showed the highest SQR inhibitory activity with a Ki value of 0.081 μM, about 4-fold more potent than penthiopyrad (Ki = 0.307 μM). In addition, compounds 43 and 62 displayed excellent fungicidal activity even at a dosage as low as 6.25 mg/L, which was superior to thifluzamide. Moreover, compound 62 exhibited excellent protection effect against R. solani and provided about 81.2% protective control efficancy after 21 days with two sprayings. The present work indicated that these two compounds could be used as potential agricultural fungicides targeting SQR.Keywords: complex II; diphenyl ether; molecular docking; structure−activity relationship; succinate ubiquinone oxidoreductase;
Co-reporter:Rui Zhang;Qiong-You Wu;Jun Tao;Jin-Huan Pan
New Journal of Chemistry (1998-Present) 2017 vol. 41(Issue 1) pp:204-211
Publication Date(Web):2016/12/19
DOI:10.1039/C6NJ02454H
The respiratory chain succinate–ubiquinone oxidoreductase (SQR or complex II) is a promising target for fungicide discovery. As a continuation of our research work on the development of new fungicides, a series of bitriazolyl compounds were designed and synthesized in excellent yields by an ionic liquid promoted 1,3-dipolar Huisgen cycloaddition reaction of azides and akynes. These newly synthesized compounds were characterized by 1H NMR, 13C NMR and HR-MS spectroscopy. The in vitro assay indicated that several compounds displayed good inhibitory effects against porcine succinate–cyctochrome reductase (SCR) with IC50 values ranging from 2.89 to 61.19 μM. Compound 1b with an IC50 value of 2.89 μM, comparable to the commercial control penthiopyrad, was identified as the most promising inhibitor. Further evaluation of the representative compounds against respective SQR and cyt bc1 indicated that their inhibitory potency against SQR was much higher than that against cyt bc1, suggesting that SQR might be a potential target of these inhibitors. Furthermore, molecular docking studies suggested that strong hydrogen bonding and π–π stacking interactions might be responsible for a higher SQR inhibitory effect of compound 1b as compared to that of compounds 1d and 2b. Consequently, bitriazolyl compounds, a totally new skeleton that is distinct from the existing commercial SQR-inhibiting fungicides, were discovered, which could potentially be a new lead for further development of SQR inhibitors.
Co-reporter:Shu-Hou Yang;Qi Sun;Hao Xiong;Shi-Yu Liu;Behrooz Moosavi;Wen-Chao Yang
Chemical Communications 2017 vol. 53(Issue 28) pp:3952-3955
Publication Date(Web):2017/04/04
DOI:10.1039/C7CC00577F
We report herein the structure-based design and application of a fluorogenic molecular probe (BChE-FP) specific to butyrylcholinesterase (BChE). This probe was rationally designed by mimicking the native substrate and optimized stepwise by manipulating the steric feature and the reactivity of the designed probe targeting the structural difference of the active pockets of BChE and AChE. The refined probe, BChE-FP, exhibits high specificity toward BChE compared to AChE, producing about 275-fold greater fluorescence enhancement upon the catalysis by BChE. Thus, BChE-FP is a specific BChE probe identified by the structure-based design and it can discriminate BChE from AChE. Furthermore, it has been successfully applied for imaging the endogenous BChE in living cells, as well as BChE inhibitor screening and characterization under physiological conditions.
Co-reporter:Shu-Hou Yang;Qi Sun;Hao Xiong;Shi-Yu Liu;Behrooz Moosavi;Wen-Chao Yang
Chemical Communications 2017 vol. 53(Issue 28) pp:3952-3955
Publication Date(Web):2017/04/04
DOI:10.1039/C7CC00577F
We report herein the structure-based design and application of a fluorogenic molecular probe (BChE-FP) specific to butyrylcholinesterase (BChE). This probe was rationally designed by mimicking the native substrate and optimized stepwise by manipulating the steric feature and the reactivity of the designed probe targeting the structural difference of the active pockets of BChE and AChE. The refined probe, BChE-FP, exhibits high specificity toward BChE compared to AChE, producing about 275-fold greater fluorescence enhancement upon the catalysis by BChE. Thus, BChE-FP is a specific BChE probe identified by the structure-based design and it can discriminate BChE from AChE. Furthermore, it has been successfully applied for imaging the endogenous BChE in living cells, as well as BChE inhibitor screening and characterization under physiological conditions.
Co-reporter:Ponnam Devendar
Topics in Current Chemistry 2017 Volume 375( Issue 6) pp:82
Publication Date(Web):09 October 2017
DOI:10.1007/s41061-017-0169-9
Modern agricultural chemistry has to support farmers by providing innovative agrochemicals. In this context, the introduction of sulfur atoms into an active ingredient is still an important tool in modulating the properties of new crop-protection compounds. More than 30% of today’s agrochemicals contain at least one sulfur atom, mainly in fungicides, herbicides and insecticides. A number of recently developed sulfur-containing agrochemical candidates represent a novel class of chemical compounds with new modes of action, so we intend to highlight the emerging interest in commercially active sulfur-containing compounds. This chapter gives a comprehensive overview of selected leading sulfur-containing pesticidal chemical families namely: sulfonylureas, sulfonamides, sulfur-containing heterocyclics, thioureas, sulfides, sulfones, sulfoxides and sulfoximines. Also, the most suitable large-scale synthetic methods of the recently launched or provisionally approved sulfur-containing agrochemicals from respective chemical families have been highlighted.
Co-reporter:Behrooz Moosavi, Bibimaryam Mousavi, Wen-Chao Yang, Guang-Fu Yang
European Journal of Cell Biology 2017 Volume 96, Issue 6(Issue 6) pp:
Publication Date(Web):1 September 2017
DOI:10.1016/j.ejcb.2017.06.003
Understanding cellular processes at molecular levels in health and disease requires the knowledge of protein–protein interactions (PPIs). In line with this, identification of PPIs at genome-wide scale is highly valuable to understand how different cellular pathways are interconnected, and it eventually facilitates designing effective drugs against certain PPIs. Furthermore, investigating PPIs at a small laboratory scale for deciphering certain biochemical pathways has been demanded for years. In this regard, yeast two hybrid system (Y2HS) has proven an extremely useful tool to discover novel PPIs, while Y2HS derivatives and novel yeast-based assays are contributing significantly to identification of protein-drug/inhibitor interaction at both large- and small-scale set-ups. These methods have been evolving over time to provide more accurate, reproducible and quantitative results. Here we briefly describe different yeast-based assays for identification of various protein-protein/drug/inhibitor interactions and their specific applications, advantages, shortcomings, and improvements. The broad range of yeast-based assays facilitates application of the most suitable method(s) for each specific need.
Co-reporter:Qi Sun, Shu-Hou Yang, Lei Wu, Wen-Chao Yang, and Guang-Fu Yang
Analytical Chemistry 2016 Volume 88(Issue 4) pp:2266
Publication Date(Web):January 20, 2016
DOI:10.1021/acs.analchem.5b04029
A benzoquinolizine coumarin-based fluorescent probe was developed for detecting thiophenols, demonstrating the superior fluorescence properties caused by the decay of the twisting effect of N,N-diethylamino group of coumarin. It discriminated thiophenols from various analytes including aliphatic thiols with good selectivity and displayed ∼700-fold fluorescence intensity enhancement and a remarkable limit of detection (4.5 nM). The new probe also can be applied to quantitative determine the concentrations of thiophenol in water samples and living cells.
Co-reporter:Qi Sun, Shu-Hou Yang, Lei Wu, Qing-Jian Dong, Wen-Chao Yang, and Guang-Fu Yang
Analytical Chemistry 2016 Volume 88(Issue 11) pp:6084
Publication Date(Web):May 10, 2016
DOI:10.1021/acs.analchem.6b01545
Selenocysteine (Sec), encoded as the 21st amino acid, is the predominant chemical form of selenium that is closely related to various human diseases. Thus, it is of high importance to identify novel probes for sensitive and selective recognition of Sec and Sec-containing proteins. Although a few probes have been reported to detect artificially introduced selenols in cells or tissues, none of them has been shown to be sensitive enough to detect endogenous selenols. We report the characterization and application of a new fluorogenic molecular probe for the detection of intracellular selenols. This probe exhibits near-zero background fluorescence but produces remarkable fluorescence enhancement upon reacting with selenols in a fast chemical reaction. It is highly specific and sensitive for intracellular selenium-containing molecules such as Sec and selenoproteins. When combined with flow cytometry, this probe is able to detect endogenous selenols in various human cancer cells. It is also able to image endogenous selenol-containing molecules in zebrafish under a fluorescence microscope. These results demonstrate that this molecular probe can function as a useful molecular tool for intracellular selenol sensing, which is valuable in the clinical diagnosis for human diseases associated with Sec-deficiency or overdose.
Co-reporter:Hu Zhang, Peng-Fei Liu, Qiong Chen, Qiong-You Wu, Anne Seville, Yu-Cheng Gu, John Clough, Shao-Lin Zhou and Guang-Fu Yang
Organic & Biomolecular Chemistry 2016 vol. 14(Issue 13) pp:3482-3485
Publication Date(Web):08 Mar 2016
DOI:10.1039/C6OB00371K
The synthesis of albucidin and its enantiomer are described. It involves a visible-light photocatalysis deiodination at the late stage. The absolute configuration of natural albucidin is determined as (1R,3S). This work provides a basis for structural modification to develop a new type of herbicidal from an old structure.
Co-reporter:Yang Zuo, Qiongyou Wu, Sun-wen Su, Cong-wei Niu, Zhen Xi, and Guang-Fu Yang
Journal of Agricultural and Food Chemistry 2016 Volume 64(Issue 3) pp:552-562
Publication Date(Web):January 5, 2016
DOI:10.1021/acs.jafc.5b05378
Protoporphyrinogen oxidase (PPO, E.C. 1.3.3.4) is known as a key action target for several structurally diverse herbicides. As a continuation of our research work on the development of new PPO-inhibiting herbicides, a series of novel 3-(2′-halo-5′-substituted-benzothiazol-1′-yl)-1-methyl-6-(trifluoromethyl)pyrimidine-2,4-diones 9 were designed and synthesized. The bioassay results indicated that a number of the newly synthesized compounds exhibited higher inhibition activity against tobacco PPO (mtPPO) than the controls, saflufenacil and sulfentrazone. Compound 9F-5 was identified as the most potent inhibitor with a Ki value of 0.0072 μM against mtPPO, showing about 4.2-fold and 1.4-fold higher potency than sulfentrazone (Ki = 0.03 μM) and saflufenacil (Ki = 0.01 μM), respectively. An additional green house assay demonstrated that compound 9F-6 (Ki = 0.012 μM) displayed the most promising postemergence herbicidal activity with a broad spectrum even at a concentration as low as 37.5 g of active ingredient (ai)/ha. Maize exhibits relative tolerance against compound 9F-6 at the dosage of 150 g ai/ha, but it is susceptible to saflufenacil even at 75 g ai/ha. Thus, compound 9F-6 exhibits the potential to be a new herbicide for weed control in maize fields.
Co-reporter:Yu-Chao Liu, Ren-Yu Qu, Qiong Chen, Jing-Fang Yang, Niu Cong-Wei, Xi Zhen, and Guang-Fu Yang
Journal of Agricultural and Food Chemistry 2016 Volume 64(Issue 24) pp:4845-4857
Publication Date(Web):June 5, 2016
DOI:10.1021/acs.jafc.6b00720
Acetohydroxyacid synthase (AHAS; also known as acetolactate synthase; EC 2.2.1.6, formerly EC 4.1.3.18) is the first common enzyme in the biosynthetic pathway leading to the branched-chain amino acids in plants and a wide range of microorganisms. Weed resistance to AHAS-inhibiting herbicides, increasing at an exponential rate, is becoming a global problem and leading to an urgent demand of developing novel compounds against both resistant and wild AHAS. In the present work, a series of novel 2-aroxyl-1,2,4-triazolopyrimidine derivatives (a total of 55) were designed and synthesized with the aim to discover an antiresistant lead compound. Fortunately, the screening results indicated that many of the newly synthesized compounds showed a better, even excellent, inhibition effect against both the wild-type Arabidopsis thaliana AHAS and P197L mutants. Among them, compounds 5-3 to 5-17, compounds 5-19 to 5-26, compounds 5-28 to 5-45, and compound 5-48 have the lower values of resistance factor (RF) and display a potential power to overcome resistance associated with the P197L mutation in the enzyme levels. Further greenhouse in vivo assay showed that compounds 5-15 and 5-20 displayed “moderate” to “good” herbicidal activity against both the wild type-and the resistant (P197L mutation) Descurainia sophia, even at a rate as low as 0.9375 (g of ai/ha). The above results indicated that these two compounds could be used as new leads for the future development of antiresistance herbicides.
Co-reporter:Li Xiong, Xiao-Lei Zhu, Hua-Wei Gao, Yu Fu, Sheng-Quan Hu, Li-Na Jiang, Wen-Chao Yang, and Guang-Fu Yang
Journal of Agricultural and Food Chemistry 2016 Volume 64(Issue 24) pp:4830-4837
Publication Date(Web):May 26, 2016
DOI:10.1021/acs.jafc.6b00325
Succinate-ubiquinone oxidoreductase (SQR) is an attractive target for fungicide discovery. Herein, we report the discovery of novel SQR inhibitors using a pharmacophore-linked fragment virtual screening approach, a new drug design method developed in our laboratory. Among newly designed compounds, compound 9s was identified as the most potent inhibitor with a Ki value of 34 nM against porcine SQR, displaying approximately 10-fold higher potency than that of the commercial control penthiopyrad. Further inhibitory kinetics studies revealed that compound 9s is a noncompetitive inhibitor with respect to the substrate cytochrome c and DCIP. Interestingly, compounds 8a, 9h, 9j, and 9k exhibited good in vivo preventive effects against Rhizoctonia solani. The results obtained from molecular modeling showed that the orientation of the R2 group had a significant effect on binding with the protein.
Co-reporter:Da-Wei Wang, Hong-Yan Lin, Bo He, Feng-Xu Wu, Tao Chen, Qiong Chen, Wen-Chao Yang, and Guang-Fu Yang
Journal of Agricultural and Food Chemistry 2016 Volume 64(Issue 47) pp:8986-8993
Publication Date(Web):November 9, 2016
DOI:10.1021/acs.jafc.6b04110
