Co-reporter:Yanxian Hou, Jingbo Shao, Qiang Fu, Jingru Li, Jin Sun, Zhonggui He
International Journal of Pharmaceutics 2017 Volume 516, Issues 1–2(Issue 1) pp:
Publication Date(Web):10 January 2017
DOI:10.1016/j.ijpharm.2016.11.043
For a highly hydrophobic and drug, it is difficult to formulate and solidify its nanocrystals with high drug loading and good redispersity. In this study, Allisartan Isoproxil was used as a model drug, and SDS was tested in combination with sugar alcohols to improve the drug loading and redispersity for its spray-dried nanocrystals, simultaneously. These spray-dried nanocrystals had high drug loading of 61.7% and good redispersity, which was mainly attributed to the addition of SDS. In addition, the nanocrystals were characterized by scanning electron microscopy, differential scanning calorimetry, X-ray power diffraction analysis, Fourier transform infrared spectroscopy and Raman spectroscopy. The results showed that Allisartan Isoproxil was unchanged in chemical structure, but was partially amorphous. Regarding the in vitro dissolution, the optimism formulation shown an increased dissolution compared with the bulk drug and aggregated nanocrystals. Importantly, the optimum formulation increased the oral bioavailability of crude ALS-3 for 4.73 times. In conclusion, we developed a method to solidify aqueous nanocrystals with increased drug loading, good redispersity and improved bioavailability for high hydrophobic drugs.Download high-res image (104KB)Download full-size image
Co-reporter:Rui Fan, Yinghua Sun, Bing Li, Ruyi Yang, ... Jin Sun
Asian Journal of Pharmaceutical Sciences 2017 Volume 12, Issue 2(Volume 12, Issue 2) pp:
Publication Date(Web):1 March 2017
DOI:10.1016/j.ajps.2016.09.003
The objective of this study was to prepare tamsulosin hydrochloride-sustained release (TSH-SR) pellets which showed good release stability with frame-controlled method. TSH was added to Eudragit®NE30D and Eudragit®L30D-55 polymers to form drug-loaded inner core. Afterwards, enteric Eudragit®L30D-55 polymer was modified on the surface of it to the final product. Dissolution studies showed that TSH-SR pellets were more stable during the coating process, different curing temperatures and storage conditions compared with TSH pellets produced by film-controlled technique. Appearances and glass transition temperatures (Tgs) of free films and surface morphologies observed by scanning electron microscopy (SEM) of blank sustained release pellets prepared by different ratios of Eudragit®NE30D and Eudragit®L30D-55 further indicated that temperature and relative humidity (RH) were the key factors when Eudragit®NE30D blended with Eudragit®L30D-55 were applied to sustained/controlled release preparations. In addition, SEM identified the surface morphologies of TSH-SR pellets before and after dissolution, which showed intact surface structure and great correlation with release curve respectively.Download high-res image (72KB)Download full-size image
Co-reporter:Jingjing Ma, Yinxian Yang, Yinghua Sun, Jin Sun
Asian Journal of Pharmaceutical Sciences 2017 Volume 12, Issue 4(Volume 12, Issue 4) pp:
Publication Date(Web):1 July 2017
DOI:10.1016/j.ajps.2016.09.008
Azilsartan (AZL), a poorly soluble drug, was considered to be fit for nanocrystals to improve its solubility. Our study intended to prepare AZL nanocrystals by means of bead milling method. Eight stabilizers or their binary combination and the milling time were set to be variable factors to optimize AZL nanosuspension formulation, and six types of freeze-drying supports were investigated to reduce the aggregation of particles during the solidification. AZL nanocrystals with or without sodium deoxycholate (NaDC) as combined stabilizer with Poloxamer 188 (F68) were prepared owning mean particle sizes of about 300 nm and 460 nm. During the screening processes, the formulation containing NaDC showed a smaller particle size and better stability during lyophilization. The irregular shape and crystal form changing in AZL nanocrystals were discovered by various characterizations. And with physical mixture as reference, nanocrystals showed its improvement about in-vitro dissolution and in-vivo bioavailability. In conclusion, the nanocrystals of AZL could be prepared well in our study. Additionally, our results suggested that NaDC was an appreciated excipient on the nanocrystals platform, which can exhibit the abilities of size-reduction and stability-maintaining on freeze-drying.Through formulation screening, the irregular-shaped azilsartan nanocrystals were prepared and optimized by bead milling method and solidified by freeze-drying. The best formulation which had the size of 300 nm was expressed in its state by the studies of morphology and crystal from. And azilsartan nanocrystals showed its improvement about in-vitro dissolution and in-vivo bioavailability with physical mixture as reference.Download high-res image (78KB)Download full-size image
Co-reporter:Xin Wang, Chang Li, Na Fan, Jing Li, Zhonggui He, Jin Sun
Materials Science and Engineering: C 2017 Volume 78(Volume 78) pp:
Publication Date(Web):1 September 2017
DOI:10.1016/j.msec.2017.04.060
•Multimodal nanoporous silica nanoparticles (M-NSNs) were successfully synthesized.•M-NSNs were amino modified (M-NSNs-NH2) using post-grafting method.•DOX loaded M-NSNS-NH2 had higher drug loading capacity than DOX loaded M-NSNs.•M-NSNs-NH2 was superior in prolonging DOX release than M-NSNs.The purpose of this study was to develop amino modified multimodal nanoporous silica nanoparticles (M-NSNs-NH2) loaded with doxorubicin hydrochloride (DOX), intended to enhance the drug loading capacity and to achieve controlled release effect. M-NSNs were functionalized with aminopropyl groups through post-synthesis. The contribution of large pore sizes and surface chemical groups on DOX loading and release were systemically studied using transmission electron microscope (TEM), nitrogen adsorption/desorption measurement, Fourier transform infrared spectroscopy (FTIR), zeta potential analysis, X-ray photoelectron spectroscopy (XPS) and ultraviolet spectrophotometer (UV). The results demonstrated that the NSNs were functionalized with aminopropyl successfully and the DOX molecules were adsorbed inside the nanopores by the hydrogen bonding. The release performance indicated that DOX loaded M-NSNs significantly controlled DOX release, furthermore DOX loaded M-NSNs-NH2 performed slower controlled release, which was mainly attributed to its stronger hydrogen bonding forces. As expected, we developed a novel carrier with high drug loading capacity and controlled release for DOX.Download high-res image (247KB)Download full-size image
Co-reporter:Qiuhua Luo, Ping Gong, Mengchi Sun, Longfa Kou, Vadivel Ganapathy, Yongkui Jing, Zhonggui He, Jin Sun
Journal of Controlled Release 2016 Volume 243() pp:370-380
Publication Date(Web):10 December 2016
DOI:10.1016/j.jconrel.2016.10.031
Rapidly proliferating tumor cells upregulate specific amino acid transporters, which hold great potential for tumor-selective drug delivery. Published reports have focused primarily on blocking these transporters as a means of starving the tumor cells of amino acids, but their potential in drug delivery remains understudied. In the present study, we developed liposomes functionalized with lysine and polyoxyethylene stearate conjugate (LPS) to interact with ATB0,+, an amino acid transporter overexpressed in hepatocarcinoma and the liver cancer cell line HepG2. The LPS modified liposomes (LPS-Lips) were ~ 100 nm in size and exhibited high drug encapsulation efficiency as 94.7%. The uptake of LPS-Lips in HepG2 cells was dependent on Na+ and Cl−. Molecular dynamic simulation showed that a sustained occluded state of the transporter upon binding to co-transported ions was formed and LPS-Lips triggered the cellular internalization of liposomes. We loaded these LPS-Lips with docetaxel and evaluated the potential of ATB0,+-mediated endocytosis of the drug-loaded LPS-Lips in HepG2 cells in vitro and in syngeneic mouse transplants in vivo. Compared with unmodified liposomes, which did not interact with ATB0,+, LPS-Lips exhibited the ability to deliver docetaxel more efficiently into tumor cells with consequent greater antitumor efficacy and less systemic toxicity. These studies provide first evidences that ATB0,+ can be used as a novel and effective target for drug delivery system in tumor cells using chemically modified liposomes for loading with chemotherapeutics and targeting them for the transporter-mediated endocytosis. As ATB0,+ is highly upregulated in several cancers, this approach holds potential for tumor-selective delivery of drugs to treat these cancer types.In the case of transporter-mediated nanoparticle endocytosis, extracellular LPS-Lips and Na+/Cl- ions bind ATB0,+ for the formation of an intermediate complex composed of co-transported ions, LPS-Lips and transporter, and then initiate the conformational switches of ATB0,+ from an outward-open state to a substrate-bound occluded state. The following conformational transition of ATB0,+ to an inward-open state was hindered sterically by the liposomes-containing complex with a large size and could induce certain changes in surface properties of the biomembrane, which triggers membrane invagination.
Co-reporter:Le Sun, Weixiang Zhang, Xiaohong Liu, Jin Sun
Asian Journal of Pharmaceutical Sciences (June 2014) Volume 9(Issue 3) pp:
Publication Date(Web):1 June 2014
DOI:10.1016/j.ajps.2014.03.003
The objective of this study was to prepare azithromycin (AZI) sustained-release products in order to allow for a high dose to be administered, reduce gastrointestinal side-effects and increase the compliance of patients. AZI sustained-release tablets with different release performance (F-I: T100% = 3 h and F-II: T100% = 8 h in pH 6.0 phosphate buffer) were successfully prepared by wet granulation. The in vitro release rate and drug release mechanism were studied. The release rate of F-I was affected by dissolution media with different pH, but not for F-II. Hixson–Crowell model was the best regression fitting model for F-I and F-II. Additionally, F-I and F-II both belonged to non-Fick diffusion. Oral pharmacokinetics of the two tablets and one AZI dispersible tablet as reference were studied in six healthy beagle dogs after oral administration. Compared with the reference, the Cmax of F-I and F-II were decreased, and the Tmax were prolonged, in that case which meet the requirement of sustained-release tablets. The relative bioavailability of F-I and F-II were 79.12% and 64.09%. T-test of AUC0–144, and AUC0–∞ for F-I and F-II indicated there was no significant difference between F-I and F-II. These mean that the extended release rate did not induce different pharmacokinetics in vivo.
Co-reporter:Jingmin Wang, Yinghua Sun, Bo Li, Rui Fan, Bing Li, Tengrui Yin, Ling Rong, Jin Sun
Asian Journal of Pharmaceutical Sciences (February 2015) Volume 10(Issue 1) pp:31-39
Publication Date(Web):February 2015
DOI:10.1016/j.ajps.2014.08.009
Co-reporter:Xinhuan Wan, Xiaohong Liu, Zheng Liu, Panqin Ma, Feitong Zhang, Xiangrong Zhang, Jin Sun
Asian Journal of Pharmaceutical Sciences (December 2014) Volume 9(Issue 6) pp:342-347
Publication Date(Web):December 2014
DOI:10.1016/j.ajps.2014.08.006

