Co-reporter:Teng Ai, Rose Willett, Jessica Williams, Rui Ding, Daniel J. Wilson, Jiashu Xie, Do-Hyung Kim, Rosa Puertollano, and Liqiang Chen
ACS Medicinal Chemistry Letters 2017 Volume 8(Issue 1) pp:
Publication Date(Web):November 28, 2016
DOI:10.1021/acsmedchemlett.6b00392
Guided by antiproliferative activity in MIA PaCa-2 cells, we have performed preliminary structure–activity relationship studies on N-(1-benzyl-3,5-dimethyl-1H-pyrazol-4-yl)benzamides. Two selected compounds showed submicromolar antiproliferative activity and good metabolic stability. Both compounds reduced mTORC1 activity and increased autophagy at the basal level. In addition, they disrupted autophagic flux by interfering with mTORC1 reactivation and clearance of LC3-II under starvation/refeed conditions, as evidenced by accumulation of LC3-II and abnormal LC3 labeled punctae. Therefore, N-(1-benzyl-3,5-dimethyl-1H-pyrazol-4-yl)benzamides may represent a new class of autophagy modulators that possesses potent anticancer activity and potentially a novel mechanism of action.Keywords: anticancer agents; Autophagy; autophagy modulator; mTOR; pancreatic cancer;
Co-reporter:Teng Ai; Daniel J. Wilson; Swati S. More; Jiashu Xie
Journal of Medicinal Chemistry 2016 Volume 59(Issue 7) pp:2928-2941
Publication Date(Web):March 16, 2016
DOI:10.1021/acs.jmedchem.5b01376
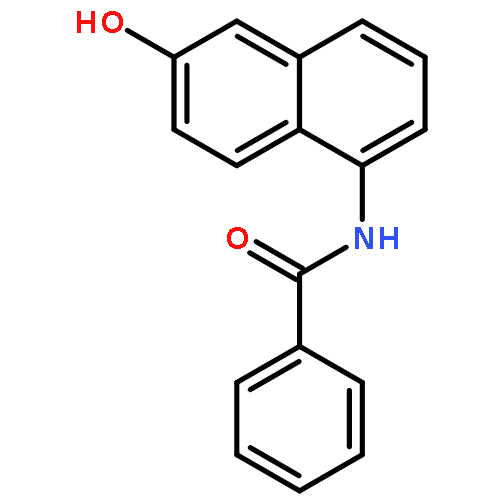
Derived from our previously reported human sirtuin 2 (SIRT2) inhibitors that were based on a 5-aminonaphthalen-1-yloxy nicotinamide core structure, 5-((3-amidobenzyl)oxy)nicotinamides offered excellent activity against SIRT2 and high isozyme selectivity over SIRT1 and SIRT3. Selected compounds also exhibited generally favorable in vitro absorption, distribution, metabolism, and excretion properties. Kinetic studies revealed that a representative SIRT2 inhibitor acted competitively against both NAD+ and the peptide substrate, an inhibitory modality that was supported by our computational study. More importantly, two selected compounds exhibited significant protection against α-synuclein aggregation-induced cytotoxicity in SH-SY5Y cells. Therefore, 5-((3-amidobenzyl)oxy)nicotinamides represent a new class of SIRT2 inhibitors that are attractive candidates for further lead optimization in our continued effort to explore selective inhibition of SIRT2 as a potential therapy for Parkinson’s disease.
Co-reporter:Teng Ai, Li Qiu, Jiashu Xie, Robert J. Geraghty, Liqiang Chen
Bioorganic & Medicinal Chemistry 2016 Volume 24(Issue 4) pp:686-692
Publication Date(Web):15 February 2016
DOI:10.1016/j.bmc.2015.12.035
In our continued effort to discover new anti-hepatitis C virus (HCV) agents, we validated the anti-replicon activity of compound 1, a potent and selective anti-HCV hydroxamic acid recently reported by us. Generally favorable physicochemical and in vitro absorption, distribution, metabolism, and excretion (ADME) properties exhibited by 1 made it an ideal parent compound from which activity-based protein profiling (ABPP) probe 3 was designed and synthesized. Evaluation of probe 3 revealed that it possessed necessary anti-HCV activity and selectivity. Therefore, we have successfully obtained compound 3 as a suitable ABPP probe to identify potential molecular targets of compound 1. Probe 3 and its improved analogs are expected to join a growing list of ABPP probes that have made important contributions to not only the studies of biochemical and cellular functions but also discovery of selective inhibitors of protein targets.