4-Hydroxyphenylpyruvate dioxygenase (EC 1.13.11.27, HPPD) is an important target for new bleaching herbicides discovery. As a continuous work to discover novel crop selective HPPD inhibitor, a series of 2-(aryloxyacetyl)cyclohexane-1,3-diones were rationally designed and synthesized by an efficient one-pot procedure using N,N′-carbonyldiimidazole (CDI), triethylamine, and acetone cyanohydrin in CH2Cl2. A total of 58 triketone compounds were synthesized in good to excellent yields. Some of the triketones displayed potent in vitro Arabidopsis thaliana HPPD (AtHPPD) inhibitory activity. 2-(2-((1-Bromonaphthalen-2-yl)oxy)acetyl)-3-hydroxycyclohex-2-en-1-one, II-13, displayed high, broad-spectrum, and postemergent herbicidal activity at the dosage of 37.5–150 g ai/ha, nearly as potent as mesotrione against some weeds. Furthermore, II-13 showed good crop safety against maize and canola at the rate of 150 g ai/ha, indicating that II-13 might have potential as a herbicide for weed control in maize and canola fields. II-13 is the first HPPD inhibitor showing good crop safety toward canola.Keywords: 4-hydroxyphenylpyruvate dioxygenase; herbicidal activity; one-pot synthesis; structure−activity relationship; triketone;
Co-reporter:Cheng Chen, Rui Zhang, Long Lin, Guang-Fu Yang, Qiong-You Wu
Tetrahedron 2016 Volume 72(27–28) pp:3917-3921
Publication Date(Web):7 July 2016
DOI:10.1016/j.tet.2016.05.007
4-Aryl-3,4-dihydrocoumarins are a class of valuable molecules demonstrating attractive pharmaceutical and biological properties. In this paper, we designed a new and facile approach to synthesis of 4-aryl-3,4-dihydrocoumarin derivatives by Brønsted acid catalyzed Friedel–Crafts alkylation and cycloaddition reaction. With this protocol, 15 examples of 4-aryl-3,4-dihydrocoumarins were successfully prepared with yields ranging from 82 to 99%.
Co-reporter:Zhi-You Huang, Jing-Fang Yang, Ke Song, Qian Chen, Shao-Lin Zhou, Ge-Fei Hao, and Guang-Fu Yang
The Journal of Organic Chemistry 2016 Volume 81(Issue 20) pp:9647-9657
Publication Date(Web):September 20, 2016
DOI:10.1021/acs.joc.6b01725
N-Quinolyl biaryl carboxamides have received tremendous attention for their notable biological properties. Here we have described a general protocol for the preparation of N-quinolyl 3′/4′-biaryl carboxamides by microwave-assisted Suzuki–Miyaura cross-coupling reaction and N-Boc deprotection in one pot. This method, which did not require acids, was used to produce a series of N-quinolyl 3′/4′-biaryl carboxamides with excellent functional group tolerance and high yields (70% to 95%).
Co-reporter:Li Xiong, Xiao-Lei Zhu, Yan-Qing Shen, Wickramabahu Kandergama Wasala Mudiyanselage Wishwajith, Kui Li, Guang-Fu Yang
European Journal of Medicinal Chemistry 2015 Volume 95() pp:424-434
Publication Date(Web):5 May 2015
DOI:10.1016/j.ejmech.2015.03.060
•A series of N-(2-benzooxazol-5-yl)-pyrazole-4-carboxamides were designed and synthesized.•All compounds showed excellent inhibitory activity against porcine SCR.•Compound 13b with a Ki value of 11 nM was identified as the most potent candidate.•13b is non-competitive inhibitor with respect to the substrate cytochrome c and DCIP.•The binding model of the title compounds was established by computational simulations.Succinate–ubiquinone oxidoreductase (SQR, EC 1.3.5.1, complex II), an essential component of cellular respiratory chain and tricarboxylic acid (or Krebs) cycle, has been identified as one of the most significant targets for pharmaceutical and agrochemical. Herein, with the aim of discovery of new antibacterial lead structure, a series of N-benzoxazol-5-yl-pyrazole-4-carboxamides were designed, synthesized, and evaluated for their SQR inhibitory effects. Very promisingly, one candidate (Ki = 11 nM, porcine SQR) was successfully identified as the most potent synthetic SQR inhibitor so far. The further inhibitory kinetics studies revealed that the candidate is non-competitive with respect to the substrate cytochrome c and DCIP. Computational simulations revealed that the titled compounds have formed hydrogen bond with D_Y91 and B_W173 and the pyrazole ring formed cation-π interaction with C_R46. In addition, in R1 position, –CHF2 group has increased the binding affinity and decreased the entropy contribution, while –CF3 group displayed completely opposite effect when bound with SQR. The results of the present work showed that N-benzoxazol-5-yl-pyrazole-4-carboxamide is a new scaffold for discovery of SQR inhibitors and worth further study.A series of N-benzooxazol-5-yl-pyrazole-4-carboxamides were designed and synthesized as potent SQR inhibitors. One candidate (Ki = 11 nM) was successfully identified as the most potent synthetic SQR inhibitor so far.
Co-reporter:Ming-Zhi Zhang, Qiong Chen, Cai-Hong Xie, Nick Mulholland, Sarah Turner, Dianne Irwin, Yu-Cheng Gu, Guang-Fu Yang, John Clough
European Journal of Medicinal Chemistry 2015 Volume 92() pp:776-783
Publication Date(Web):6 March 2015
DOI:10.1016/j.ejmech.2015.01.043
•A series of novel indole-modified analogues of streptochlorin were synthesized.•Streptochlorin analogues were screened against seven phytopathogenic fungi.•Some compounds showed good antifungal activity in primary assays.•The SAR of the streptochlorin analogues were summarized.Streptochlorin, first isolated as a new antibiotic in 1988 from the lipophilic extracts of the mycelium of a Streptomyces sp, is an indole natural products with a variety of biological activities. Based on the methods developed for the synthesis of pimprinine in our laboratory, we have synthesized a series of indole-modified streptochlorin analogues and measured their activities against seven phytopathogenic fungi. Some of the analogues displayed good activity in the primary assays, and the seven compounds 10b, 10c, 11e, 13e, 21, 22c and 22e (shown in Figure 1) were identified as the most promising candidates for further study. Structural optimization is still ongoing with the aim of discovering synthetic analogues with improved antifungal activity.A series of novel streptochlorin analogues were designed and synthesized by modifying the indole moiety. Antifungal activity screening against seven phytopathogenic fungi led to the identification of seven compounds as the most promising candidate for further study.
Co-reporter:Ming-Zhi Zhang, Qiong Chen, Guang-Fu Yang
European Journal of Medicinal Chemistry 2015 Volume 89() pp:421-441
Publication Date(Web):7 January 2015
DOI:10.1016/j.ejmech.2014.10.065
•The recent development of indole derivatives as antiviral agents was reviewed.•A comprehensive list of indole antiviral agents on market or in clinical trials was provided.•The future of indole-based antiviral agents was prospected.Indole represents one of the most important privileged scaffolds in drug discovery. Indole derivatives have the unique property of mimicking the structure of peptides and to bind reversibly to enzymes, which provide tremendous opportunities to discover novel drugs with different modes of action. There are seven indole-containing commercial drugs in the Top-200 Best Selling Drugs by US Retail Sales in 2012. There are also an amazing number of approved indole-containing drugs in the market as well as compounds currently going through different clinical phases or registration statuses. This review focused on the recent development of indole derivatives as antiviral agents with the following objectives: 1) To present one of the most comprehensive listings of indole antiviral agents, drugs on market or compounds in clinical trials; 2) To focus on recent developments of indole compounds (including natural products) and their antiviral activities, summarize the structure property, hoping to inspire new and even more creative approaches; 3) To offer perspectives on how indole scaffolds as a privileged structure might be exploited in the future.The recent developments of indole compounds for antiviral drug discovery were comprehensively reviewed.
Co-reporter:Yu-Ling Xu, Hong-Yan Lin, Xu Ruan, Sheng-Gang Yang, Ge-Fei Hao, Wen-Chao Yang, Guang-Fu Yang
European Journal of Medicinal Chemistry 2015 Volume 92() pp:427-438
Publication Date(Web):6 March 2015
DOI:10.1016/j.ejmech.2015.01.018
•A series of pyrazole-benzimidazolone hybrids were designed by scaffold hopping strategy.•Most of the new compounds showed good activity against recombinant human HPPD.•Compound 9l was identified as the most potent candidate (IC50 = 21 nM), about 3-fold higher than NTBC.4-Hydroxyphenylpyruvate dioxygenase (HPPD), an essential enzyme in tyrosine catabolism, is an important target for treating type I tyrosinemia. Inhibition of HPPD can effectively alleviate the symptoms of type I tyrosinemia. However, only one commercial HPPD inhibitor, 2-(2-nitro-4-trifluoromethylbenzoyl) cyclohexane-1,3-dione (NTBC), has been available for clinical use so far. In the present study, a series of novel pyrazole-benzimidazolone hybrids were designed, synthesized and evaluated as potent human HPPD inhibitors. Most of the new compounds displayed significant inhibitory activity against the recombinant human HPPD. Moreover, compound 9l was identified as the most potent candidate with IC50 value of 0.021 μM against recombinant human HPPD, about 3-fold more potent than NTBC. Thus the pyrazole-benzimidazolone hybrid has great potential to be further developed for the treatment of type I tyrosinemia.A series of pyrazole-benzimidazolone hybrids were designed and synthesized as potent 4-hydroxyphenylpyruvate dioxygenase inhibitors. Compound 9l (IC50 = 21 nM) was the most potent candidate.
Co-reporter:Hua Cheng, Yan-Qing Shen, Xia-Yan Pan, Yi-Ping Hou, Qiong-You Wu and Guang-Fu Yang
New Journal of Chemistry 2015 vol. 39(Issue 9) pp:7281-7292
Publication Date(Web):15 Jul 2015
DOI:10.1039/C5NJ00215J
Respiratory chain succinate-ubiquinone oxidoreductase (SQR or complex II) and ubihydroquinone-cytochrome (cyt) c oxidoreductase (cyt bc1 or complex III) have been demonstrated as the promising targets of numerous antibiotics and fungicides. As a continuation of our research work on the development of new fungicides, a series of 1,2,4-triazole-1,3-disulfonamide derivatives with dual functions targeting both SQR and cyt bc1 were designed and synthesized by coupling diverse diphenyl ether moieties with triazolesulfonamide units. These newly synthesized compounds were characterized by elemental analyses, 1H NMR and ESI-MS spectrometry. The in vitro assay indicated that most of the synthesized compounds displayed good inhibition against porcine succinate-cytochrome reductase (SCR) with IC50 values ranging from 3.2 to 81.8 μM, revealing much higher activity than that of the commercial control amisulbrom whose IC50 value is 93.0 μM. Further evaluation against the respective SQR and cyt bc1 indicated that most compounds exhibited SQR-inhibiting activity as well as cyt bc1-inhibiting activity, but the inhibition potency against SQR is much higher than that against cyt bc1, showing that the SCR inhibition might be contributed greatly by the SQR inhibition. The further antibacterial evaluation against Xanthomonas oryzae pv. oryzae revealed that four compounds showed excellent potency at the concentration of 20 μg mL−1. In particular, compounds 6h and 6j exhibited much better antibacterial activity than the commercial control bismerthiazol in terms of their EC50. Impressively, 6j has an EC90 of 33.62 μg mL−1, more than 10-fold higher than that of bismerthiazol.
Co-reporter:Xiaolei Zhu, Mengmeng Zhang, Jingjing Liu, Jingming Ge, and Guangfu Yang
Journal of Agricultural and Food Chemistry 2015 Volume 63(Issue 13) pp:3377-3386
Publication Date(Web):March 17, 2015
DOI:10.1021/acs.jafc.5b00228
Ametoctradin is a new Oomycete-specific fungicide under development by BASF. It is a potent inhibitor of the bc1 complex in mitochondrial respiration. However, its detailed action mechanism remains unknown. In the present work, the binding mode of ametoctradin was first uncovered by integrating molecular docking, MD simulations, and MM/PBSA calculations, which showed that ametoctradin should be a Qo site inhibitor of bc1 complex. Subsequently, a series of new 1,2,4-triazolo[1,5-a]pyrimidine derivatives were designed and synthesized to further understand the substituent effects on the 5- and 6-position of 1,2,4-triazolo[1,5-a]pyrimidine. The calculated binding free energies (ΔGcal) of newly synthesized analogues as Qo site inhibitors correlated very well (R2 = 0.96) with their experimental binding free energies (ΔGexp). Two compounds (4a and 4c) with higher inhibitory activity against porcine SQR than ametoctradin were successfully identified. The structural and mechanistic insights obtained from the present study will provide a valuable clue for future designing of a new promising bc1 inhibitor.
Co-reporter:Zhi-You Huang, Jing-Fang Yang, Qian Chen, Run-Jie Cao, Wei Huang, Ge-Fei Hao and Guang-Fu Yang
RSC Advances 2015 vol. 5(Issue 92) pp:75182-75186
Publication Date(Web):24 Aug 2015
DOI:10.1039/C5RA13302E
An efficient one-pot, Pd(PPh3)4 catalyzed, water-promoted method for the synthesis of N-(pyridin-2-ylmethyl) biphenyl-4-sulfonamides was developed under microwave irradiation. This methodology is acid free, has good substrate scope, excellent functional group compatibility, and excellent product yields, and is superior to the existing procedures for the synthesis of biphenyl-4-sulfonamides bearing a pyridin-2-ylmethyl group.
Co-reporter:Fengxiang Zhu;Ling Zhu-Ge;Dr. Guangfu Yang;Dr. Shaolin Zhou
ChemSusChem 2015 Volume 8( Issue 4) pp:609-612
Publication Date(Web):
DOI:10.1002/cssc.201403234
Abstract
The catalytic hydrogenation of carbon dioxide and bicarbonate to formate has been explored extensively. The vast majority of the known active catalyst systems are based on precious metals. Herein, we describe an effective, phosphine-free, air- and moisture-tolerant catalyst system based on Knölker’s iron complex for the hydrogenation of bicarbonate and carbon dioxide to formate. The catalyst system can hydrogenate bicarbonate at remarkably low hydrogen pressures (1–5 bar).
Co-reporter:Jun Li, Chun-Fang Zhang, Shu-Hou Yang, Wen-Chao Yang, and Guang-Fu Yang
Analytical Chemistry 2014 Volume 86(Issue 6) pp:3037
Publication Date(Web):February 10, 2014
DOI:10.1021/ac403885n