![2H-Pyrrol-2-one,1,5-dihydro-3-(4-methylphenyl)-4-[4-(methylsulfonyl)phenyl]-1-propyl-](http://img.cochemist.com/ccimg/395700/395683-14-4.png)
![2H-Pyrrol-2-one,1,5-dihydro-3-(4-methylphenyl)-4-[4-(methylsulfonyl)phenyl]-1-propyl-](http://img.cochemist.com/ccimg/395700/395683-14-4_b.png)

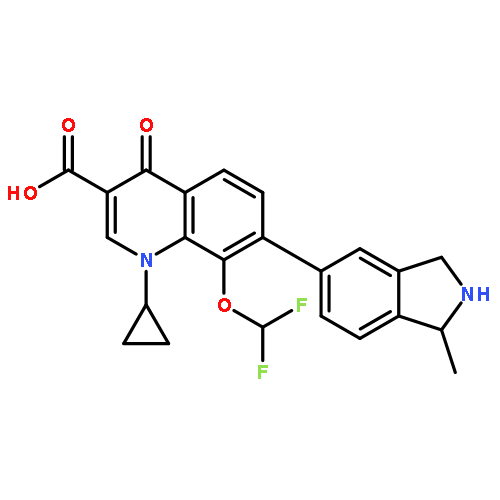
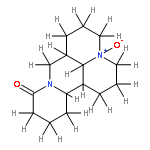











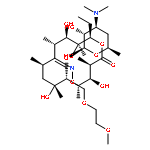
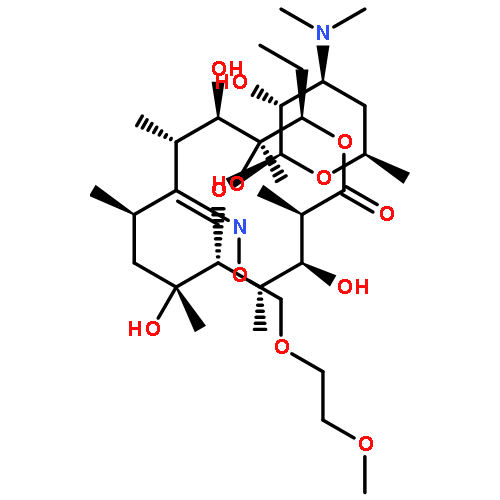

![2H-Indol-2-one,3-[(3,5-dimethyl-1H-pyrrol-2-yl)methylene]-1,3-dihydro-, (3Z)-](http://img.cochemist.com/ccimg/194500/194413-58-6.png)
![2H-Indol-2-one,3-[(3,5-dimethyl-1H-pyrrol-2-yl)methylene]-1,3-dihydro-, (3Z)-](http://img.cochemist.com/ccimg/194500/194413-58-6_b.png)


![(6R,7R)-7-{[(2Z)-2-(2-amino-1,3-thiazol-4-yl)-2-(hydroxyimino)acetyl]amino}-8-oxo-3-{(E)-[2-oxo-1-(2,2,2-trifluoroethyl)pyrrolidin-3-ylidene]methyl}-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid](http://img.cochemist.com/ccimg/161700/161672-76-0.png)
![(6R,7R)-7-{[(2Z)-2-(2-amino-1,3-thiazol-4-yl)-2-(hydroxyimino)acetyl]amino}-8-oxo-3-{(E)-[2-oxo-1-(2,2,2-trifluoroethyl)pyrrolidin-3-ylidene]methyl}-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid](http://img.cochemist.com/ccimg/161700/161672-76-0_b.png)


![Glycine,N-[(1R)-2-[(2S)-2-[[[3-[(aminoiminomethyl)amino]propyl]amino]carbonyl]-1-piperidinyl]-1-(cyclohexylmethyl)-2-oxoethyl]-](http://img.cochemist.com/ccimg/155500/155415-08-0.png)
![Glycine,N-[(1R)-2-[(2S)-2-[[[3-[(aminoiminomethyl)amino]propyl]amino]carbonyl]-1-piperidinyl]-1-(cyclohexylmethyl)-2-oxoethyl]-](http://img.cochemist.com/ccimg/155500/155415-08-0_b.png)
![Glycine,N-[[(3S)-1-(aminoiminomethyl)-3-piperidinyl]methyl]-N2-(2-naphthalenylsulfonyl)-L-asparaginyl-N-cyclopropyl-](http://img.cochemist.com/ccimg/154400/154397-77-0.png)
![Glycine,N-[[(3S)-1-(aminoiminomethyl)-3-piperidinyl]methyl]-N2-(2-naphthalenylsulfonyl)-L-asparaginyl-N-cyclopropyl-](http://img.cochemist.com/ccimg/154400/154397-77-0_b.png)
![1H-Indole-2-carboxylicacid, 4,6-dichloro-3-[(1E)-3-oxo-3-(phenylamino)-1-propen-1-yl]-](http://img.cochemist.com/ccimg/153500/153436-22-7.png)
![1H-Indole-2-carboxylicacid, 4,6-dichloro-3-[(1E)-3-oxo-3-(phenylamino)-1-propen-1-yl]-](http://img.cochemist.com/ccimg/153500/153436-22-7_b.png)
![Benzoic acid,2-hydroxy-5-[2-[4-[[(3-methyl-2-pyridinyl)amino]sulfonyl]phenyl]ethynyl]-](http://img.cochemist.com/ccimg/149600/149556-49-0.png)
![Benzoic acid,2-hydroxy-5-[2-[4-[[(3-methyl-2-pyridinyl)amino]sulfonyl]phenyl]ethynyl]-](http://img.cochemist.com/ccimg/149600/149556-49-0_b.png)
![(2S,3S)-2-(diphenylmethyl)-N-[2-methoxy-5-(propan-2-yl)benzyl]-1-azabicyclo[2.2.2]octan-3-amine](http://img.cochemist.com/ccimg/147200/147116-64-1.png)
![(2S,3S)-2-(diphenylmethyl)-N-[2-methoxy-5-(propan-2-yl)benzyl]-1-azabicyclo[2.2.2]octan-3-amine](http://img.cochemist.com/ccimg/147200/147116-64-1_b.png)