Co-reporter:Rui Ding, Ting Zhang, Jiashu Xie, Jessica Williams, Yihong Ye, Liqiang Chen
Bioorganic & Medicinal Chemistry Letters 2016 Volume 26(Issue 21) pp:5177-5181
Publication Date(Web):1 November 2016
DOI:10.1016/j.bmcl.2016.09.068
Inhibition of p97 (also known as valosin-containing protein (VCP)), has been validated as a promising strategy for cancer therapy. Eeyarestatin I (EerI) blocks p97 through a novel mechanism of action and has favorable anti-cancer activities against cultured cancer cells. However, its poor aqueous solubility severely limits its in vivo applications. To circumvent this problem, we have identified EerI derivatives that possess improved aqueous solubility by introducing a single solubilizing group. These modified compounds preserved endoplasmic reticulum (ER) stress-inducing and antiproliferative activities as well as generally good in vitro metabolic properties, suggesting that these EerI derivatives could serve as candidates for further optimization.Download high-res image (109KB)Download full-size image
Co-reporter:Teng Ai; Yanli Xu; Li Qiu; Robert J. Geraghty
Journal of Medicinal Chemistry 2015 Volume 58(Issue 2) pp:785-800
Publication Date(Web):December 9, 2014
DOI:10.1021/jm501330g
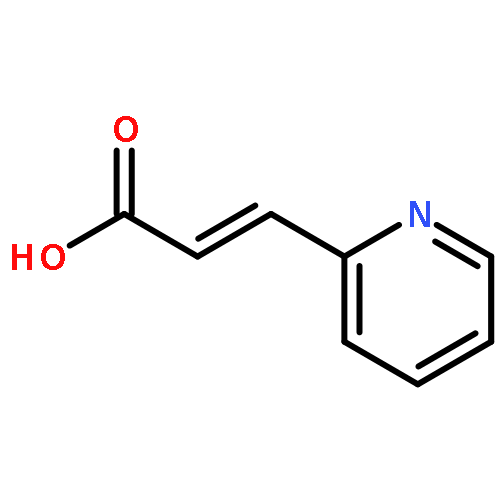
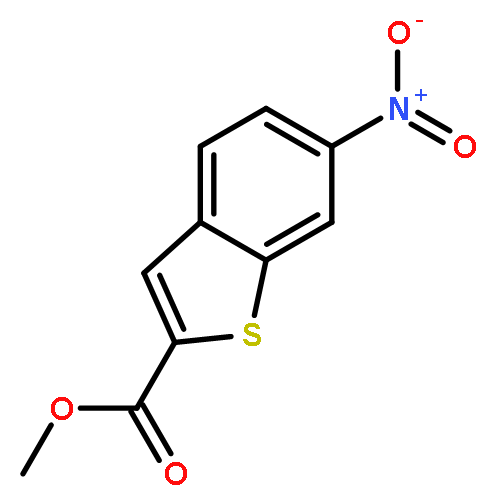
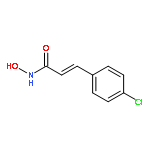
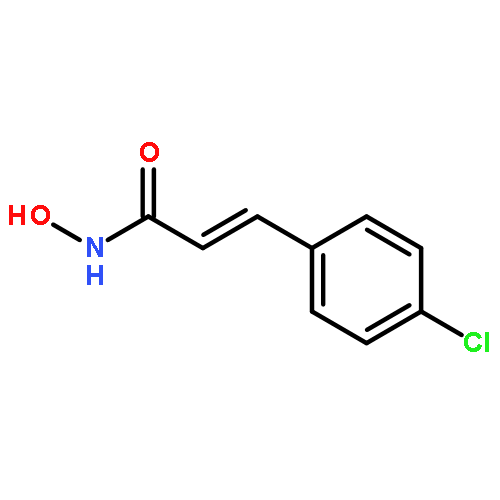
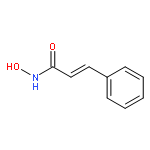
Intrigued by the role of protein acetylation in hepatitis C virus (HCV) replication, we tested known histone deacetylase (HDAC) inhibitors and a focused library of structurally simple hydroxamic acids for inhibition of a HCV subgenomic replicon. While known HDAC inhibitors with varied inhibitory profiles proved to be either relatively toxic or ineffective, structure–activity relationship (SAR) studies on cinnamic hydroxamic acid and benzo[b]thiophen-2-hydroxamic acid gave rise to compounds 22 and 53, which showed potent and selective anti-HCV activity and therefore are promising starting points for further structural optimization and mechanistic studies.