The development of probes for specific thiophenol detection is of great importance, due to the toxicity of thiophenols and their derivatives in the environment. In the present study, a novel fluorescent probe was rationally designed for detecting thiophenols via an intramolecular charge transfer mechanism. The developed probe selectively and sensitively distinguished thiophenols from aliphatic thiols. It displayed a large Stokes shift (145 nm) and >280-fold fluorescence enhancement. Moreover, the new probe not only displayed excellent cell permeability for the successful detection of thiophenol in HEK293 cells but also quantitatively measured thiophenols in water samples with good recovery (more than 90%), indicating that it has promising prospects for application for thiophenol sensing in environmental and biological sciences.
Co-reporter:Ge-Fei Hao, Ying Tan, Wei-Fang Xu, Run-Jie Cao, Zhen Xi, and Guang-Fu Yang
Journal of Agricultural and Food Chemistry 2014 Volume 62(Issue 29) pp:7209-7215
Publication Date(Web):July 1, 2014
DOI:10.1021/jf5018115
The potential of protoporphyrinogen oxidase (PPO) to develop resistance against five PPO-inhibiting herbicides has been studied using computational mutation scanning (CMS) protocol, leading to valuable insights into the resistance mechanisms and structure-resistance relationship of the PPO inhibitors. The calculated shifts in the binding free energies caused by the mutations correlated very well with those derived from the corresponding experimental data obtained from site-directed mutagenesis of PPO, leading to valuable insights into the resistance mechanisms of PPO inhibitors. The calculated entropy change was related to the conformational flexibility of the inhibitor, which demonstrated that inhibitors with appropriate conformational flexibility may inhibit both the wild type and mutants simultaneously. The reasonable correlation between the computational and experimental data further validate that CMS protocol is valuable for predicting resistance associated with amino acid mutations on target proteins.
Co-reporter:Qiong-You Wu, Li-Li Jiang, Sheng-Gang Yang, Yang Zuo, Zhi-Fang Wang, Zhen Xi and Guang-Fu Yang
New Journal of Chemistry 2014 vol. 38(Issue 9) pp:4510-4518
Publication Date(Web):02 Jul 2014
DOI:10.1039/C4NJ00636D
Protoporphyrinogen oxidase (PPO, EC 1.3.3.4) has attracted continuous interest during the last few decades not only because of its unique biochemical characteristics but also because of its biomedical significance. As a continuation of our research work on the development of new PPO inhibitors, N-(benzothiazol-5-yl)-hexahydro-2H-isoindole-1,3-dithione (1a–j) and N-(benzothiazol-5-yl)-octahydro-3-thioxoisoindol-1-one derivatives (2a–i) were designed and synthesized. These newly prepared compounds were characterized by elemental analyses, 1H NMR and ESI-MS spectroscopy. The in vitro assay indicated that these compounds displayed good inhibition activity against human PPO (hPPO) with Ki values ranging from 0.38 μM to 6.83 μM. Notably, most of the monothionated products (1a–j) displayed a higher or comparable PPO-inhibition activity compared with the commercial control sulfentrazone. The comparison of the dihedral angles of the representative compound with that of acifluorfen (ACF) complexed with hPPO clearly indicated that the dihedral angle between the thionyl amide or carbonyl amide ring and the benzothiazole ring was closely related to the variation of the PPO inhibition activity of different types of inhibitors.
Co-reporter:Hui Li;Xiao-Lei Zhu;Wen-Chao Yang
Chemical Biology & Drug Design 2014 Volume 83( Issue 1) pp:71-80
Publication Date(Web):
DOI:10.1111/cbdd.12199
Antimycin and cyazofamid are specific inhibitors of the mitochondrial respiratory chain and bind to the Qi site of the cytochrome bc1 complex. With the aim to understand the detailed molecular inhibition mechanism of Qi inhibitors, we performed a comparative investigation of the inhibitory kinetics of them against the porcine bc1 complex. The results showed that antimycin is a slow tight-binding inhibitor of succinate–cytochrome c reductase (SCR) with Ki = 0.033 ± 0.00027 nm and non-competitive inhibition with respect to cytochrome c. Cyazofamid is a classical inhibitor of SCR with Ki = 12.90 ± 0.91 μm and a non-competitive inhibitor with respect to cytochrome c. Both of them show competitive inhibition with respect to substrate DBH2. Further molecular docking and quantum mechanics calculations were performed. The results showed that antimycin underwent significant conformational change upon the binding. The energy barrier between the conformations in the crystal and in the binding pocket is ~13.63 kcal/mol. Antimycin formed an H-bond with Asp228 and two water-bridged H-bonds with Lys227 and His201, whereas cyazofamid formed only one H-bond with Asp228. The conformational change and the different hydrogen bonding network might account for why antimycin is a slow tight-binding inhibitor, whereas cyazofamid is a classic inhibitor.
Co-reporter:Qiong-You Wu;Li-Li Jiang;Yang Zuo;Zhi-Fang Wang;Zhen Xi
Chemical Biology & Drug Design 2014 Volume 84( Issue 4) pp:431-442
Publication Date(Web):
DOI:10.1111/cbdd.12331
Protoporphyrinogen oxidase (EC 1.3.3.4) is one of the most significant targets for a large family of herbicides. As part of our continuous efforts to search for novel protoporphyrinogen oxidase-inhibiting herbicides, N-(benzothiazol-5-yl)tetrahydroisoindole-1,3-dione was selected as a lead compound for structural optimization, leading to the syntheses of a series of novel N-(benzothiazol-5-yl)hexahydro-1H-isoindole-1,3-diones (1a–o) and N-(benzothiazol-5-yl)hexahydro-1H-isoindol-1-ones (2a–i). These newly prepared compounds were characterized by elemental analyses, 1H NMR, and ESI-MS, and the structures of 1h and 2h were further confirmed by X-ray diffraction analyses. The bioassays indicated that some compounds displayed comparable or higher protoporphyrinogen oxidase inhibition activities in comparison with the commercial control. Very promising, compound 2a, ethyl 2-((6-fluoro-5-(4,5,6,7-tetrahydro-1-oxo-1H-isoindol-2(3H)-yl)benzo[d]thiazol-2-yl)-sulfanyl)acetate, was recognized as the most potent candidate with Ki value of 0.0091 μm. Further greenhouse screening results demonstrated that some compounds exhibited good herbicidal activity against Chenopodium album at the dosage of 150 g/ha.
Co-reporter:Da-Wei Wang, Hong-Yan Lin, Run-Jie Cao, Sheng-Gang Yang, Qiong Chen, Ge-Fei Hao, Wen-Chao Yang, and Guang-Fu Yang
Journal of Agricultural and Food Chemistry 2014 Volume 62(Issue 49) pp:11786-11796
Publication Date(Web):November 18, 2014
DOI:10.1021/jf5048089
Exploring novel 4-hydroxyphenylpyruvate dioxygenase (EC 1.13.11.27, HPPD) inhibitors is one of the most promising research directions in herbicide discovery. To discover new triketone herbicides with broad-spectrum weed control as well as excellent crop selectivity, a series of (total 52) novel triketone-containing quinazoline-2,4-dione derivatives were synthesized and further bioevaluated. The greenhouse testing indicated that many of the newly synthesized compounds showed better or excellent herbicidal activity against broadleaf and monocotyledonous weeds at the dosages of 37.5–150 g of active ingredient (ai)/ha. The structure and activity relationship in this study indicated that the triketone-containing quinazoline-2,4-dione motif has possessed great impact on herbicide activity and may be used for further optimization. Among the new compounds, III-b and VI-a–VI-d displayed a broader spectrum of weed control than mesotrione. In addition, the compound III-b also demonstrated comparatively superior crop selectivity to mesotrione, thus possessing great potential for weed control in the field.
Co-reporter:Qi Sun, Da-Yong Peng, Sheng-Gang Yang, Xiao-Lei Zhu, Wen-Chao Yang, Guang-Fu Yang
Bioorganic & Medicinal Chemistry 2014 Volume 22(Issue 17) pp:4784-4791
Publication Date(Web):1 September 2014
DOI:10.1016/j.bmc.2014.06.057
Exploring small-molecule acetylcholinesterase (AChE) inhibitors to slow the breakdown of acetylcholine (Ach) represents the mainstream direction for Alzheimer’s disease (AD) therapy. As the first acetylcholinesterase inhibitor approved for the clinical treatment of AD, tacrine has been widely used as a pharmacophore to design hybrid compounds in order to combine its potent AChE inhibition with other multi-target profiles. In present study, a series of novel tacrine–coumarin hybrids were designed, synthesized and evaluated as potent dual-site AChE inhibitors. Moreover, compound 1g was identified as the most potent candidate with about 2-fold higher potency (Ki = 16.7 nM) against human AChE and about 2-fold lower potency (Ki = 16.1 nM) against BChE than tacrine (Ki = 35.7 nM for AChE, Ki = 8.7 nM for BChE), respectively. In addition, some of the tacrine–coumarin hybrids showed simultaneous inhibitory effects against both Aβ aggregation and β-secretase. We therefore conclude that tacrine–coumarin hybrid is an interesting multifunctional lead for the AD drug discovery.
Co-reporter:Hua Cheng, Qiong-You Wu, Fan Han, Guang-Fu Yang
Chinese Chemical Letters 2014 Volume 25(Issue 5) pp:705-709
Publication Date(Web):May 2014
DOI:10.1016/j.cclet.2014.03.013
Pyrazoles and their derivatives are important heterocycles found in nature and present in numerous bioactive compounds. In contrast to 3 or 5-aryl pyrazole, the preparation of 4-aryl pyrazole is fairly rare. Utilizing microwave irradiation, the synthesis of 4-substituted-arylpyrazole via Suzuki cross-coupling has been developed with a wide range of substrates. The remarkable advantages of this method are mild reaction conditions, simple operation, high yield, and short reaction time. Product structures were identified by MS, 1H NMR, 13C NMR, and elemental analysis.Utilizing microwave irradiation, the synthesis of 4-substituted pyrazole via Suzuki cross-coupling has been developed with a wide range of substrates. The easy availability and the broad structural diversity of substrates make the reaction useful for the construction of 4-substituted pyrazole libraries.
Co-reporter:Pingyuan Wang, Jian Li, Xue Jiang, Zhiqing Liu, Na Ye, Youjun Xu, Guangfu Yang, Yechun Xu, Ao Zhang
Tetrahedron 2014 70(35) pp: 5666-5673
Publication Date(Web):
DOI:10.1016/j.tet.2014.06.064
Co-reporter:Xiao-Lei Zhu;Li Xiong;Hui Li;Xin-Ya Song;Jing-Jing Liu; Guang-Fu Yang
ChemMedChem 2014 Volume 9( Issue 7) pp:1512-1521
Publication Date(Web):
DOI:10.1002/cmdc.201300456
Abstract
Succinate-ubiquinone oxidoreductase (SQR, EC 1.3.5.1), also known as mitochondrial respiratory complex II or succinate dehydrogenase (SDH), catalyzes the oxidation of succinate to fumarate as part of the tricarboxylic acid cycle. SQR has been identified as a novel target of a large family of agricultural fungicides. However, the detailed mechanism of action between the fungicides and SQR is still unclear, and the bioactive conformation of fungicides in the SQR binding pocket has not been identified. In this study, the kinetics of porcine SQR inhibition by ten commercial carboxamide fungicides were measured, and noncompetitive inhibition was observed with respect to succinate, DCIP, and cytochrome c, while competitive inhibition was observed with respect to ubiquinone. With the aim to uncover the binding conformation of these fungicides, molecular docking, molecular dynamics simulation, and molecular mechanics/Poisson–Boltzmann surface area (MM/PBSA) calculations were then performed. The excellent correlation (r2=0.94) between the calculated (ΔGcal) and experimental (ΔGexp) binding free energies indicates that the obtained docking conformation could be the bioactive conformation. The acid moiety of carboxamide fungicides inserts into the ubiquinone binding site (Q-site) of SQR, forming van der Waals (vdW) interactions with C_R46, C_S42, B_I218, and B_P169, while the amine moiety extends to the mouth of the Q-site, forming vdW interactions with C_W35, C_I43, and C_I30. The carbonyl oxygen atom of the carboxamide forms hydrogen bonds with B_W173 and D_Y91. These findings provide valuable information for the design of more potent and specific inhibitors of SQR.
Co-reporter:Yu-Ling Xu, Hong-Yan Lin, Run-Jie Cao, Ze-Zhong Ming, Wen-Chao Yang, Guang-Fu Yang
Bioorganic & Medicinal Chemistry 2014 22(19) pp: 5194-5211
Publication Date(Web):
DOI:10.1016/j.bmc.2014.08.011
Co-reporter:Pingyuan Wang, Shanshan Song, Zehong Miao, Guangfu Yang, and Ao Zhang
Organic Letters 2013 Volume 15(Issue 15) pp:3852-3855
Publication Date(Web):July 15, 2013
DOI:10.1021/ol401489x
An efficient one-pot synthesis of polyhydroxyalkyl-substituted pyrroles from 1,2-cyclopropa-3-pyranones with primary amines is reported. With 10% of InBr3 as the catalyst, both aryl- and alkylamines as well as various 1,2-cyclopropa-3-pyranones are well tolerated. This method is highly appealing because of its one-pot process, mild reaction conditions, substrate simplicity, and broad substrate scope.
Co-reporter:Qi Sun, Jun Li, Wan-Nian Liu, Qing-Jian Dong, Wen-Chao Yang, and Guang-Fu Yang
Analytical Chemistry 2013 Volume 85(Issue 23) pp:11304
Publication Date(Web):October 28, 2013
DOI:10.1021/ac402097g
Human neutrophil elastase (HNE) has been identified as a potential therapeutic target for the discovery of anti-inflammatory drugs for decades. However, little progress has been made on assays measuring the activity of HNE, especially on synthetic substrates which play essential role in determination of HNE activity. Herein, a small-molecule compound, 2,2,3,3,3-pentafluoro-N-(2-oxo-4-(trifluoromethyl)-2H-chromen-7-yl)-propanamide (compound 4), has been successfully designed as the first ever non-peptide-based fluorogenic substrate for HNE. A “turn-on” fluorometric assay based on 4 has been successfully developed for rapid determination of HNE activity and the inhibitory kinetic study. Most importantly, the probe 4 shows highly specific response for HNE among seven tested hydrolases or proteins and can be directly used to detect the elevated HNE activity in the serum of chronic obstructive pulmonary disease (COPD) patients compared to that of healthy controls. This specific and cost-effective probe will facilitate future high-throughput discovery of HNE inhibitors and clinical diagnosis of elastase-related diseases.
Co-reporter:Wei Huang, Qiong Chen, Wen-Chao Yang, Guang-Fu Yang
European Journal of Medicinal Chemistry 2013 Volume 66() pp:161-170
Publication Date(Web):August 2013
DOI:10.1016/j.ejmech.2013.05.037