![Acetic acid,2-[[1-[(2S)-2-[[4-(aminoiminomethyl)benzoyl]amino]-3-(4-hydroxyphenyl)-1-oxopropyl]-4-piperidinyl]oxy]-](http://img.cochemist.com/ccimg/144500/144412-49-7.png)
![Acetic acid,2-[[1-[(2S)-2-[[4-(aminoiminomethyl)benzoyl]amino]-3-(4-hydroxyphenyl)-1-oxopropyl]-4-piperidinyl]oxy]-](http://img.cochemist.com/ccimg/144500/144412-49-7_b.png)
![Benzenepropanoic acid,2-[[(1S,2R,3S,4R)-3-[4-[(pentylamino)carbonyl]-2-oxazolyl]-7-oxabicyclo[2.2.1]hept-2-yl]methyl]-](http://img.cochemist.com/ccimg/143500/143443-90-7.png)
![Benzenepropanoic acid,2-[[(1S,2R,3S,4R)-3-[4-[(pentylamino)carbonyl]-2-oxazolyl]-7-oxabicyclo[2.2.1]hept-2-yl]methyl]-](http://img.cochemist.com/ccimg/143500/143443-90-7_b.png)
![1H-Imidazole-5-propanamide,N-[(1S,2R,3S)-1-(cyclohexylmethyl)-3-cyclopropyl-2,3-dihydroxypropyl]-a-[[(2S)-2-[[(1,1-dimethylethyl)sulfonyl]methyl]-1-oxo-3-phenylpropyl]amino]-,(aS)-](http://img.cochemist.com/ccimg/126300/126222-34-2.png)
![1H-Imidazole-5-propanamide,N-[(1S,2R,3S)-1-(cyclohexylmethyl)-3-cyclopropyl-2,3-dihydroxypropyl]-a-[[(2S)-2-[[(1,1-dimethylethyl)sulfonyl]methyl]-1-oxo-3-phenylpropyl]amino]-,(aS)-](http://img.cochemist.com/ccimg/126300/126222-34-2_b.png)



![Acetic acid, methoxy-,(1S,2S)-2-[2-[[3-(1H-benzimidazol-2-yl)propyl]methylamino]ethyl]-6-fluoro-1,2,3,4-tetrahydro-1-(1-methylethyl)-2-naphthalenylester](http://img.cochemist.com/ccimg/116700/116644-53-2.png)
![Acetic acid, methoxy-,(1S,2S)-2-[2-[[3-(1H-benzimidazol-2-yl)propyl]methylamino]ethyl]-6-fluoro-1,2,3,4-tetrahydro-1-(1-methylethyl)-2-naphthalenylester](http://img.cochemist.com/ccimg/116700/116644-53-2_b.png)
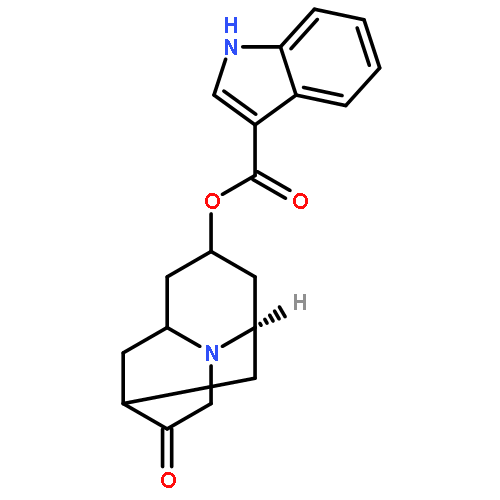
![Propanoic acid,3-[[[3-[(1E)-2-(7-chloro-2-quinolinyl)ethenyl]phenyl][[3-(dimethylamino)-3-oxopropyl]thio]methyl]thio]-](http://img.cochemist.com/ccimg/115200/115104-28-4.png)
![Propanoic acid,3-[[[3-[(1E)-2-(7-chloro-2-quinolinyl)ethenyl]phenyl][[3-(dimethylamino)-3-oxopropyl]thio]methyl]thio]-](http://img.cochemist.com/ccimg/115200/115104-28-4_b.png)
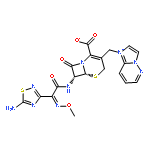
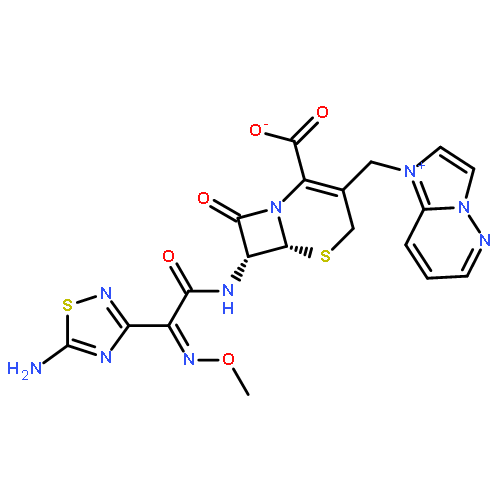
![5-Hexyn-1-one,1-[3,5-bis(1,1-dimethylethyl)-4-hydroxyphenyl]-](http://img.cochemist.com/ccimg/112100/112018-00-5.png)
![5-Hexyn-1-one,1-[3,5-bis(1,1-dimethylethyl)-4-hydroxyphenyl]-](http://img.cochemist.com/ccimg/112100/112018-00-5_b.png)
![1-Azoniabicyclo[2.2.2]octane,4-(aminocarbonyl)-1-[[(6R,7R)-7-[[(2Z)-2-(5-amino-1,2,4-thiadiazol-3-yl)-2-(methoxyimino)acetyl]amino]-2-carboxy-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-en-3-yl]methyl]-,inner salt](http://img.cochemist.com/ccimg/105300/105239-91-6.png)
![1-Azoniabicyclo[2.2.2]octane,4-(aminocarbonyl)-1-[[(6R,7R)-7-[[(2Z)-2-(5-amino-1,2,4-thiadiazol-3-yl)-2-(methoxyimino)acetyl]amino]-2-carboxy-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-en-3-yl]methyl]-,inner salt](http://img.cochemist.com/ccimg/105300/105239-91-6_b.png)
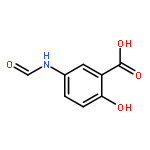
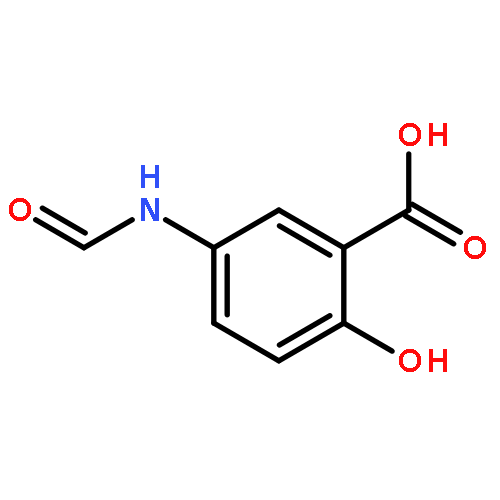
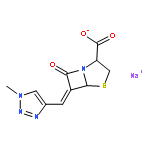
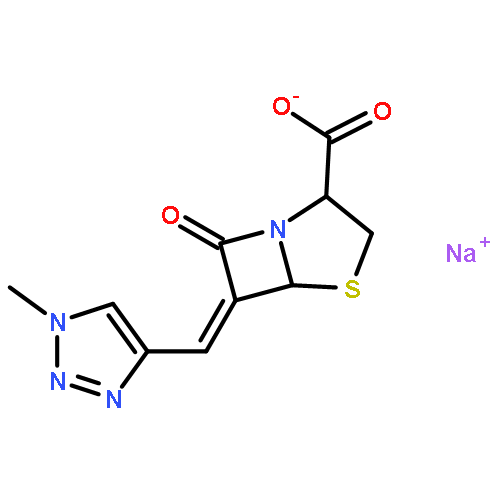
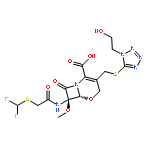
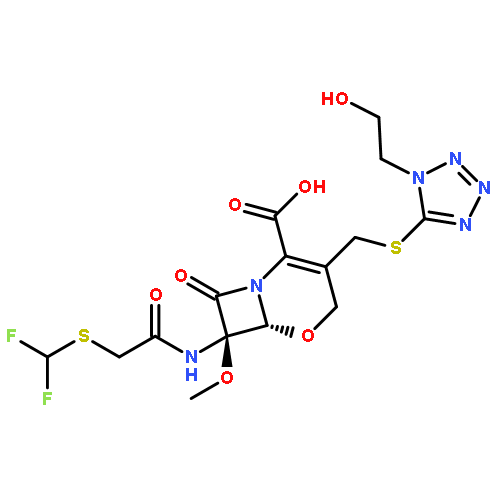
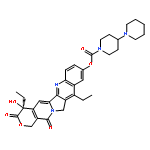
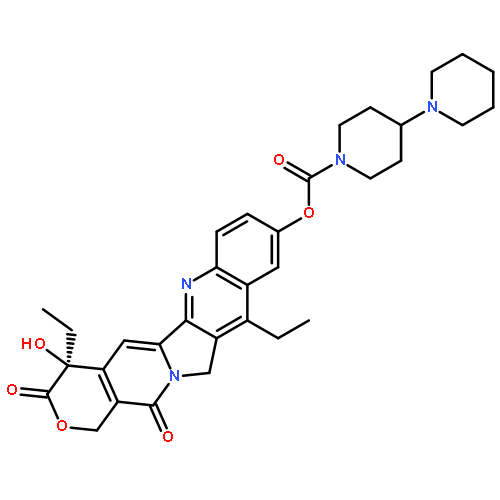
![(5R,6S)-3-{[2-(carbamoyloxy)ethyl]sulfanyl}-6-[(1R)-1-hydroxyethyl]-7-oxo-4-thia-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylic acid](http://img.cochemist.com/ccimg/95500/95415-91-1.png)
![(5R,6S)-3-{[2-(carbamoyloxy)ethyl]sulfanyl}-6-[(1R)-1-hydroxyethyl]-7-oxo-4-thia-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylic acid](http://img.cochemist.com/ccimg/95500/95415-91-1_b.png)