Co-reporter:Huaqing Cui ; Zeeshan Kamal ; Teng Ai ; Yanli Xu ; Swati S. More ; Daniel J. Wilson
Journal of Medicinal Chemistry 2014 Volume 57(Issue 20) pp:8340-8357
Publication Date(Web):September 13, 2014
DOI:10.1021/jm500777s
Sirtuin 2 (SIRT2) is one of the sirtuins, a family of NAD+-dependent deacetylases that act on a variety of histone and non-histone substrates. Accumulating biological functions and potential therapeutic applications have drawn interest in the discovery and development of SIRT2 inhibitors. Herein we report our discovery of novel SIRT2 inhibitors using a fragment-based approach. Inspired by the purported close binding proximity of suramin and nicotinamide, we prepared two sets of fragments, namely, the naphthylamide sulfonic acids and the naphthalene–benzamides and −nicotinamides. Biochemical evaluation of these two series provided structure–activity relationship (SAR) information, which led to the design of (5-benzamidonaphthalen-1/2-yloxy)nicotinamide derivatives. Among these inhibitors, one compound exhibited high anti-SIRT2 activity (48 nM) and excellent selectivity for SIRT2 over SIRT1 and SIRT3. In vitro, it also increased the acetylation level of α-tubulin, a well-established SIRT2 substrate, in both concentration- and time-dependent manners. Further kinetic studies revealed that this compound behaves as a competitive inhibitor against the peptide substrate and most likely as a noncompetitive inhibitor against NAD+. Taken together, these results indicate that we have discovered a potent and selective SIRT2 inhibitor whose novel structure merits further exploration.
Co-reporter:Liqiang Chen ; Daniel J. Wilson ; Yanli Xu ; Courtney C. Aldrich ; Krzysztof Felczak ; Yuk Y. Sham ;Krzysztof W. Pankiewicz
Journal of Medicinal Chemistry 2010 Volume 53(Issue 12) pp:4768-4778
Publication Date(Web):May 19, 2010
DOI:10.1021/jm100424m
The modular nature of nicotinamide adenine dinucleotide (NAD)-mimicking inosine monophsophate dehydrogenase (IMPDH) inhibitors has prompted us to investigate novel mycophenolic adenine dinucleotides (MAD) in which 1,2,3-triazole linkers were incorporated as isosteric replacements of the pyrophosphate linker. Synthesis and evaluation of these inhibitors led to identification of low nanomolar inhibitors of human IMPDH and more importantly the first potent inhibitor of IMPDH from Mycobacterium tuberculosis (mtIMPDH). Computational studies of these IMPDH enzymes helped rationalize the observed structure−activity relationships. Additionally, the first cloning, expression, purification and characterization of mtIMPDH is reported.
Co-reporter:Liqiang Chen, Riccardo Petrelli, Guangyao Gao, Daniel J. Wilson, Garrett T. McLean, Hiremagalur N. Jayaram, Yuk Y. Sham, Krzysztof W. Pankiewicz
Bioorganic & Medicinal Chemistry 2010 Volume 18(Issue 16) pp:5950-5964
Publication Date(Web):15 August 2010
DOI:10.1016/j.bmc.2010.06.081
Small molecules that act on multiple biological targets have been proposed to combat the drug resistance commonly observed for cancer chemotherapy. By combining the structural features of known inhibitors of inosine monophosphate dehydrogense (IMPDH) and histone deacetylase (HDAC), dual inhibitors of IMPDH and HDAC based on the scaffold of cinnamic hydroxamic acid (CHA) have been designed, synthesized, and evaluated in biological assays. Key features, including the linker length, linker functionality, substitution position, and interacting groups, have been explored. Their individual contribution to the inhibitory activities against human IMPDH1 and IMPDH2 as well as HDAC has been assessed.
Co-reporter:Liqiang Chen, Daniel J. Wilson, Nicholas P. Labello, Hiremagalur N. Jayaram, Krzysztof W. Pankiewicz
Bioorganic & Medicinal Chemistry 2008 Volume 16(Issue 20) pp:9340-9345
Publication Date(Web):15 October 2008
DOI:10.1016/j.bmc.2008.08.062
Mycophenolic acid (MPA), a clinically used immunosuppressant, is extensively metabolized into an inactive C7-glucuronide and removed from circulation. To circumvent the metabolic liability imposed by the C7-hydroxyl group, we have designed a series of hybrid MPA analogs based on the pharmacophores present in MPA and new generations of inosine monophosphate dehydrogenase (IMPDH) inhibitors. The synthesis of MPA analogs has been accomplished by an allylic substitution of a common lactone. Biological evaluations of these analogs and a preliminary structure–activity relationship (SAR) are presented.Aiming to circumvent the metabolic liability imposed by mycophenolic acid (MPA), a series of MPA analogs have been designed by combining the structural elements of MPA and known IMPDH inhibitors.