•A microwave-assisted synthesis of thioether-substituted flavonoids was developed.•57 novel flavonoids were synthesized and screened against six tumor cell lines.•Three candidates with promising antitumor activity were successfully identified.As widely occurring natural products, flavonoids are an important source for drug discovery, due to their structural diversity and broad-spectrum biological activity. In this work, a library of novel, thioether-substituted flavonoids with diverse heterocyclic groups was synthesized via a microwave-assisted procedure with the advantages of good yields, short times, mild conditions and ready isolation of the products. Their antiproliferative activities were evaluated against six cancer cell lines, HCCLM-7, Hela, MDA-MB-435S, SW-480, Hep-2, and MCF-7 by the MTT-based assay. Compared with the positive control 5-fluorouracil, three compounds, 6a, 6b and 6j were successfully identified as the most promising candidates, due to their higher potency and broad-spectrum bioactivity with IC50 values in the range of 0.43 μM–6.7 μM.A library of 57 novel flavonoids bearing diverse heterocyclic groups was efficiently synthesized via microwave-assisted procedure. Three candidates with promising potent and broad-spectrum antitumor activity were successfully identified.
Co-reporter:Zuo Yang, Yang Sheng-Gang, Luo Yan-Ping, Tan Ying, Hao Ge-Fei, Wu Qiong-You, Xi Zhen, Yang Guang-Fu
Bioorganic & Medicinal Chemistry 2013 Volume 21(Issue 11) pp:3245-3255
Publication Date(Web):1 June 2013
DOI:10.1016/j.bmc.2013.03.056
Protoporphyrinogen oxidase (PPO, E.C. 1.3.3.4) is the action target for several structurally diverse herbicides. A series of novel 4-(difluoromethyl)-1-(6-halo-2-substituted-benzothiazol-5-yl)-3-methyl-1H-1,2,4-triazol-5(4H)-ones 2a–z were designed and synthesized via the ring-closure of two ortho-substituents. The in vitro bioassay results indicated that the 26 newly synthesized compounds exhibited good PPO inhibition effects with Ki values ranging from 0.06 to 17.79 μM. Compound 2e, ethyl 2-{[5-(4-(difluoromethyl)-3-methyl-5-oxo-4,5-dihydro-1H-1,2,4-triazol-1-yl)-6-fluorobenzo-thiazol-2-yl]thio}acetate, was the most potent inhibitor with Ki value of 0.06 μM against mtPPO, comparable to (Ki = 0.03 μM) sulfentrazone. Further green house assays showed that compound 2f (Ki = 0.24 μM, mtPPO), ethyl 2-{[5-(4-(difluoromethyl)-3-methyl-5-oxo-4,5-dihydro-1H-1,2,4-triazol-1-yl)-6-fluorobenzothiazol-2-yl]thio}propanoate, showed the most promising post-emergence herbicidal activity with broad spectrum even at concentrations as low as 37.5 g ai/ha. Soybean exhibited tolerance to compound 2f at the dosages of 150 g ai/ha, whereas they are susceptible to sulfentrazone even at 75 g ai/ha. Thus, compound 2f might be a potential candidate as a new herbicide for soybean fields.
Co-reporter:Jun Li, Chun-Fang Zhang, Ze-Zhong Ming, Wen-Chao Yang and Guang-Fu Yang
RSC Advances 2013 vol. 3(Issue 48) pp:26059-26065
Publication Date(Web):24 Oct 2013
DOI:10.1039/C3RA45002C
Two novel coumarin-derived fluorescent probes were designed and synthesized for the quantitative determination of biothiols, such as cysteine (Cys), glutathione (GSH) and homocysteine (Hcy). Both probes selectively and sensitively detected biothiols in vitro, and successfully sensed biothiols in vivo with low cytotoxicity, suggesting their potential application for biothiol detection.
Co-reporter:Shenggang Yang;Gefei Hao;Franck E. Dayan;Patrick J. Tranel;Guangfu Yang
Chinese Journal of Chemistry 2013 Volume 31( Issue 9) pp:1153-1158
Publication Date(Web):
DOI:10.1002/cjoc.201300449
Abstract
Protoporphyrinogen oxidase (PPO, EC 1.3.3.4) is one of the most significant targets for a large family of inhibitors that may be used as herbicide, bactericide, fungicide, or photosensitizing activator to treat cancer through photodynamic therapy (PDT). Molecular docking and CoMFA were combined in a multistep framework with the ultimate goal of identifying important factor contributing to the activity of PPO inhibitors. As a continuation of the previous research work on the development of new PPO inhibitors, the bioassay results indicated that good PPO inhibitors were discovered in all of the three chemical series with IC50 values ranging from 0.010 to 0.061 µmol·L−1. Using the crystal structure of tobacco mitochondrial PPO (mtPPO) as template, all the compounds were docked into the enzyme active site. The docking pose of each compound was subsequently used in a receptor-based alignment, leading to the development of a significant CoMFA model with r2 value of 0.98 and q2 (cross validation r2) value of 0.63. This novel multistep framework gives insight into the structural characteristics for the binding of inhibitors, and it can be extended to other classes of PPO inhibitors. In addition, the simplicity of the proposed approach may be particularly applicable in virtual screening procedures.
Co-reporter:Shenggang Yang;Gefei Hao;Franck E. Dayan;Patrick J. Tranel;Guangfu Yang
Chinese Journal of Chemistry 2013 Volume 31( Issue 9) pp:
Publication Date(Web):
DOI:10.1002/cjoc.201390021
Co-reporter:Guangfu Yang;Gefei Hao
Science 2013 Volume 342(Issue 6160) pp:
Publication Date(Web):
DOI:10.1126/science.342.6160.799-b
Co-reporter:Jun Li, Chun-Fang Zhang, Ze-Zhong Ming, Ge-Fei Hao, Wen-Chao Yang, Guang-Fu Yang
Tetrahedron 2013 69(23) pp: 4743-4748
Publication Date(Web):
DOI:10.1016/j.tet.2013.03.032
Co-reporter:Xiao-Lei Zhu, Ning-Xi Yu, Ge-Fei Hao, Wen-Chao Yang, Guang-Fu Yang
Journal of Molecular Graphics and Modelling 2013 Volume 41() pp:55-60
Publication Date(Web):April 2013
DOI:10.1016/j.jmgm.2013.01.004
Acetylcholinesterase (AChE) is a key enzyme of the cholinergic nervous system. More than one gene encodes the synaptic AChE target. As the most potent known AChE inhibitor, the syn1-TZ2PA6 isomer was recently shown to have higher affinity as a reversible organic inhibitor of acetylcholinesterase1 (AChE1) than the anti1-TZ2PA6 isomer. Opposite selectivity has been shown for acetylcholinesterase2 (AChE2). In an attempt to understand the selectivity of the syn1-TZ2PA6 and anti1-TZ2PA6 isomers for AChE1 and AChE2, six molecular dynamics (MD) simulations were carried out with mouse AChE (mAChE, type of AChE1), Torpedo californica AChE (TcAChE, type of AChE1), and Drosophila melanogaster AChE (DmAChE, type of AChE2) bound with syn1-TZ2PA6 and anti1-TZ2PA6 isomers. Within the structure of the inhibitor, the 3,8-diamino-6-phenylphenanthridinium subunit and 9-amino-1,2,3,4-tetrahydroacridine subunit, via π–π interactions, made more favorable contributions to syn1-TZ2PA6 or anti1-TZ2PA6 isomer binding in the mAChE/TcAChE enzyme than the 1,2,3-triazole subunit. Compared to AChE1, the triazole subunit had increased binding energy with AChE2 due to a greater negative charge in the active site. The binding free energy calculated using the MM/PBSA method suggests that selectivity between AChE1 and AChE2 is mainly attributed to decreased binding affinity for the inhibitor.Graphical abstractAcetylcholinesterase (AChE) is a key enzyme of the cholinergic nervous system and has two subtypes, AChE1 and AChE2. Taking the most potent AChE inhibitor, the syn1-TZ2PA6 and its anti-isomer, as representative, comprehensive molecular dynamics (MD) simulations was performed to uncover the selectivity mechanism of the syn1-TZ2PA6 and anti1-TZ2PA6 isomers for AChE1 and AChE2. This computational insight could guide the design of drugs that selectively inhibit AChE1 or AChE2.Highlights► We report the selectivity mechanism of the syn1-TZ2PA6 and anti1-TZ2PA6 isomers for AChE1 and AChE2. ► We employed molecular dynamic simulations and binding free energy calculation. ► Calculated data was in good agreement with experimental values.
Co-reporter:Ge-Fei Hao ; Fu Wang ; Hui Li ; Xiao-Lei Zhu ; Wen-Chao Yang ; Li-Shar Huang ; Jia-Wei Wu ; Edward A. Berry
Journal of the American Chemical Society 2012 Volume 134(Issue 27) pp:11168-11176
Publication Date(Web):June 12, 2012
DOI:10.1021/ja3001908
A critical challenge to the fragment-based drug discovery (FBDD) is its low-throughput nature due to the necessity of biophysical method-based fragment screening. Herein, a method of pharmacophore-linked fragment virtual screening (PFVS) was successfully developed. Its application yielded the first picomolar-range Qo site inhibitors of the cytochrome bc1 complex, an important membrane protein for drug and fungicide discovery. Compared with the original hit compound 4 (Ki = 881.80 nM, porcine bc1), the most potent compound 4f displayed 20 507-fold improved binding affinity (Ki = 43.00 pM). Compound 4f was proved to be a noncompetitive inhibitor with respect to the substrate cytochrome c, but a competitive inhibitor with respect to the substrate ubiquinol. Additionally, we determined the crystal structure of compound 4e (Ki = 83.00 pM) bound to the chicken bc1 at 2.70 Å resolution, providing a molecular basis for understanding its ultrapotency. To our knowledge, this study is the first application of the FBDD method in the discovery of picomolar inhibitors of a membrane protein. This work demonstrates that the novel PFVS approach is a high-throughput drug discovery method, independent of biophysical screening techniques.
Co-reporter:Ming-Zhi Zhang, Qiong Chen, Nick Mulholland, David Beattie, Dianne Irwin, Yu-Cheng Gu, Guang-Fu Yang, John Clough
European Journal of Medicinal Chemistry 2012 Volume 53() pp:283-291
Publication Date(Web):July 2012
DOI:10.1016/j.ejmech.2012.04.012
A simple and efficient synthetic protocol for 5-(3-indolyl)-oxazoles has been developed and further used to synthesize a series of novel analogues of natural product pimprinine. All new compounds were identified by 1H NMR, high resolution mass spectrometry, and the structures of 10 and 18o were further confirmed by X-ray crystallographic diffraction analysis. Bioassay conducted at Syngenta showed that several of the synthesized compounds exhibited fungicidal activity. Compounds 10, 17, 18h, 18o, 19h, 19i and 19l all showed effective control of three out of the seven tested phytopathogenic fungi at the highest rate screened. Compounds 17 and 19h in particular showed activity against the four pathogens screened in artificial media; Pythium dissimile, Alternaria solani, Botryotinia fuckeliana and Gibberella zeae.Graphical abstractA simple and efficient synthetic protocol for 5-(3-indolyl)-oxazoles has been developed. By this new protocol, a series of novel pimprinine analogues were synthesized and screened for the fungicidal activity.Highlights► A simple and efficient synthesis of 5-(3-indolyl)-oxazoles has been developed. ► Thirty six novel analogues of natural product pimprinine were synthesized and screened. ► The structure–activity relationships of pimprinine analogues were summarized for the first time.
Co-reporter:Da-Yong Peng, Qi Sun, Xiao-Lei Zhu, Hong-Yan Lin, Qiong Chen, Ning-Xi Yu, Wen-Chao Yang, Guang-Fu Yang
Bioorganic & Medicinal Chemistry 2012 Volume 20(Issue 22) pp:6739-6750
Publication Date(Web):15 November 2012
DOI:10.1016/j.bmc.2012.09.016
Alzheimer’s disease (AD) is a multifactorial syndrome with several target proteins contributing to its etiology. In this study, we conducted a structure-based design and successfully produced a series of new multi-site AChE inhibitors with a novel framework. Compound 2e, characterized by a central benzamide moiety linked to an isoquinoline at one side and acetophenone at the other, was the most potent candidate with Ki of 6.47 nM against human AChE. Particularly, it showed simultaneous inhibitory effects against BChE, Aβ aggregation, and β-secretase. We therefore conclude that compound 2e is a very promising multi-function lead for the treatment of AD.
Co-reporter:Yang Zuo, Sheng-Gang Yang, Li-Li Jiang, Ge-Fei Hao, Zhi-Fang Wang, Qiong-You Wu, Zhen Xi, Guang-Fu Yang
Bioorganic & Medicinal Chemistry 2012 Volume 20(Issue 1) pp:296-304
Publication Date(Web):1 January 2012
DOI:10.1016/j.bmc.2011.10.079
Protoporphyrinogen oxidase (Protox, EC 1.3.3.4) has attracted great interest during the last decades due to its unique biochemical characteristics and biomedical significance. As a continuation of our research work on the development of new PPO inhibitors, 23 new 1,3,4-thiadiazol-2(3H)-ones bearing benzothiazole substructure were designed and synthesized. The in vitro assay indicated that the newly synthesized compounds 1a–w displayed good inhibition activity against human PPO (hPPO) with Ki values ranging from 0.04 μM to 245 μM. To the knowledge, compound 1a, O-ethyl S-(5-(5-(tert-butyl)-2-oxo-1,3,4-thiadiazol-3(2H)-yl)-6-fluorobenzothiazol-2-yl)carbonothioate, with the Ki value of 40 nM, is so far known as the most potent inhibitor against hPPO. Based on the molecular docking and modified molecular mechanics/Poisson–Boltzmann surface area (MM-PBSA) calculations, the quantitative structure–activity relationships of 1,3,4-thiadiazol-2(3H)-ones and 1,3,4-oxadiazol-2(3H)-one derivatives were established with excellent correlation relationships (r2 = 0.81) between the calculated and experimental binding free energies. Some important insights were also concluded for guiding the future rational design of new hPPO inhibitors with improved potency.
Co-reporter:Long Lin, Nick Mulholland, Qiong-You Wu, David Beattie, Shao-Wei Huang, Dianne Irwin, John Clough, Yu-Cheng Gu, and Guang-Fu Yang
Journal of Agricultural and Food Chemistry 2012 Volume 60(Issue 18) pp:4480-4491
Publication Date(Web):March 22, 2012
DOI:10.1021/jf300610j
Sclerotiorin 1, first isolated from Penicillium sclerotiorum, has weak antifungal activity and belongs to the azaphilone-type family of natural products. Several series of sclerotiorin analogues were designed and synthesized with the aim of discovering novel fungicides with improved activity. The syntheses involved two key steps, cycloisomerization and then oxidation, and used a simple and efficient Sonogashira cross-coupling reaction to construct the required functionalized precursor. With sclerotiorin as a control, the activities of the newly synthesized analogues were evaluated against seven fungal pathogens, and several promising candidates (compounds 3a1, 3d2, 3e2, 3f2 and 3k2) with greater activity and simpler structures than sclerotiorin were discovered. In addition, preliminary structure–activity relationships were studied, which revealed that not only the chlorine or bromine substituent at the 5-position of the nucleus but also the phenyl group at the 3-position and the substituent pattern on it contributed crucially to the observed antifungal activity. Analogues with a methyl substituent at the 1-position have reduced levels of activity, while those with a free hydroxyl group in place of acetoxy at the quaternary center of the bicyclic ring system retain activity.
Co-reporter:Wen-Chao Yang, Jing Li, Jun Li, Qiong Chen, Guang-Fu Yang
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 3) pp:1455-1458
Publication Date(Web):1 February 2012
DOI:10.1016/j.bmcl.2011.11.115