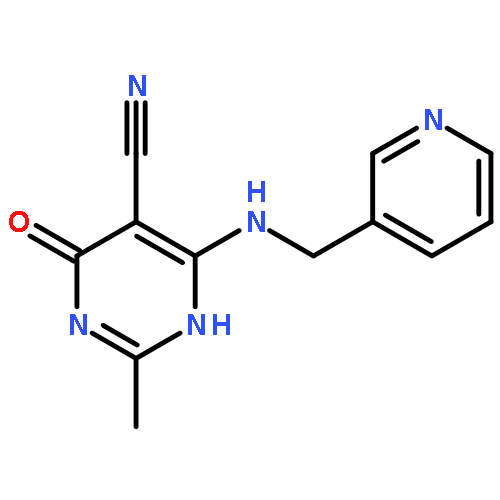



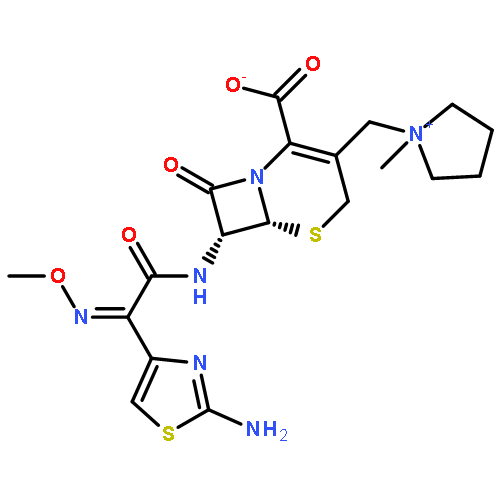

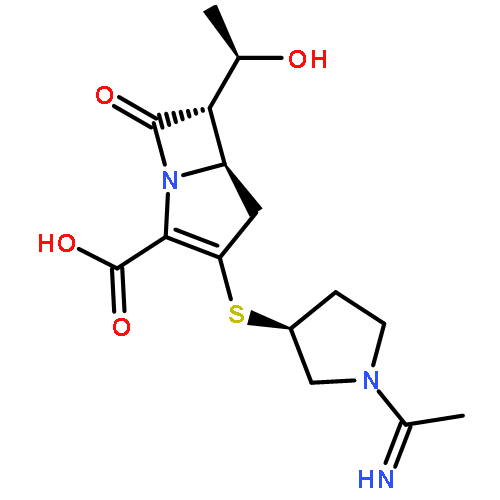
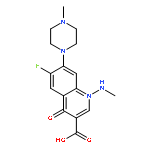
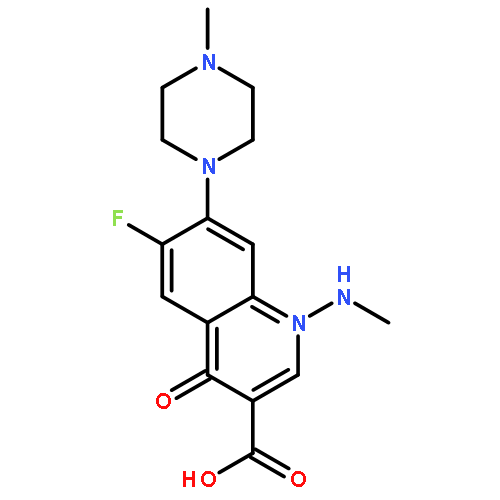
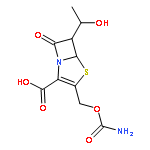


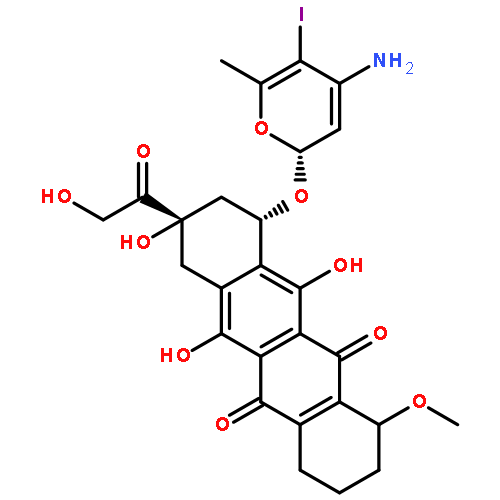




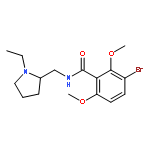
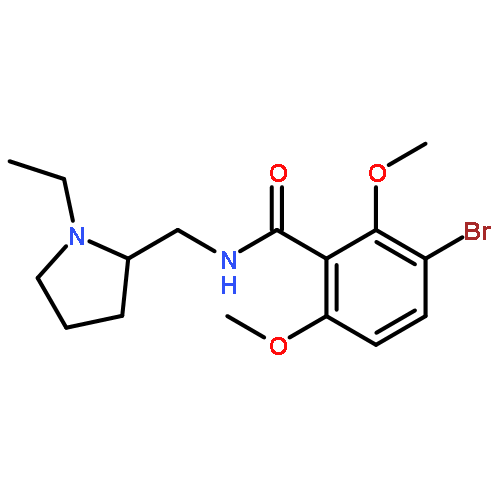
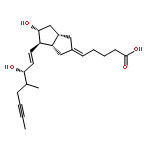
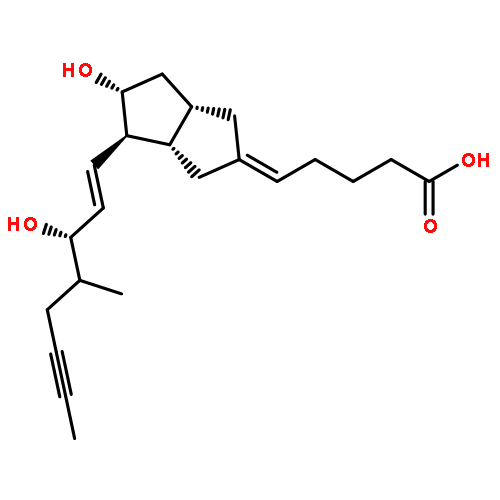
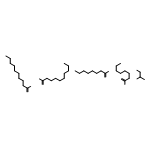
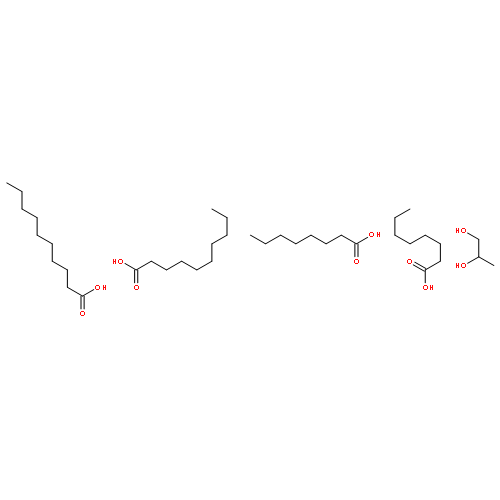
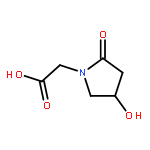
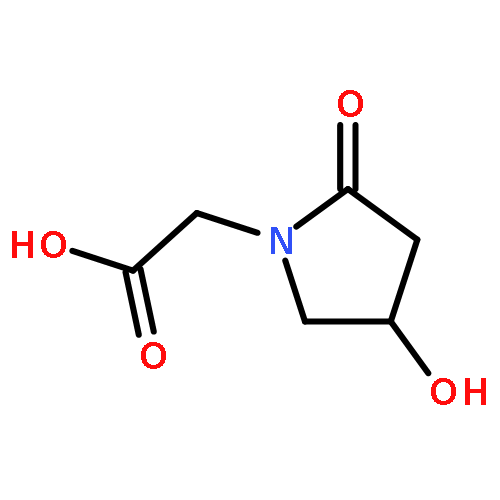