A novel synthetic method of N-cyanocarboxamides has been developed with advantages of mild reaction condition, simpler procedure and easy reactant-product isolation compared with the existing methods. Using this novel method, 16 new N-cyano-1H-imidazole-4-carboxamide derivatives were synthesized and their structures were characterized by spectrum analysis. Further antifungal activity study showed that most of the newly synthesized compounds have good antifungal activity selectively against Rhizoctonia solani among the six fungi tested. Particularly, compound 12h was identified as the most promising candidate with an EC50 of 2.63 μg/mL against R. solani.A novel synthetic method of N-cyanocarboxamides was developed and applied to the syntheses of antifungal N-cyano-1H-imidazole-4-carboxamide derivatives.
Co-reporter:Long Lin;Qiongyou Wu;Shaowei Huang ;Guangfu Yang
Chinese Journal of Chemistry 2012 Volume 30( Issue 5) pp:1075-1082
Publication Date(Web):
DOI:10.1002/cjoc.201100560
Abstract
A novel three-component reaction of o-bromobenzaldehyde, terminal alkynes and tert-butyl amine has been established, which proceeded smoothly to give 3-substituted isoquinolines in good yields in the presence of palladium/copper catalysts under microwave irradiation.
Co-reporter:Xiaolei Zhu;Fu Wang;Hui Li;Wenchao Yang;Qiong Chen ;Guangfu Yang
Chinese Journal of Chemistry 2012 Volume 30( Issue 9) pp:1999-2008
Publication Date(Web):
DOI:10.1002/cjoc.201200607
Abstract
Strobilurins are one of the most important natural products with fungicidal activities and well known for their novel action mode, broad fungicidal spectrum, lower toxicity against mammalian cells, and environmentally benign characteristics. Design and syntheses of strobilurin analogues therefore have attracted great attention in the field of agrochemistry. Previously, we successfully developed a new molecular design method of pharmacophore-linked fragment virtual screening (PFVS) and discovered a lead compound (E)-methyl-2-(2-(((3-(imino-(phenyl)methyl)phenyl)thio)methyl)phenyl)-3-methoxyacrylate (1). To discover new strobilurin analogues with higher fungicidal activity, the structural modification of compound 1 was carried out guided by bioisosterism. A series of benzophenone derivatives 2a–2j were synthesized, among which compound 2j with a Ki value of 1.89 nmol/L was identified as the most promising inhibitor of porcine cytochrome bc1 complex, 157-fold improved binding affinity compared to the commercially available bc1 inhibitor Azoxystrobin (AZ). In addition, most of the new compounds displayed excellent fungicidal activity against Sphaerotheca fuliginea at the concentration of 200 µmol/L. The present work indicates that strobilurin analogues containing benzophenone side chains may be the ideal leads for future fungicide discovery.
Co-reporter:Dr. Wen-Chao Yang;Hui Li;Fu Wang;Dr. Xiao-Lei Zhu ; Dr. Guang-Fu Yang
ChemBioChem 2012 Volume 13( Issue 11) pp:1542-1551
Publication Date(Web):
DOI:10.1002/cbic.201200295
Abstract
The cytochrome bc1 complex (complex III, cyt bc1) is an essential component of cellular respiration. Cyt bc1 has three core subunits that are required for its catalytic activity: cytochrome b, cytochrome c1, and the Rieske iron–sulfur protein (ISP). Although most fungicides inhibit this enzyme by binding to the cytochrome b subunit, resistance to these fungicides has developed rapidly due to their widespread application. Resistance is mainly associated with mutations in cytochrome b, the only subunit encoded by mitochondrial DNA. Recently, the flexibility and motion of the ISP and its essential role in electron transfer have received intense attention; this leads us to propose a new classification of cyt bc1 inhibitors (three types of Qo inhibitors) that mobilize, restrict, or fix the rotation of the ISP. Importantly, the strengths of the ISP–inhibitor interactions correlate with inhibitor activity and the development of resistance to Qo inhibitors, thereby offering clues for designing novel cyt bc1 inhibitors with high potency and a low risk of resistance.
Co-reporter:Fu Wang, Hui Li, Le Wang, Wen-Chao Yang, Jia-Wei Wu, Guang-Fu Yang
Bioorganic & Medicinal Chemistry 2011 Volume 19(Issue 15) pp:4608-4615
Publication Date(Web):1 August 2011
DOI:10.1016/j.bmc.2011.06.008
The cytochrome bc1 complex (EC 1.10.2.2, bc1) is one of the most promising targets for new drugs and agricultural fungicides. Among the existing bc1 complex inhibitors specifically binding to the Qo site, oxazolidinedione derivatives have attracted great attention. With the aim to understand the substituent effects of oxazolidinedione derivatives on the inhibition activity against the bc1 complex, a series of new oxazolidinedione derivatives were designed, synthesized, and biologically evaluated. The further inhibitory kinetics studies against porcine succinate–cytochrome c reductase (SCR) revealed that the representative compound 8d and famoxadone are both non-competitive inhibitors with respect to the substrate cytochrome c, but competitive inhibitors with respect to substrate decylubiquinol (DBH2). In addition, compound 8d and famoxadone showed, respectively, 35-fold and 15-fold greater inhibitory activity against the porcine SCR than the porcine bc1 complex, indicating that these two inhibitors not only inhibited the activity of the bc1 complex, but possibly affect the interaction between the complex II and the bc1 complex. To our knowledge, this is the first report that famoxadone and its analogs have effects on the interaction between the complex II and the bc1 complex.Graphical abstract
Co-reporter:Li-Li Jiang, Yang Zuo, Zhi-Fang Wang, Yin Tan, Qiong-You Wu, Zhen Xi, and Guang-Fu Yang
Journal of Agricultural and Food Chemistry 2011 Volume 59(Issue 11) pp:6172-6179
Publication Date(Web):April 25, 2011
DOI:10.1021/jf200616y
Discovery of protoporphyrinogen oxidase (PPO, EC 1.3.3.4) inhibitors has been one of the hottest research areas in the field of herbicide development for many years. As a continuation of our research work on the development of new PPO-inhibiting herbicides, a series of novel N-(benzothiazol-5-yl)-4,5,6,7-tetrahydro-1H-isoindole-1,3(2H)-diones (1a–p) and N-(benzothiazol-5-yl)isoindoline-1,3-diones (2a–h) were designed and synthesized according to the ring-closing strategy of two ortho-substituents. The bioassay results indicated that some newly synthesized compounds exhibited higher PPO inhibition activity than the control of sulfentrazone. Compound 1a, S-(5-(1,3-dioxo-4,5,6,7-tetrahydro-1H-isoindol-2(3H)-yl)-6-fluorobenzothiazol-2-yl) O-methyl carbonothioate, was identified as the most potent inhibitor with ki value of 0.08 μM, about 9 times higher than that of sulfentrazone (ki = 0.72 μM). Further green house assay showed that compound 1b, methyl 2-((5-(1,3-dioxo-4,5,6,7-tetrahydro-1H-isoindol-2(3H)-yl)-6-fluorobenzothiazol-2-yl)thio)acetate, exhibited herbicidal activity comparable to that of sulfentrazone even at a concentration of 37.5 g ai/ha. In addition, among six tested crops, wheat exhibited high tolerance to compound 1b even at a dosage of 300 g ai/ha. These results indicated that compound 1b might have the potential to be developed as a new herbicide for weed control of wheat field.
Co-reporter:Ge-Fei Hao;Ying Tan;Ning-Xi Yu
Journal of Computer-Aided Molecular Design 2011 Volume 25( Issue 3) pp:213-222
Publication Date(Web):2011 March
DOI:10.1007/s10822-011-9412-6
Protoporphyrinogen oxidase (PPO, EC 1.3.3.4), which has been identified as a significant target for a great family of herbicides with diverse chemical structures, is the last common enzyme responsible for the seventh step in the biosynthetic pathway to heme and chlorophyll. Among the existing PPO inhibitors, diphenyl-ether is the first commercial family of PPO inhibitors and used as agriculture herbicides for decades. Most importantly, diphenyl-ether inhibitors have been found recently to possess the potential in Photodynamic therapy (PDT) to treat cancer. Herein, molecular dynamics simulations, approximate free energy calculations and hydrogen bond energy calculations were integrated together to uncover the structure–activity relationships of this type of PPO inhibitors. The calculated binding free energies are correlated very well with the values derived from the experimental ki data. According to the established computational models and the results of approximate free energy calculation, the substitution effects at different position were rationalized from the view of binding free energy. Some outlier (e.g. LS) in traditional QSAR study can also be explained reasonably. In addition, the hydrogen bond energy calculation and interaction analysis results indicated that the carbonyl oxygen on position-9 and the NO2 group at position-8 are both vital for the electrostatic interaction with Arg98, which made a great contribution to the binding free energy. These insights from computational simulations are not only helpful for understanding the molecular mechanism of PPO-inhibitor interactions, but also beneficial to the future rational design of novel promising PPO inhibitors.
Co-reporter:Xiao-Lei Zhu;Wen-Chao Yang;Ning-Xi Yu;Sheng-Gang Yang
Journal of Molecular Modeling 2011 Volume 17( Issue 3) pp:495-503
Publication Date(Web):2011 March
DOI:10.1007/s00894-010-0742-4
Herbicides targeting grass plastidic acetyl-CoA carboxylase (ACCase, EC 6.4.1.2) are selectively effective against graminicides. The intensive worldwide use of this herbicide family has selected for resistance genes in a number of grass weed species. Recently, the active-site W374C mutation was found to confer multi-drug resistance toward haloxyfop (HF), fenoxaprop (FR), Diclofop (DF), and clodinafop (CF) in A. myosuroides. In order to uncover the resistance mechanism due to W374C mutation, the binding of above-mentioned four herbicides to both wild-type and the mutant-type ACCase was investigated in the current work by molecular docking and molecular dynamics (MD) simulations. The binding free energies were calculated by molecular mechanics-Poisson-Boltzmann surface area (MM/PBSA) method. The calculated binding free energy values for four herbicides were qualitatively consistent with the experimental order of IC50 values. All the computational model and energetic results indicated that the W374C mutation has great effects on the conformational change of the binding pocket and the ligand-protein interactions. The most significant conformational change was found to be associated with the aromatic amino acid residues, such as Phe377, Tyr161′ and Trp346. As a result, the π-π interaction between the ligand and the residue of Phe377 and Tyr161′, which make important contributions to the binding affinity, was decreased after mutation and the binding affinity for the inhibitors to the mutant-type ACCase was less than that to the wild-type enzyme, which accounts for the molecular basis of herbicidal resistance. The structural role and mechanistic insights obtained from computational simulations will provide a new starting point for the rational design of novel inhibitors to overcome drug resistance associated with W374C mutation.
Co-reporter:Li Zhang, Yin Tan, Neng-Xue Wang, Qiong-You Wu, Zhen Xi, Guang-Fu Yang
Bioorganic & Medicinal Chemistry 2010 Volume 18(Issue 22) pp:7948-7956
Publication Date(Web):15 November 2010
DOI:10.1016/j.bmc.2010.09.036
The characteristics of low application rates, good crop selectivity, low residue and environmental safety exhibited by Protoporphyrinogen oxidase (PPO, EC 1.3.3.4)-inhibiting herbicides have attracted a world-wide research interests. As continuation of our research work on the development of new PPO inhibitors, a series of mono-carbonyl analogues of cyclic imides, N-phenyl pyrrolidin-2-ones and N-phenyl-1H-pyrrol-2-ones, were designed and synthesized based on previously established DFT-QSAR results. The PPO inhibition activities of 29 newly synthesized compounds were tested and a predictive comparative molecular field analysis (CoMFA) model was established with the conventional correlation coefficient r2 = 0.980 and the cross-validated coefficient q2 = 0.518. According to the CoMFA model, the substituent effects on the PPO inhibition activity were explained reasonably. Further greenhouse assay showed that 2-(4-chloro-2-fluoro-5-propoxy-phenyl)-2,3,4,5,6,7-hexahydro-isoindol-1-one (C6, ki = 0.095 μM) and 2-(5-allyloxy-4-chloro-2-fluorophenyl)-2,3,4,5,6,7-hexahydro-isoindol-1-one (C7, ki = 0.12 μM) displayed excellent post-emergency herbicidal activity at the concentration of 150 g.ai/ha against seven tested weeds. Due to their high PPO inhibition effect and broad spectrum herbicidal activity, these two compounds have the potential for further study on crop selectivity and field trial. These results confirmed once again that only one of the carbonyl groups of cyclic imides is essential to the PPO inhibition activity.
Co-reporter:Li-Li Jiang, Ying Tan, Xiao-Lei Zhu, Zhi-Fang Wang, Yang Zuo, Qiong Chen, Zhen Xi and Guang-Fu Yang
Journal of Agricultural and Food Chemistry 2010 Volume 58(Issue 5) pp:2643-2651
Publication Date(Web):December 2, 2009
DOI:10.1021/jf9026298
Protoporphyrinogen oxidase (PPO, EC 1.3.3.4) has been identified as one of the most significant action targets for a large chemically diverse family of herbicides that exhibit some interesting characteristics, such as low use rate, low toxicity to mammals, and low environmental impact. As a continuation of research work on the development of new PPO inhibitors, some benzothiazole analogues of oxadiargyl, an important PPO-inhibiting commercial herbicide, were designed and synthesized by ring-closing of the substituents at the C-4 and C-5 positions. The bioassay results indicated that the series 8, 9, and 10 have good PPO inhibition activity with ki values ranging from 0.25 to 18.63 μM. Most interestingly, 9l, ethyl 2-((5-(5-tert-butyl-2-oxo-1,3,4-oxadiazol-2(3H)-yl)-6-fluorobenzothiazol-2-yl)sulfanyl) propanoate, was identified as the most promising candidate due to its high PPO inhibition effect (ki = 1.42 μM) and broad spectrum postemergence herbicidal activity at the concentration of 37.5 g of ai/ha.
Co-reporter:Chao-Nan Chen, Qiong Chen, Yu-Chao Liu, Xiao-Lei Zhu, Cong-Wei Niu, Zhen Xi, Guang-Fu Yang
Bioorganic & Medicinal Chemistry 2010 Volume 18(Issue 14) pp:4897-4904
Publication Date(Web):15 July 2010
DOI:10.1016/j.bmc.2010.06.015