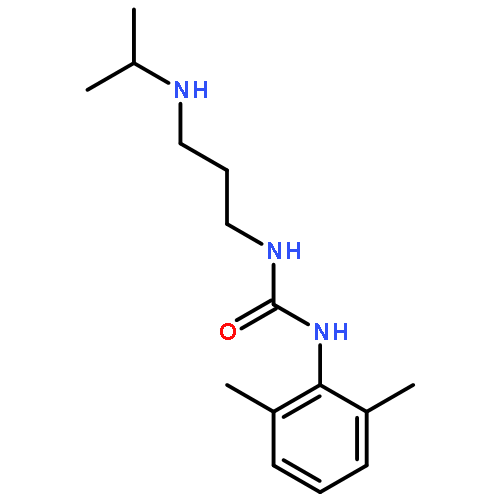
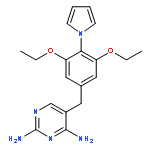
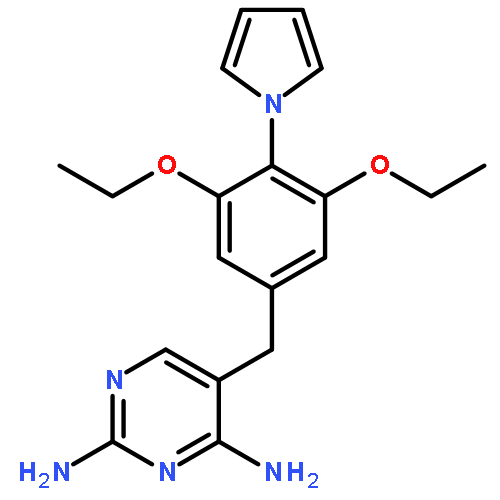




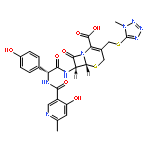


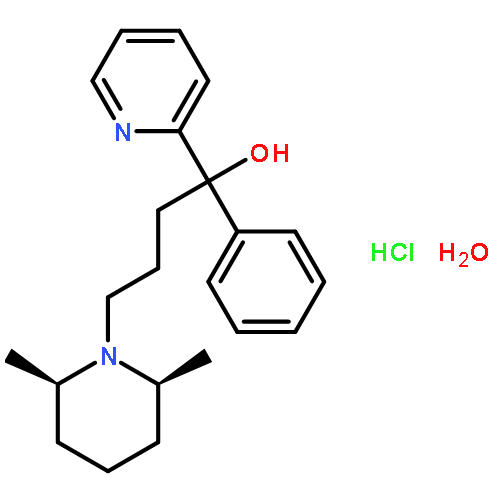


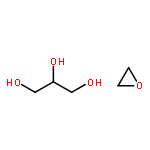


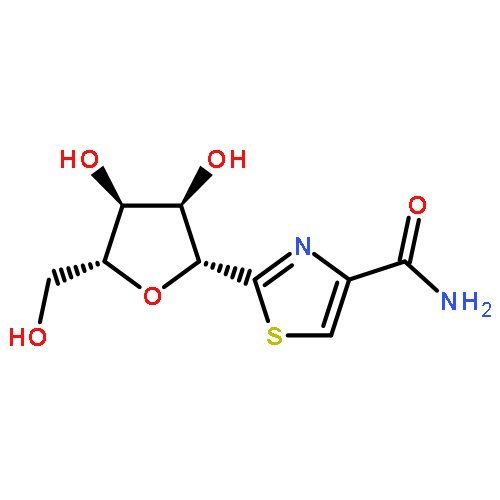
![Benzeneacetamide,N-(4-chlorophenyl)-N-[1-(1-methylethyl)-4-piperidinyl]-](http://img.cochemist.com/ccimg/59800/59729-31-6.png)
![Benzeneacetamide,N-(4-chlorophenyl)-N-[1-(1-methylethyl)-4-piperidinyl]-](http://img.cochemist.com/ccimg/59800/59729-31-6_b.png)
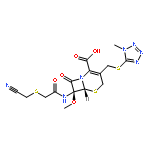
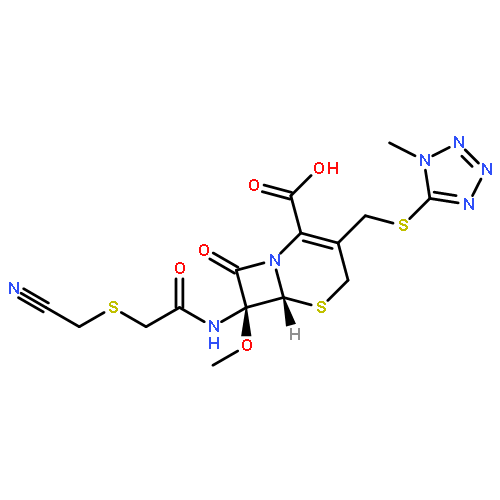


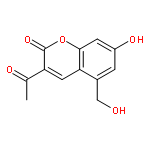
![Rifamycin,3-[[(4-methyl-1-piperazinyl)oxidoimino]methyl]-](http://img.cochemist.com/ccimg/52000/51963-55-4.png)
![Rifamycin,3-[[(4-methyl-1-piperazinyl)oxidoimino]methyl]-](http://img.cochemist.com/ccimg/52000/51963-55-4_b.png)






![Pyridinium,1-[[[4-(aminocarbonyl)pyridinio]methoxy]methyl]-2-[(hydroxyimino)methyl]-,chloride (1:2)](http://img.cochemist.com/ccimg/34500/34433-31-3.png)
![Pyridinium,1-[[[4-(aminocarbonyl)pyridinio]methoxy]methyl]-2-[(hydroxyimino)methyl]-,chloride (1:2)](http://img.cochemist.com/ccimg/34500/34433-31-3_b.png)

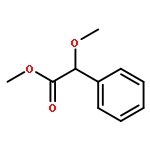
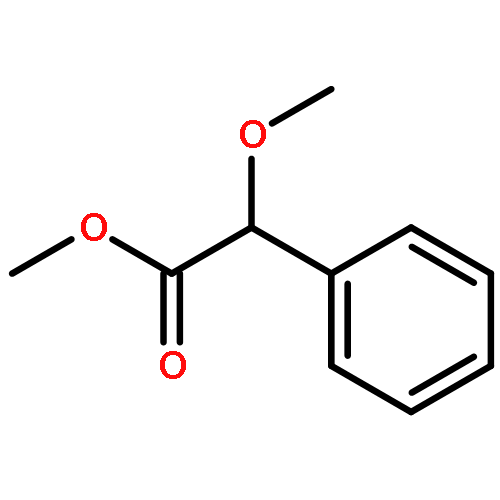






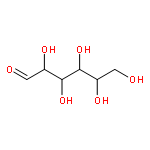
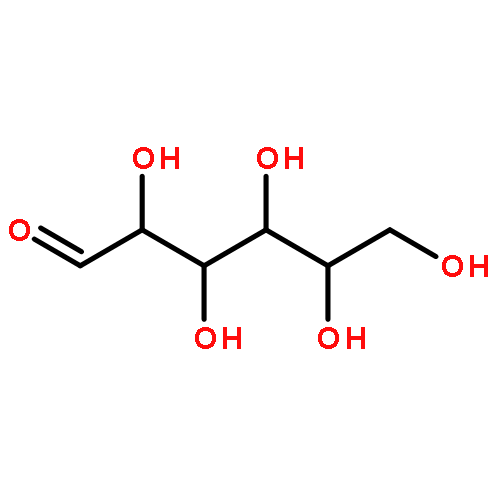