The triazolopyrimidine-2-sulfonanilide, discovered from preparing bioisosteres of the sulfonylurea herbicides, is an important class of acetohydroxyacid synthase (AHAS, EC 4.1.3.18) inhibitors. At least over ten triazolopyrimidine sulfonanilides have been commercialized as herbicides for the control of broadleaf weeds and grass with cereal crop selectivity. Herein, a series of triazolopyrimidine-2-sulfonanilides were designed and synthesized with the aim of discovery of new herbicides with higher activity. The assay results of the inhibition activity of the synthesized compounds against Arabidopsis thatiana AHAS indicated that some compounds showed a little higher activity against flumetsulam (FS), the first commercial triazolopyrimidine-2-sulfonanilide-type herbicide. The ki values of two promising compounds 3d and 8h are respectively, 1.61 and 1.29 μM, while that of FS is 1.85 μM. Computational simulation results indicated the ester group of compound 3d formed hydrogen bonds with the surrounding residues Arg’198 and Ser653, which accounts for its 11.5-folds higher AHAS inhibition activity than Y6610. Further green house assay showed that compound 3d has comparable herbicidal activity as FS. Even at the concentration of 37.5 g.ai/ha, 3d showed excellent herbicidal activity against Galium aparine, Cerastium arvense, Chenopodium album, Amaranthus retroflexus, and Rμmex acetasa, moderate herbicidal activity against Polygonum humifusum, Cyperus iria, and Eclipta prostrate. The combination of in vitro and in vivo assay indicated that 3d could be regarded as a new potential acetohydroxyacid synthase-inhibiting herbicide candidate for further study.
Co-reporter:Ge-Fei Hao, Guang-Fu Yang and Chang-Guo Zhan
The Journal of Physical Chemistry B 2010 Volume 114(Issue 29) pp:9663-9676
Publication Date(Web):July 6, 2010
DOI:10.1021/jp102546s
The drug resistance of various clinically available HIV-1 protease inhibitors has been studied using a new computational protocol, that is, computational mutation scanning (CMS), leading to valuable insights into the resistance mechanisms and structure-resistance correction of the HIV-1 protease inhibitors associated with a variety of active site and nonactive site mutations. By using the CMS method, the calculated mutation-caused shifts of the binding free energies linearly correlate very well with those derived from the corresponding experimental data, suggesting that the CMS protocol may be used as a generalized approach to predict drug resistance associated with amino acid mutations. Because it is essentially important for understanding the structure−resistance correlation and for structure-based drug design to develop an effective computational protocol for drug resistance prediction, the reasonable and computationally efficient CMS protocol for drug resistance prediction should be valuable for future structure-based design and discovery of antiresistance drugs in various therapeutic areas.
Co-reporter:Pei-Liang Zhao ; Le Wang ; Xiao-Lei Zhu ; Xiaoqin Huang ; Chang-Guo Zhan ; Jia-Wei Wu
Journal of the American Chemical Society 2009 Volume 132(Issue 1) pp:185-194
Publication Date(Web):November 23, 2009
DOI:10.1021/ja905756c
Cytochrome bc1 complex (EC 1.10.2.2, bc1), an essential component of the cellular respiratory chain and the photosynthetic apparatus in photosynthetic bacteria, has been identified as a promising target for new drugs and agricultural fungicides. X-ray diffraction structures of the free bc1 complex and its complexes with various inhibitors revealed that the phenyl group of Phe274 in the binding pocket exhibited significant conformational flexibility upon different inhibitors binding to optimize respective π−π interactions, whereas the side chains of other hydrophobic residues showed conformational stability. Therefore, in the present study, a strategy of optimizing the π−π interaction with conformationally flexible residues was proposed to design and discover new bc1 inhibitors with a higher potency. Eight new compounds were designed and synthesized, among which compound 5c, with a Ki value of 570 pM, was identified as the most promising drug or fungicide candidate, significantly more potent than the commercially available bc1 inhibitors, including azoxystrobin (AZ), kresoxim-methyl (KM), and pyraclostrobin (PY). To our knowledge, this is the first bc1 inhibitor discovered from structure-based design with a potency of subnanomolar Ki value. For all of the compounds synthesized and assayed, the calculated binding free energies correlated reasonably well with the binding free energies derived from the experimental Ki values, with a correlation coefficient of r2 = 0.89. The further inhibitory kinetics studies revealed that 5c is a noncompetitive inhibitor with respect to substrate cytochrome c, but it is a competitive inhibitor with respect to substrate ubiquinol. Due to its subnanomolar Ki potency and slow dissociation rate constant (k−0 = 0.00358 s−1), 5c could be used as a specific probe for further elucidation of the mechanism of bc1 function and as a new lead compound for future drug discovery.
Co-reporter:Xiao-Lei Zhu, Hao Ge-Fei, Chang-Guo Zhan and Guang-Fu Yang
Journal of Chemical Information and Modeling 2009 Volume 49(Issue 8) pp:1936-1943
Publication Date(Web):July 13, 2009
DOI:10.1021/ci900174d
Grass weed populations resistant to acetyl-CoA carboxylase-inhibiting (ACCase; EC 6.4.1.2) herbicides represent a major problem for the sustainable development of modern agriculture. In the present study, extensive computational simulations, including homology modeling, molecular dynamics (MD) simulations, and molecular mechanics-Poisson−Boltzmann surface area (MM/PBSA) calculations, have been carried out to uncover the detailed molecular mechanism of Alopecurus myosuroides resistance to clodinafop, a commercial herbicide targeting ACCase. All the computational model and energetic results indicated that W374C, I388N, D425G, and G443A mutations have great effects on the conformational change of the binding pocket and the hydrogen-bonding interactions. The π−π interaction between ligand and the residue of Phe377 and Tyr161′, playing an important contribution to the binding affinity, were decreased after mutations. In addition, the hydrogen-bonding interactions between clodinafop and the residues (Ile158′ and Ala54′) disappeared or decreased significantly upon mutation. As a result, the mutant-type ACCase has a lower affinity for the inhibitor binding than the wild-type enzyme, which accounts for the molecular basis of herbicidal resistance. The structural and mechanistic insights obtained from the present study will provide a valuable clue for future designing of a promising inhibitor to reduce drug resistance associated with both active and nonactive site mutations.
Co-reporter:Wei Huang, Yu Ding, Yan Miao, Ming-Zhen Liu, Yan Li, Guang-Fu Yang
European Journal of Medicinal Chemistry 2009 Volume 44(Issue 9) pp:3687-3696
Publication Date(Web):September 2009
DOI:10.1016/j.ejmech.2009.04.004
A series of chromone derivatives bearing diverse dithiocarbamate moieties were designed and synthesized via a three-component reaction protocol. Their in vitro antitumor activities were evaluated by MTT method against HCCLM-7, Hela, MDA-MB-435S, SW-480, Hep-2 and MCF-7. Two compounds (3-chloro-4-oxo-4H-chromen-2-yl)methyl piperidine-1-carbodithioate (Iq) and (6-chloro-4-oxo-4H-chromen-3-yl)methyl piperidine-1-carbodithioate (IIu), were identified as the most promising candidate due to their high potency and broad-spectrum. Further flow-activated cell sorting analysis revealed that compounds Iq and IIu arrest the cell cycle of SW-480 and MDA-MB-435s both in G2/M phase with dose-dependent effect and might display apoptosis-inducing effect on these tumor cell lines.A series of dithiocarbamate substituted chromones were synthesized and evaluated for their antitumor activities against cancer cell lines including HCCLM-7, Hela, MDA-MB-435S, SW-480, Hep-2 and MCF-7.
Co-reporter:Li Zhang, Ge-Fei Hao, Yin Tan, Zhen Xi, Ming-Zhi Huang, Guang-Fu Yang
Bioorganic & Medicinal Chemistry 2009 Volume 17(Issue 14) pp:4935-4942
Publication Date(Web):15 July 2009
DOI:10.1016/j.bmc.2009.06.003
Bioactive conformation of drugs is one of the key points for understanding the ligand–receptor interactions. In the present study, by combining density functional theory-based (DFT-based) conformation analysis with quantitative structure–activity relationship analysis (QSAR), we developed successfully a new approach (DFT/QSAR) to carry out bioactive conformation analyses for a series of 25 cyclic imide derivatives as protoporphyrinogen oxidase (PPO) inhibitors. Further potential energy surface scan, molecular docking and molecular dynamic simulation calculations validated that the DFT/QSAR-derived conformation is indeed very similar to the ‘real’ bioactive conformation. We believe the DFT/QSAR approach provides a simple alternative for the bioactive conformation of small molecules, especially in the case that the three-dimensional structure of protein is unknown.
Co-reporter:Chao-Nan Chen, Li-Li Lv, Feng-Qin Ji, Qiong Chen, Hui Xu, Cong-Wei Niu, Zhen Xi, Guang-Fu Yang
Bioorganic & Medicinal Chemistry 2009 Volume 17(Issue 8) pp:3011-3017
Publication Date(Web):15 April 2009
DOI:10.1016/j.bmc.2009.03.018
Triazolopyrimidine-2-sulfonamide belongs to a herbicide group called acetohydroxyacid synthase inhibitors. With the aim to discover new triazolopyrimidine sulfonanilide compounds with high herbicidal activity and faster degradation rate in soil, the methyl group of Flumetsulam (FS) was modified into a methoxy group to produce a new herbicidal compound, N-2,6-difluorophenyl-5-methoxy-1,2,4-triazolo[1,5-a]pyrimidine-2-sulfonamide (experimental code: Y6610). The enzymatic kinetic results indicated that compound Y6610 and FS have ki values of 3.31 × 10−6 M and 3.60 × 10−7 M against Arabidopsis thaliana AHAS, respectively. The 10-fold lower enzyme-inhibiting activity of Y6610 was explained rationally by further computational simulations and binding free energy calculations. In addition, compound Y6610 was found to display the same level in vivo post-emergent herbicidal activity as FS against some broad-leaf weeds and good safety to rice, maize, and wheat at the dosages of 75–300 g ai/ha. Further determination of the half-lives in soil revealed that the half-life in soil of Y6610 is 3.9 days shorter than that of FS. The experimental results herein showed that compound Y6610 could be regarded as a new potential acetohydroxyacid synthase-inhibiting herbicide candidate for further study.
Co-reporter:Qiong Chen;Zu-Ming Liu;Chao-Nan Chen;Li-Li Jiang
Chemistry & Biodiversity 2009 Volume 6( Issue 8) pp:1254-1265
Publication Date(Web):
DOI:10.1002/cbdv.200800168
Abstract
A series of new acetohydrazone-containing 1,2,4-triazolo[1,5-a]pyrimidine derivatives were designed and synthesized for the purpose of searching for novel agrochemicals with higher fungicidal activity. Their in vitro fungicidal activities against Rhizoctonia solani were evaluated, and the most promising compound, 2-[(5,7-dimethyl[1,2,4]triazolo[1,5-a]pyrimidin-2-yl)sulfanyl]-2′-[(2-hydroxyphenyl)methylidene]acetohydrazide (2-17), showed a lower EC50 value (5.34 μg ml−1) than that of commercial carbendazim (EC50=7.62 μg ml−1). Additionally, compound 2-17 was also found to display broad-spectrum fungicidal activities, and its EC50 value (4.56 μg ml−1) against Botrytis cinereapers was very similar to that of carbendazim. Qualitative structure–activity relationships (QSARs) of the synthesized compounds were also discussed.
Co-reporter:Qiong Chen;Li-Li Jiang;Chao-Nan Chen
Journal of Heterocyclic Chemistry 2009 Volume 46( Issue 2) pp:139-148
Publication Date(Web):
DOI:10.1002/jhet.1
Co-reporter:Ge-Fei Hao, Xiao-Lei Zhu, Feng-Qin Ji, Li Zhang, Guang-Fu Yang and Chang-Guo Zhan
The Journal of Physical Chemistry B 2009 Volume 113(Issue 14) pp:4865-4875
Publication Date(Web):March 16, 2009
DOI:10.1021/jp807442n
Protoporphyrinogen oxidase (PPO; EC 1.3.3.4) is the last common enzyme for the enzymatic transformation of protoporphyrinogen-IX to protoporphyrin-IX, which is the key common intermediate leading to heme and chlorophyll. Hence, PPO has been identified as one of the most importance action targets for the treatment of some important diseases including cancer and variegated porphyria (VP). In the agricultural field, PPO inhibitors have been used as herbicides for many years. Recently, a unique drug resistance was found to be associated with a nonactive site residue (Gly210) deletion rather than substitution in A. tuberculatus PPO. In the present study, extensive computational simulations, including homology modeling, molecular dynamics (MD) simulations, and molecular mechanics-Poisson−Boltzmann surface area (MM-PBSA) calculations, have been carried out to uncover the detailed molecular mechanism of drug resistance associated with Gly210 deletion. Although Gly210 in the wild-type A. tuberculatus PPO has no direct interaction with the inhibitors, all the computational models and energetic results indicated that Gly210 deletion has great effects on the hydrogen-bonding network and the conformational change of the binding pocket. An interchain hydrogen bond between Gly210 with Ser424, playing an important role in stabilizing the local conformation of the wild-type enzyme, disappeared after Gly210 deletion. As a result, the mutant-type PPO has a lower affinity than the wild-type enzyme, which accounts for the molecular mechanism of drug resistance. The structural and mechanistic insights obtained from the present study provide a new starting point for future rational design of novel PPO inhibitors to overcome drug resistance associated with Gly210 deletion.
Co-reporter:Guorui Li, Jing Huang, Ming Zhang, Yangyang Zhou, Dan Zhang, Zhiguo Wu, Shaoru Wang, Xiaocheng Weng, Xiang Zhou and Guangfu Yang
Chemical Communications 2008 (Issue 38) pp:4564-4566
Publication Date(Web):04 Aug 2008
DOI:10.1039/B807916A
Two new bis(benzimidazole)aryl derivatives have been prepared and one of them has been shown to induce and stabilize formation of a G-quadruplex.
Co-reporter:Pei-Liang Zhao, Fu Wang, Ming-Zhi Zhang, Zu-Ming Liu, Wei Huang and Guang-Fu Yang
Journal of Agricultural and Food Chemistry 2008 Volume 56(Issue 22) pp:10767-10773
Publication Date(Web):October 30, 2008
DOI:10.1021/jf802343p
1-Acetyl-3,-5-diarylpyrazolines have received considerable interests from the fields of medicinal and agricultural chemistry due to their broad spectrum of biological activities. To discover new lead compounds exhibiting both fungicidal and insecticidal activities, a series of pyrazoline derivatives were designed and synthesized by introducing the β-methoxyacrylate pharmacophore into the scaffold of 1-acetyl-3,5-diarylpyrazoline. The fungicidal activities against Pseudoperoniospora cubensis, Sphaerotheca fuliginea, Botrytis cinerea, and Rhizoctonia solani and the insecticidal activities against Aphis medicagini, Nilaparvata legen, Mythima separata, and Tetranychus cinnabarnus were screened. The most potent compound 13, 1-aceto-3-{m-[o-(E-1-methoxycarboxyl-2-methoxy)-1-yl]benzyloxy}phenyl-5-(benzo-[1,3]-dioxolyl)-4,5-dihydro- pyrazoline, was identified. Its fungicidal IC50 values against P. cubensis and S. fuliginea are 26.6 and 57.6 μg mL−1, respectively, while its insecticidal LC50 value against M. separata is 26.6 μg mL−1. These results indicated that compound 13 could be used as a lead for further developing new pyrazoline type products exhibiting both fungicidal and insecticidal activities.