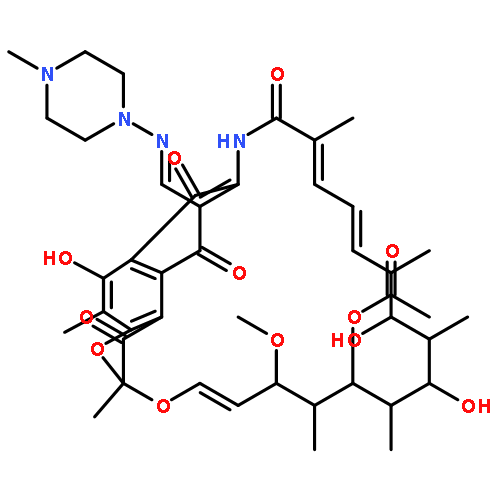




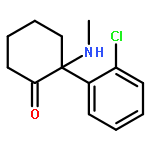
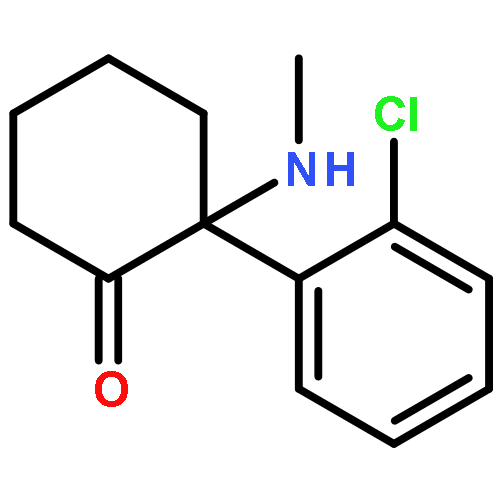
![(3r,4r,5s,6r)-3,4-dihydroxy-6-(hydroxymethyl)-5-[(2s,3r,4s,5r,6r)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxyoxan-2-one](http://img.cochemist.com/ccimg/6000/5965-65-1.png)
![(3r,4r,5s,6r)-3,4-dihydroxy-6-(hydroxymethyl)-5-[(2s,3r,4s,5r,6r)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxyoxan-2-one](http://img.cochemist.com/ccimg/6000/5965-65-1_b.png)


![(6a,11b,16a)-6-fluoro-11,21-dihydroxy-16,17-[(1-methylethylidene)bis(oxy)]-pregna-1,4-diene-3,20-dione](http://img.cochemist.com/ccimg/3400/3385-03-3.png)
![(6a,11b,16a)-6-fluoro-11,21-dihydroxy-16,17-[(1-methylethylidene)bis(oxy)]-pregna-1,4-diene-3,20-dione](http://img.cochemist.com/ccimg/3400/3385-03-3_b.png)

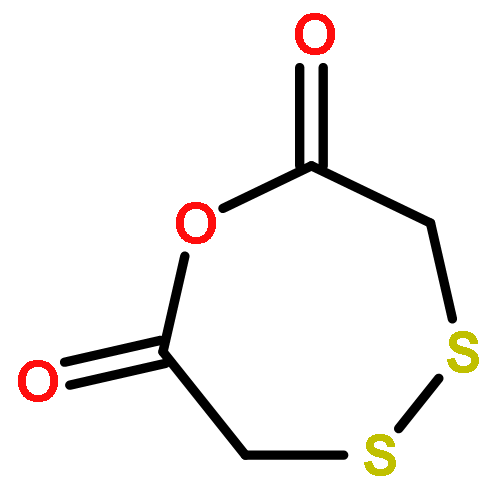
![3-(Dibenzo[b,e]oxepin-11(6H)-ylidene)-N,N-dimethylpropan-1-amine](http://img.cochemist.com/ccimg/1700/1668-19-5.png)
![3-(Dibenzo[b,e]oxepin-11(6H)-ylidene)-N,N-dimethylpropan-1-amine](http://img.cochemist.com/ccimg/1700/1668-19-5_b.png)





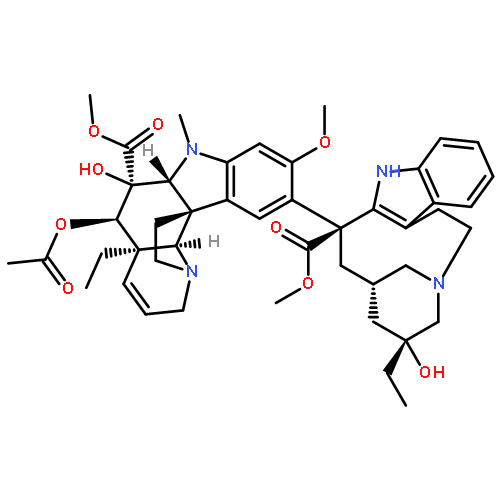




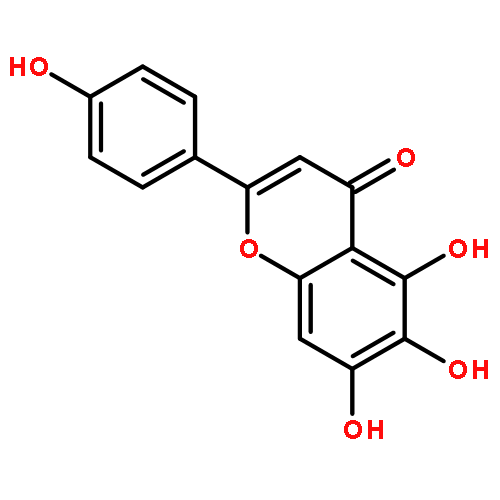


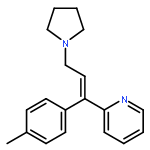

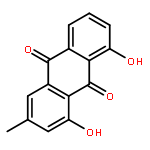
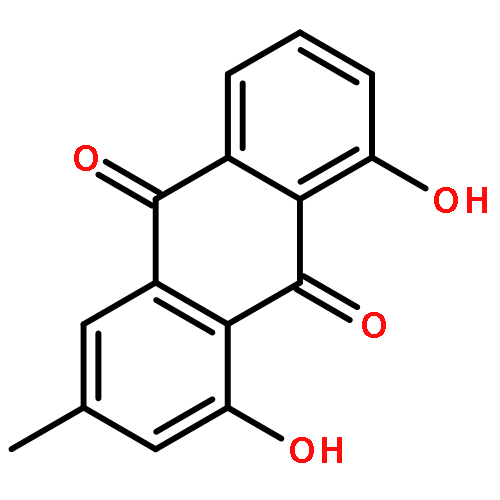
![N-phenyl-n-[1-(2-phenylethyl)piperidin-4-yl]propanamide](http://img.cochemist.com/ccimg/500/437-38-7.png)
![N-phenyl-n-[1-(2-phenylethyl)piperidin-4-yl]propanamide](http://img.cochemist.com/ccimg/500/437-38-7_b.png)
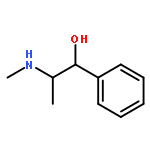
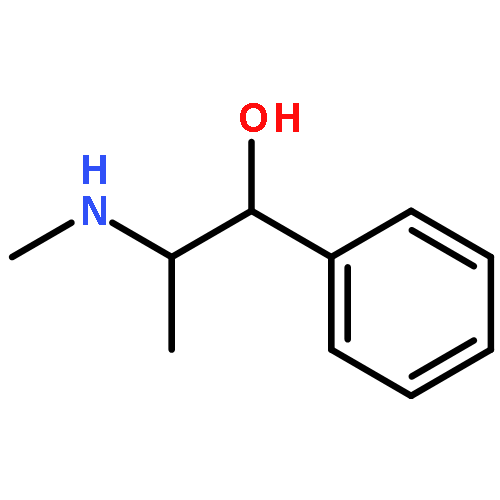




![Morpholine,4-[3-(4-butoxyphenoxy)propyl]-](http://img.cochemist.com/ccimg/200/140-65-8.png)
![Morpholine,4-[3-(4-butoxyphenoxy)propyl]-](http://img.cochemist.com/ccimg/200/140-65-8_b.png)










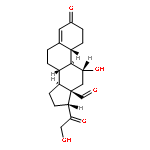
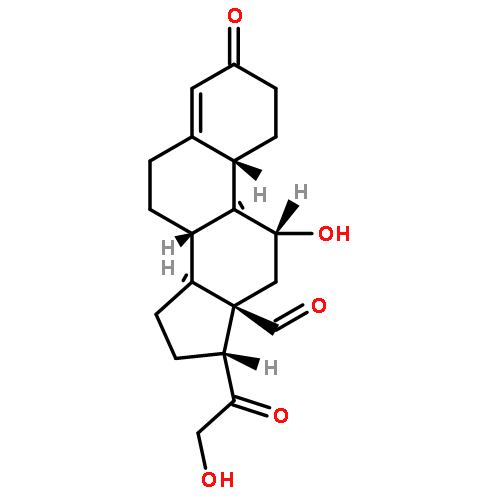
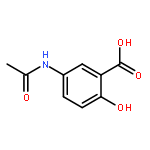
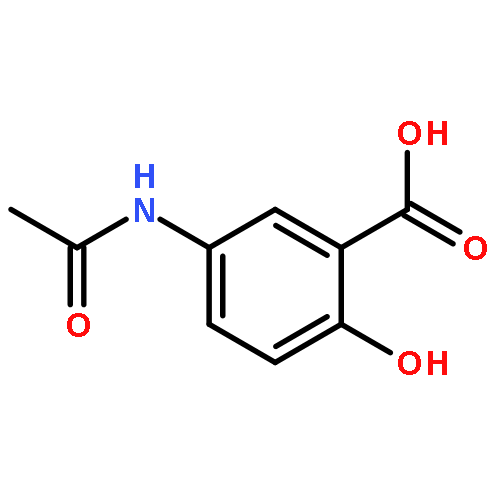


![3-(10,11-Dihydro-5H-dibenzo[b,f]azepin-5-yl)-N-methylpropan-1-amine](http://img.cochemist.com/ccimg/100/50-47-5.png)
![3-(10,11-Dihydro-5H-dibenzo[b,f]azepin-5-yl)-N-methylpropan-1-amine](http://img.cochemist.com/ccimg/100/50-47-5_b.png)
![Methyl (3s,4r)-3-benzoyloxy-8-methyl-8-azabicyclo[3.2.1]octane-4-carboxylate](http://img.cochemist.com/ccimg/100/50-36-2.png)
![Methyl (3s,4r)-3-benzoyloxy-8-methyl-8-azabicyclo[3.2.1]octane-4-carboxylate](http://img.cochemist.com/ccimg/100/50-36-2_b.png)
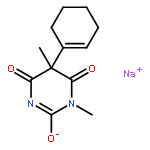
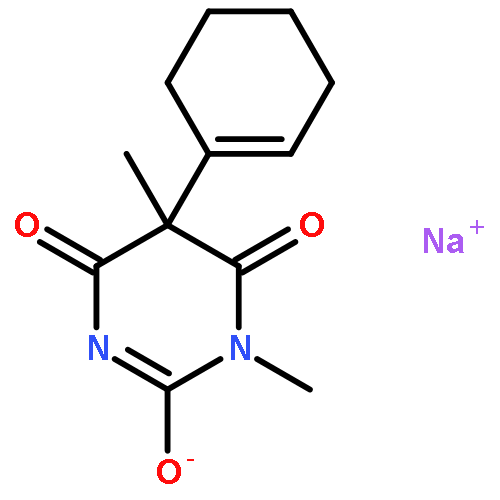
![(S)-5-Amino-7-(7-amino-5-azaspiro[2.4]heptan-5-yl)-1-cyclopropyl-6-fluoro-8-methyl-4-oxo-1,4-dihydroquinoline-3-carboxylic acid](http://img.cochemist.com/ccimg/167900/167887-97-0.png)
![(S)-5-Amino-7-(7-amino-5-azaspiro[2.4]heptan-5-yl)-1-cyclopropyl-6-fluoro-8-methyl-4-oxo-1,4-dihydroquinoline-3-carboxylic acid](http://img.cochemist.com/ccimg/167900/167887-97-0_b.png)

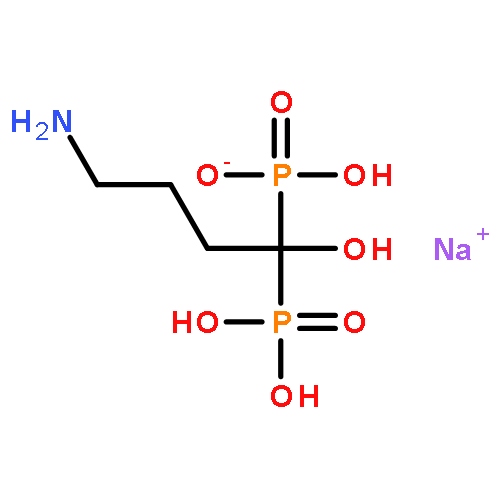




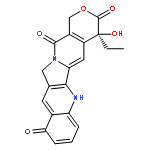

![8-(4-(4-(Pyrimidin-2-yl)piperazin-1-yl)butyl)-8-azaspiro[4.5]decane-7,9-dione](http://img.cochemist.com/ccimg/36600/36505-84-7.png)
![8-(4-(4-(Pyrimidin-2-yl)piperazin-1-yl)butyl)-8-azaspiro[4.5]decane-7,9-dione](http://img.cochemist.com/ccimg/36600/36505-84-7_b.png)
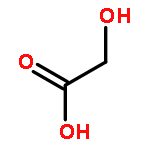
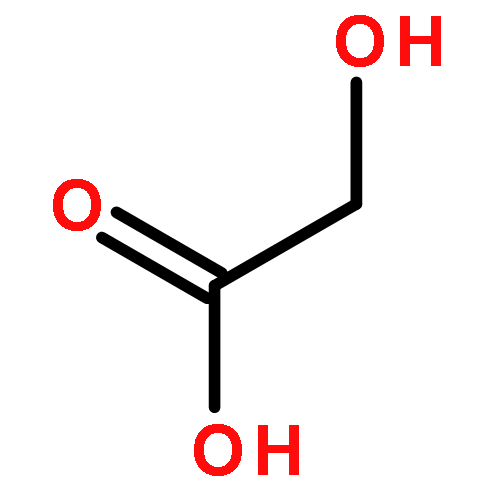
![Dibenzo[c,f]pyrazino[1,2-a]azepine,1,2,3,4,10,14b-hexahydro-2-methyl-](http://img.cochemist.com/ccimg/24300/24219-97-4.png)
![Dibenzo[c,f]pyrazino[1,2-a]azepine,1,2,3,4,10,14b-hexahydro-2-methyl-](http://img.cochemist.com/ccimg/24300/24219-97-4_b.png)
![Butanedioic acid,1-[3,4-dihydro-2,5,7,8-tetramethyl-2-(4,8,12-trimethyltridecyl)-2H-1-benzopyran-6-yl]ester](http://img.cochemist.com/ccimg/17500/17407-37-3.png)
![Butanedioic acid,1-[3,4-dihydro-2,5,7,8-tetramethyl-2-(4,8,12-trimethyltridecyl)-2H-1-benzopyran-6-yl]ester](http://img.cochemist.com/ccimg/17500/17407-37-3_b.png)
![5-Methyl-1-phenyl-3,4,5,6-tetrahydro-1H-benzo[f][1,4]oxazocine](http://img.cochemist.com/ccimg/13700/13669-70-0.png)
![5-Methyl-1-phenyl-3,4,5,6-tetrahydro-1H-benzo[f][1,4]oxazocine](http://img.cochemist.com/ccimg/13700/13669-70-0_b.png)








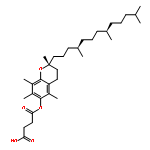

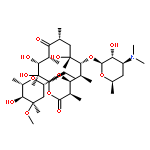
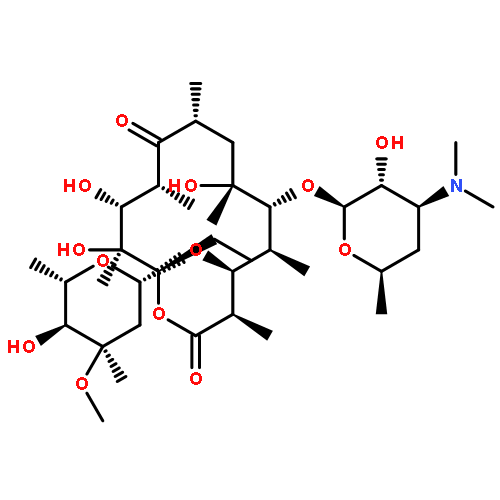