Co-reporter:Yan-Ping Luo, Li-Li Jiang, Guo-Dong Wang, Qiong Chen and Guang-Fu Yang
Journal of Agricultural and Food Chemistry 2008 Volume 56(Issue 6) pp:2118-2124
Publication Date(Web):February 26, 2008
DOI:10.1021/jf703654g
Protoporphyrinogen oxidase (Protox, EC 1.3.3.4) has been identified as one of the most important action targets of herbicides. To search for novel Protox inhibitors, a series of title compounds 1, 2, and 3 were designed and synthesized by introducing three types of pharmacophores, cyclic imide, phenylurea, and (E)-methyl 2-methoxyimino-2-o-tolylacetate, into the scaffold of triazolinone. The bioassay results indicated that the resulting cyclic imide-type triazolinones 1 displayed much better herbicidal activities than phenylurea-type triazolinones 2. Most fortunately, compound 3, methyl 2-[3-methyl-(2-fluoro-4-chloro-5-ethylsulfonamidephenyl)-4,5-dihydro-5-oxo-1H-1,2,4-triazol-4-yl]methylenephenyl-2-(E)-methoxyiminoacetate, was found to be the most promising candidate due to its comparable herbicidal activity at 75−150 g of active ingredient/ha with the commercial product sulfentrazone. On the basis of test results of herbicidal spectrum and crop selectivity, compound 3 could be developed as a postemergent herbicide used for the control of broadleaf weeds in rice fields.
Co-reporter:Feng-Qin Ji;Cong-Wei Niu;Chao-Nan Chen;Qiong Chen ;Zhen Xi ;Chang-Guo Zhan
ChemMedChem 2008 Volume 3( Issue 8) pp:1203-1206
Publication Date(Web):
DOI:10.1002/cmdc.200800103
Co-reporter:Boqiao Fu, Jing Huang, Lige Ren, Xiaocheng Weng, Yangyang Zhou, Yuhao Du, Xiaojun Wu, Xiang Zhou and Guangfu Yang
Chemical Communications 2007 (Issue 31) pp:3264-3266
Publication Date(Web):06 Jun 2007
DOI:10.1039/B704599A
Water-soluble cationic corrole derivatives were designed and synthesized, and the first observation of their interactions with the telomeric G-quadruplex was made.
Co-reporter:Hui Xu, Wenhui Pan, Dandan Song and Guangfu Yang
Journal of Agricultural and Food Chemistry 2007 Volume 55(Issue 23) pp:9351-9356
Publication Date(Web):October 23, 2007
DOI:10.1021/jf0718345
An improved liquid phase microextraction (LPME) technique has been developed. As part of this technique, analytes were extracted into an extractant microdrop which was laid on the cone-shaped bottom of a PCR tube (polychloroprene rubber tube) but not at the needle tip of a microsyringe, and the sample vial and PCR tube were horizontally placed so that the extractant was not affected by the force of vertical orientation (gravity and floating force). The stability of the extractant microdrop increased greatly, and the selection of extractant was extended. In this work, flumetsulam and its two analogous herbicides were chosen as model analytes in investigating the feasibility of the new pretreatment method by coupling it to high-performance liquid chromatography (HPLC). Under the optimized experimental conditions, the linear range and the limits of detection (S/N = 3) were 0.01–5 µg/mL (r = 0.9997) and 0.8 ng/mL for flumetsulam, 0.002–5 µg/mL (r = 0.9994) and 0.5 ng/mL for analogue 1, and 0.002–1 µg/mL (r = 0.9993) and 0.5 ng/mL for analog 2, respectively. The inter- and intraday reproducibilities (RSD) were below 5.3 and 4.5%, respectively. Good recoveries that ranged from 79.4 to 115.0% were obtained in the analysis of real soil samples. The extraction efficiency of the improved method was 4–8 times higher than that of the conventional liquid phase microextraction method. The novel, simple, rapid, sensitive technique is very suitable for extraction of apolar and medium polar analyte in complex environmental samples.
Co-reporter:Wei Huang, Ming-Zhen Liu, Yan Li, Ying Tan, Guang-Fu Yang
Bioorganic & Medicinal Chemistry 2007 Volume 15(Issue 15) pp:5191-5197
Publication Date(Web):1 August 2007
DOI:10.1016/j.bmc.2007.05.022
A series of new chromone analogues bearing heterocyclic thioether moiety and aurone analogues bearing cyclic tertiary amine moiety were designed and synthesized under microwave irradiation. The synthetic protocol was found to present many advantages, such as higher yields, shorter reaction time (10–20 min), mild condition, and readily isolation of the products. The synthesized compounds were assayed for their antitumor activity against four kinds of human solid tumor cell lines including HCCLM-7, Hep-2, MDA-MB-435S, and SW-480. Two compounds, (Z)-2-((4-benzyl-piperazin-1-yl)methylene)benzofuran-3(2H)-one 5e and (Z)-2-((4-(bis(4-fluorophenyl)methyl)piperazin-1-yl)methylene)benzofuran-3(2H)-one 5f, were identified as the most promising candidates with the IC50 values in the range of 4.1–13.1 μM. Further cell cycle studies revealed that compounds 5e and 5f arrest the cell cycle in G0/G1 phase and displayed apoptosis-inducing effect on Hep-2 cells.
Co-reporter:Long Lin;Yan-Ping Luo
Journal of Heterocyclic Chemistry 2007 Volume 44(Issue 4) pp:937-943
Publication Date(Web):13 MAR 2009
DOI:10.1002/jhet.5570440433
A novel series of 1-(4-chlorophenyl)-4-{[5-(alkylthio)-4-phenyl-4H-1,2,4-triazol-3-yl]methyl}-1,4-dihydro-5H-tetrazol-5-ones 6a-y were synthesized in good to excellent yields and their structures were identified by 1H nmr, 13C nmr, ms and elemental analysis. Determining the X-ray crystallography of compound 6j indicated that there were strong intermolecular hydrogen bonds in the stacking interactions. The bioassay results showed that compound 6k exhibited good insecticidal activities against T cinnabarinus at the dosage of 250 mg.L−1. To our knowledge, this is the first report about the insecticidal activity of tetrazolinone derivatives.
Co-reporter:Ming-Wu Ding;Zhong-Zheng Zhou;Feng-Qin Ji;Wei Huang
Heteroatom Chemistry 2007 Volume 18(Issue 4) pp:381-389
Publication Date(Web):2 MAY 2007
DOI:10.1002/hc.20309
A one-pot liquid-phase combinatorial synthesis of 2-(4-oxo-4H-1-benzopyran-3-yl)-4-thiazolidinones bearing diverse substituents at the 3-position under microwave irradiation was successfully performed using 3-formyl chromone, primary amine, and mercaptoacetic acid as reactants. Compared to an identical library generated by conventional parallel synthesis, the microwave-assisted parallel synthesis approach dramatically decreased the reaction time from an average of 9 h to 5 min, and substantially increased the product yields. The coupling of microwave technology with liquid-phase combinatorial synthesis constitutes a novel and particularly attractive avenue for the rapid generation of structurally diverse libraries. © 2007 Wiley Periodicals, Inc. Heteroatom Chem 18:381–389, 2007; Published online in Wiley InterScience (www.interscience.wiley.com). DOI 10.1002/hc.20309
Co-reporter:Zhong-Zhen Zhou;Feng-Qing Ji;Min Cao
Advanced Synthesis & Catalysis 2006 Volume 348(Issue 14) pp:
Publication Date(Web):12 SEP 2006
DOI:10.1002/adsc.200606156
This communication describes the first report of a microwave-assisted intramolecular Stetter reaction using imidazolium-type room temperature ionic liquids (RTILs) as solvents, with thiazolium salts and Et3N as catalysts. The features such as good to excellent yields, shorter reaction time (5–20 min), and recyclable and reusable ionic liquid and catalyst make this method an environmentally benign and highly efficient procedure for the preparation of chromanone derivatives.
Co-reporter:Zhongzhen Zhou;Peiliang Zhao;Wei Huang;Guangfu Yang
Advanced Synthesis & Catalysis 2006 Volume 348(Issue 1-2) pp:
Publication Date(Web):19 JAN 2006
DOI:10.1002/adsc.200505223
This paper presents the first report of a highly selective transformation of flavanones to 3-bromoflavones or flavones by microwave irradiation of the corresponding flavanone reactants and N-bromosuccinimide (NBS) in the presence of a catalytic amount of 2,2′-azobis(isobutyronitrile) (AIBN). The combination of good to excellent yields, shorter reaction time (10 min), and high levels of functional group compatibility make this an attractive synthetic approach to 3-bromoflavones and flavones.
Co-reporter:Ju-Zhen Yuan;Bo-Qiao Fu;Ming-Wu Ding
European Journal of Organic Chemistry 2006 Volume 2006(Issue 18) pp:
Publication Date(Web):19 JUL 2006
DOI:10.1002/ejoc.200600201
Iminophosphorane 1 reacted with an aromatic isocyanate to unexpectedly give a mixture of carbodiimides 2, 3 and 4 through both the normal and the abnormal aza-Wittig reactions. 3-Aminoimidazolone 10 was obtained from the reaction of hydrazine hydrate with carbodiimide 2. Reaction of 10 with triphenyphosphane, hexachloroethane and triethylamine produced iminophosphorane 11 in good yield. A tandem aza-Wittig reaction of iminophosphorane 11 with isocyanate or CS2 generated 3,5-dihydro-6H-imidazo[1,2-b]-1,2,4-triazol-6-ones 13 or 15 in satisfactory yield. Carbodiimides 18, obtained from normal aza-Wittig reactions of vinyl iminophosphorane 17 with aromatic isocyanates, reacted with hydrazine to give 2-arylamino-3-amino-4H-imidazol-4-ones 20. One-pot reactions of 2-arylamino-3-amino-4H-imidazol-4-ones 20 with isocyanates (or acyl chlorides), triphenyphosphane, hexachloroethane and triethylamine produced 3,5-dihydro-6H-imidazo[1,2-b]-1,2,4-triazol-6-ones 22 (or 23) in good yields. (© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2006)
Co-reporter:Ge-Fei Hao, Guang-Fu Yang, Chang-Guo Zhan
Drug Discovery Today (October 2012) Volume 17(Issues 19–20) pp:1121-1126
Publication Date(Web):1 October 2012
DOI:10.1016/j.drudis.2012.06.018
Drug resistance has become one of the biggest challenges in drug discovery and/or development and has attracted great research interests worldwide. During the past decade, computational strategies have been developed to predict target mutation-induced drug resistance. Meanwhile, various molecular design strategies, including targeting protein backbone, targeting highly conserved residues and dual/multiple targeting, have been used to design novel inhibitors for combating the drug resistance. In this article we review recent advances in development of computational methods for target mutation-induced drug resistance prediction and strategies for rational design of novel inhibitors that could be effective against the possible drug-resistant mutants of the target.Highlights► Recently developed structure-based computational methods are capable of predicting target mutation-induced drug resistance. ► It is possible to account for possible mutation-induced drug resistance during the drug discovery. ► Various molecular design strategies have been used to rationally design novel drugs with reduced resistance risk. ► The use of structure-based methods will become more and more popular in the battle against drug resistance.
Co-reporter:Hu Zhang, Peng-Fei Liu, Qiong Chen, Qiong-You Wu, Anne Seville, Yu-Cheng Gu, John Clough, Shao-Lin Zhou and Guang-Fu Yang
Organic & Biomolecular Chemistry 2016 - vol. 14(Issue 13) pp:NaN3485-3485
Publication Date(Web):2016/03/08
DOI:10.1039/C6OB00371K
The synthesis of albucidin and its enantiomer are described. It involves a visible-light photocatalysis deiodination at the late stage. The absolute configuration of natural albucidin is determined as (1R,3S). This work provides a basis for structural modification to develop a new type of herbicidal from an old structure.
Co-reporter:Shu-Hou Yang, Qi Sun, Hao Xiong, Shi-Yu Liu, Behrooz Moosavi, Wen-Chao Yang and Guang-Fu Yang
Chemical Communications 2017 - vol. 53(Issue 28) pp:NaN3955-3955
Publication Date(Web):2017/03/14
DOI:10.1039/C7CC00577F
We report herein the structure-based design and application of a fluorogenic molecular probe (BChE-FP) specific to butyrylcholinesterase (BChE). This probe was rationally designed by mimicking the native substrate and optimized stepwise by manipulating the steric feature and the reactivity of the designed probe targeting the structural difference of the active pockets of BChE and AChE. The refined probe, BChE-FP, exhibits high specificity toward BChE compared to AChE, producing about 275-fold greater fluorescence enhancement upon the catalysis by BChE. Thus, BChE-FP is a specific BChE probe identified by the structure-based design and it can discriminate BChE from AChE. Furthermore, it has been successfully applied for imaging the endogenous BChE in living cells, as well as BChE inhibitor screening and characterization under physiological conditions.
Co-reporter:Shu-Hou Yang, Qi Sun, Hao Xiong, Shi-Yu Liu, Behrooz Moosavi, Wen-Chao Yang and Guang-Fu Yang
Chemical Communications 2017 - vol. 53(Issue 28) pp:NaN3955-3955
Publication Date(Web):2017/03/14
DOI:10.1039/C7CC00577F
We report herein the structure-based design and application of a fluorogenic molecular probe (BChE-FP) specific to butyrylcholinesterase (BChE). This probe was rationally designed by mimicking the native substrate and optimized stepwise by manipulating the steric feature and the reactivity of the designed probe targeting the structural difference of the active pockets of BChE and AChE. The refined probe, BChE-FP, exhibits high specificity toward BChE compared to AChE, producing about 275-fold greater fluorescence enhancement upon the catalysis by BChE. Thus, BChE-FP is a specific BChE probe identified by the structure-based design and it can discriminate BChE from AChE. Furthermore, it has been successfully applied for imaging the endogenous BChE in living cells, as well as BChE inhibitor screening and characterization under physiological conditions.
Co-reporter:Boqiao Fu, Jing Huang, Lige Ren, Xiaocheng Weng, Yangyang Zhou, Yuhao Du, Xiaojun Wu, Xiang Zhou and Guangfu Yang
Chemical Communications 2007(Issue 31) pp:NaN3266-3266
Publication Date(Web):2007/06/06
DOI:10.1039/B704599A
Water-soluble cationic corrole derivatives were designed and synthesized, and the first observation of their interactions with the telomeric G-quadruplex was made.
Co-reporter:Guorui Li, Jing Huang, Ming Zhang, Yangyang Zhou, Dan Zhang, Zhiguo Wu, Shaoru Wang, Xiaocheng Weng, Xiang Zhou and Guangfu Yang
Chemical Communications 2008(Issue 38) pp:NaN4566-4566
Publication Date(Web):2008/08/04
DOI:10.1039/B807916A
Two new bis(benzimidazole)aryl derivatives have been prepared and one of them has been shown to induce and stabilize formation of a G-quadruplex.



![ETHANONE, 2,2,2-TRIFLUORO-1-[4-(TRIFLUOROMETHOXY)PHENYL]-](http://img.cochemist.com/ccimg/107800/107713-64-4.png)
![ETHANONE, 2,2,2-TRIFLUORO-1-[4-(TRIFLUOROMETHOXY)PHENYL]-](http://img.cochemist.com/ccimg/107800/107713-64-4_b.png)


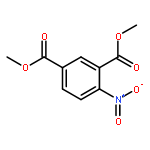


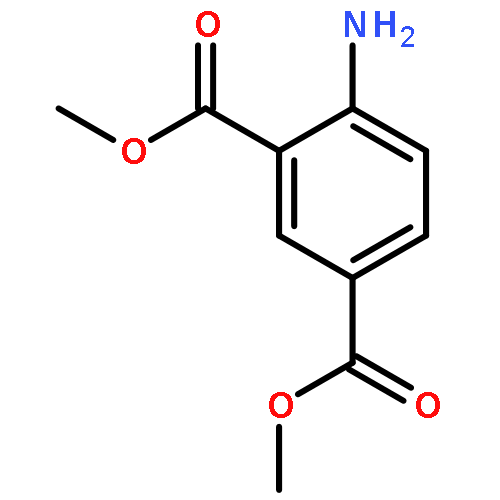









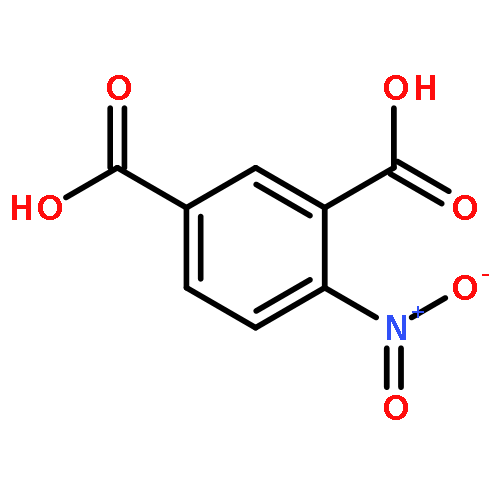




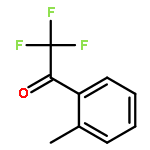





![5-ethyl-6-octyltriazolo[1,5-a]pyrimidin-7-amine](http://img.cochemist.com/ccimg/865400/865318-97-4.png)
![5-ethyl-6-octyltriazolo[1,5-a]pyrimidin-7-amine](http://img.cochemist.com/ccimg/865400/865318-97-4_b.png)

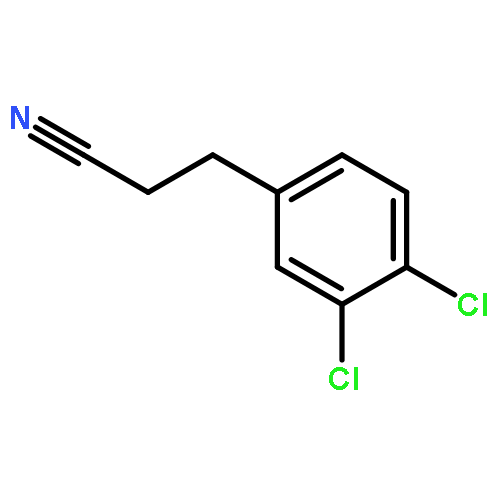




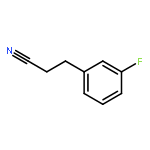



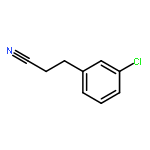

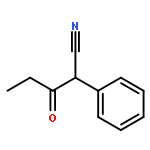
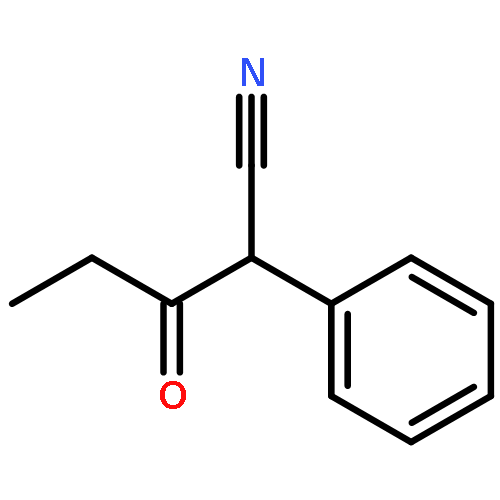




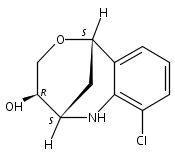
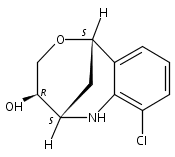
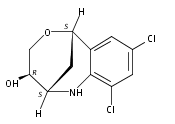
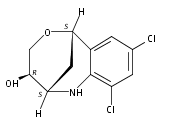
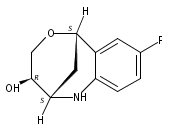
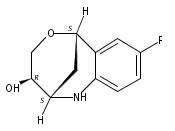
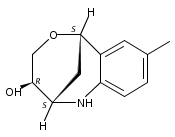
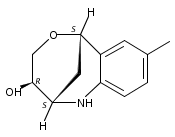
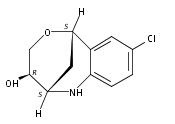
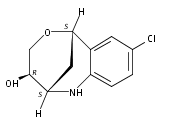
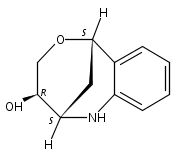

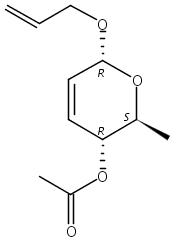
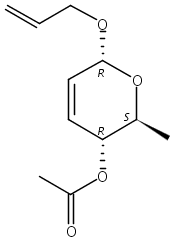


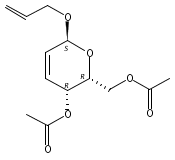
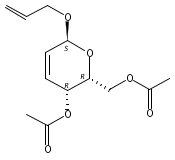
![Carbamic acid, [2-(bromomethyl)phenyl]methoxy-, methyl ester](http://img.cochemist.com/ccimg/151900/151827-83-7.png)
![Carbamic acid, [2-(bromomethyl)phenyl]methoxy-, methyl ester](http://img.cochemist.com/ccimg/151900/151827-83-7_b.png)
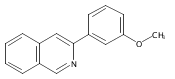
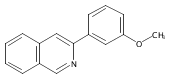


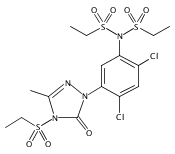

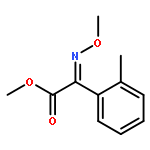
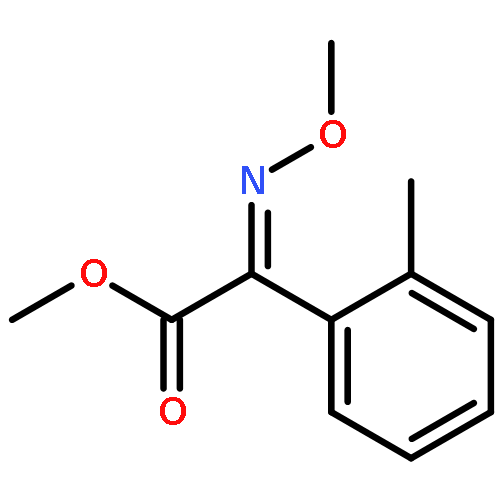
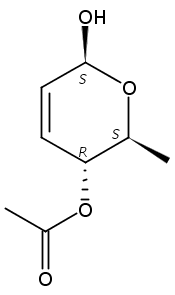
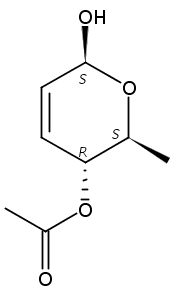
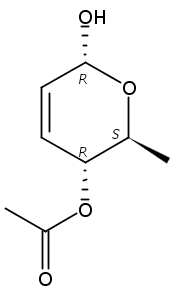
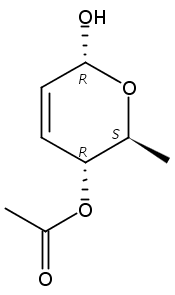
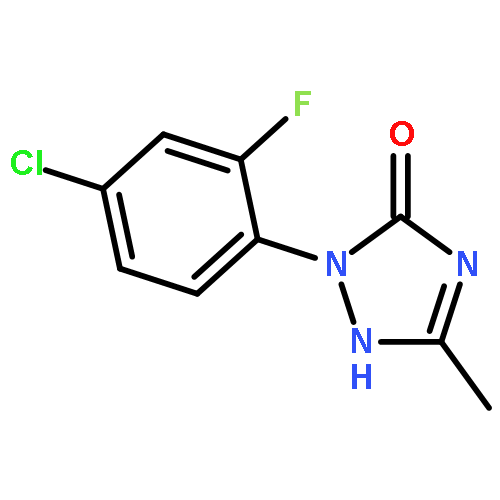
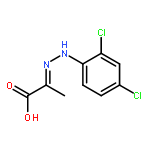
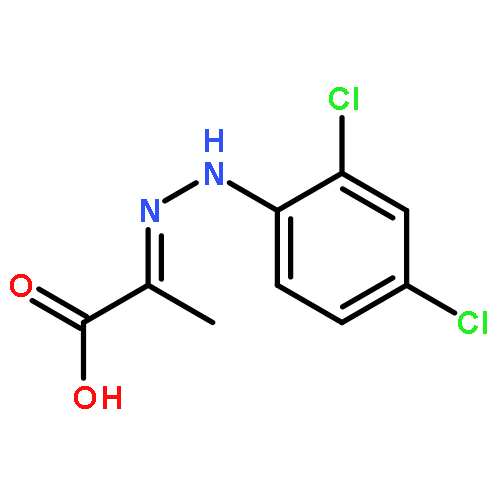
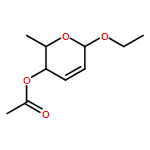
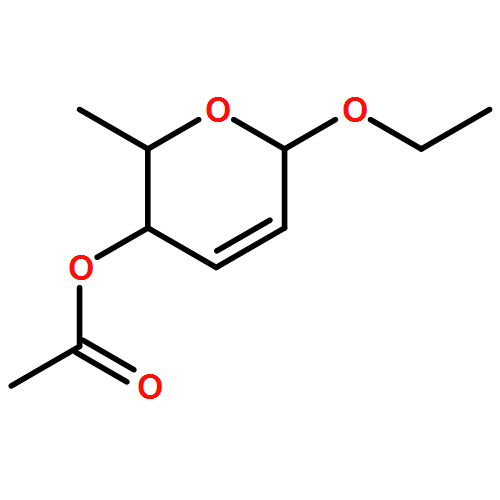
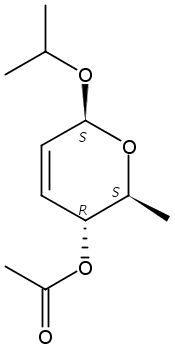
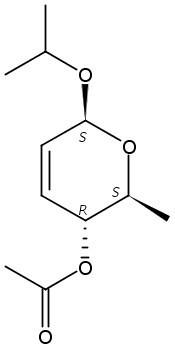
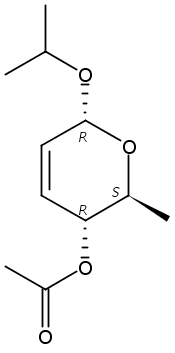
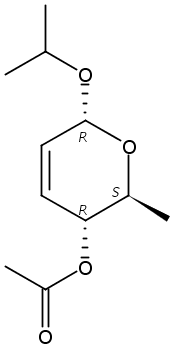







{kind=link}