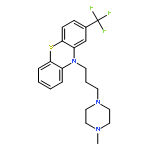
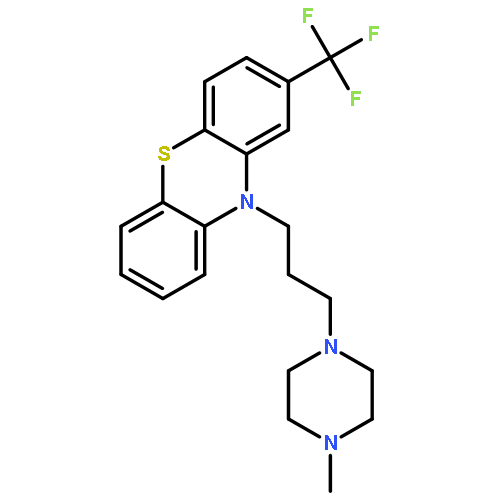




![3-Quinolinecarboxylicacid, 5-amino-7-[(7S)-7-amino-5-azaspiro[2.4]hept-5-yl]-1-cyclopropyl-6-fluoro-1,4-dihydro-8-methyl-4-oxo-,methanesulfonate (1:1)](http://img.cochemist.com/ccimg/167900/167888-07-5.png)
![3-Quinolinecarboxylicacid, 5-amino-7-[(7S)-7-amino-5-azaspiro[2.4]hept-5-yl]-1-cyclopropyl-6-fluoro-1,4-dihydro-8-methyl-4-oxo-,methanesulfonate (1:1)](http://img.cochemist.com/ccimg/167900/167888-07-5_b.png)






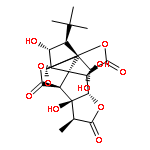



![(3aS,8aR)-1,3a,8-Trimethyl-1,2,3,3a,8,8a-hexahydropyrrolo[2,3-b]indol-5-yl methylcarbamate](http://img.cochemist.com/ccimg/100/57-47-6.png)
![(3aS,8aR)-1,3a,8-Trimethyl-1,2,3,3a,8,8a-hexahydropyrrolo[2,3-b]indol-5-yl methylcarbamate](http://img.cochemist.com/ccimg/100/57-47-6_b.png)




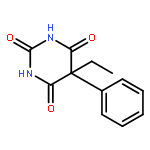





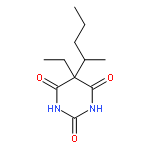
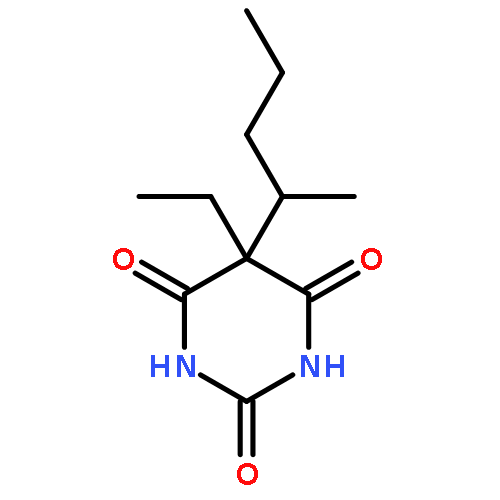




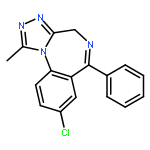

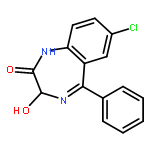
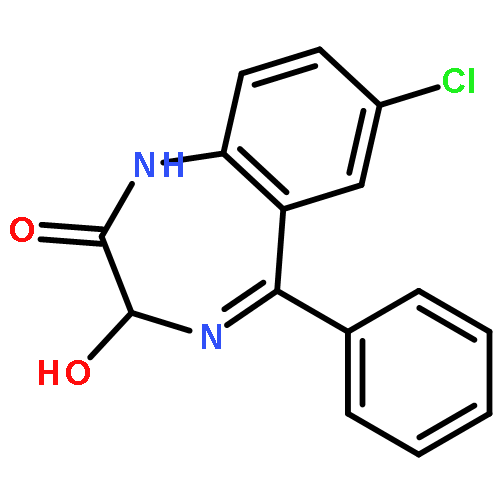




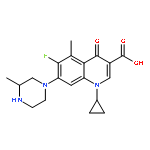
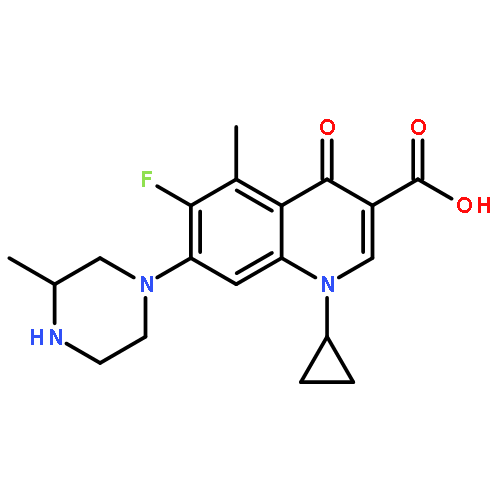
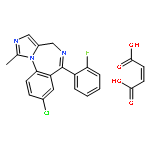

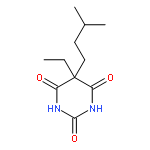


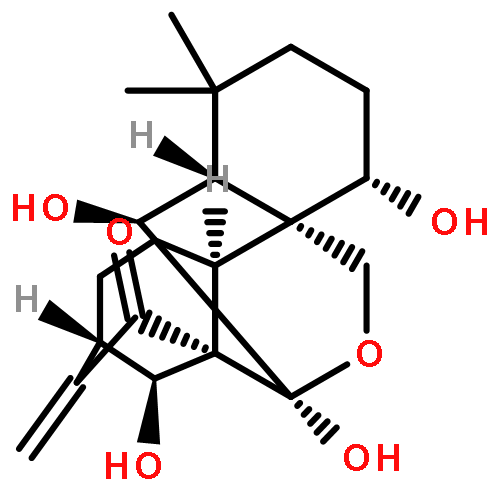
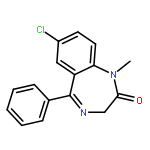
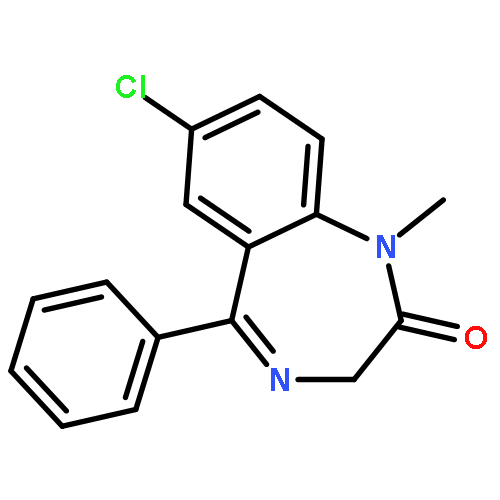
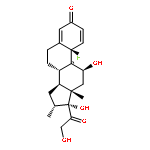
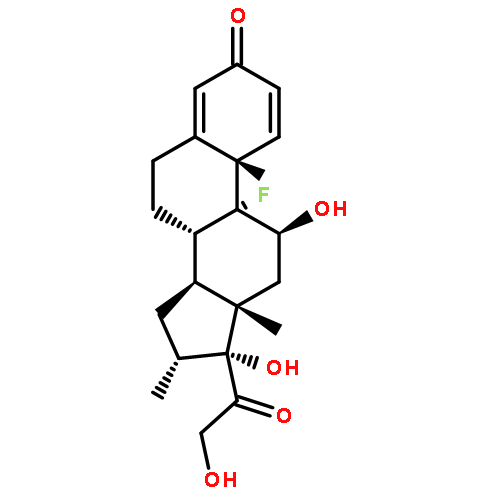
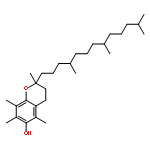




![2-Propanol, 1-[(1-methylethyl)amino]-3-(1-naphthalenyloxy)-](/data/chemimg/935900/525-66-6.png)
![2-Propanol, 1-[(1-methylethyl)amino]-3-(1-naphthalenyloxy)-](/data/chemimg/935900/525-66-6_b.png)