![N-{[4-(4-phenyl-1,3-thiazol-2-yl)tetrahydro-2h-pyran-4-yl]methyl} -3-[5-(trifluoromethyl)-1,2,4-oxadiazol-3-yl]benzamide](http://img.cochemist.com/ccimg/1314900/1314890-29-3.png)
![N-{[4-(4-phenyl-1,3-thiazol-2-yl)tetrahydro-2h-pyran-4-yl]methyl} -3-[5-(trifluoromethyl)-1,2,4-oxadiazol-3-yl]benzamide](http://img.cochemist.com/ccimg/1314900/1314890-29-3_b.png)
![PROPAN-2-YL (2S)-2-[[[(2R,3R,4R,5R)-5-(2,4-DIOXOPYRIMIDIN-1-YL)-4-FLUORO-3-HYDROXY-4-METHYLOXOLAN-2-YL]METHOXY-PHENOXYPHOSPHORYL]AMINO]PROPANOATE](http://img.cochemist.com/ccimg/1190400/1190307-88-0.png)
![PROPAN-2-YL (2S)-2-[[[(2R,3R,4R,5R)-5-(2,4-DIOXOPYRIMIDIN-1-YL)-4-FLUORO-3-HYDROXY-4-METHYLOXOLAN-2-YL]METHOXY-PHENOXYPHOSPHORYL]AMINO]PROPANOATE](http://img.cochemist.com/ccimg/1190400/1190307-88-0_b.png)




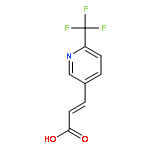

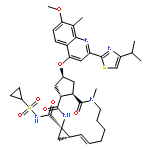
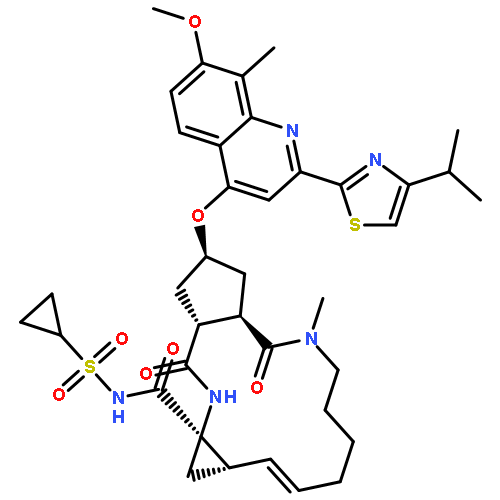
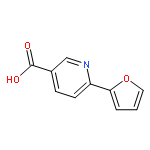
![2-Propenamide, N-hydroxy-3-[3-[(phenylamino)sulfonyl]phenyl]-, (2E)-](http://img.cochemist.com/ccimg/866400/866323-14-0.png)
![2-Propenamide, N-hydroxy-3-[3-[(phenylamino)sulfonyl]phenyl]-, (2E)-](http://img.cochemist.com/ccimg/866400/866323-14-0_b.png)
![BENZO[B]THIOPHENE-2-CARBOXAMIDE, N-HYDROXY-6-NITRO-](http://img.cochemist.com/ccimg/850100/850074-78-1.png)
![BENZO[B]THIOPHENE-2-CARBOXAMIDE, N-HYDROXY-6-NITRO-](http://img.cochemist.com/ccimg/850100/850074-78-1_b.png)

![2-Propenoic acid, 3-[2-chloro-4-(trifluoromethyl)phenyl]-, (2E)-](http://img.cochemist.com/ccimg/689400/689300-40-1.png)
![2-Propenoic acid, 3-[2-chloro-4-(trifluoromethyl)phenyl]-, (2E)-](http://img.cochemist.com/ccimg/689400/689300-40-1_b.png)
![2-Propenoic acid, 3-[2-fluoro-4-(trifluoromethyl)phenyl]-, (2E)-](http://img.cochemist.com/ccimg/545400/545393-87-1.png)
![2-Propenoic acid, 3-[2-fluoro-4-(trifluoromethyl)phenyl]-, (2E)-](http://img.cochemist.com/ccimg/545400/545393-87-1_b.png)




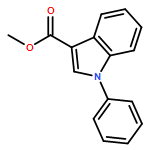
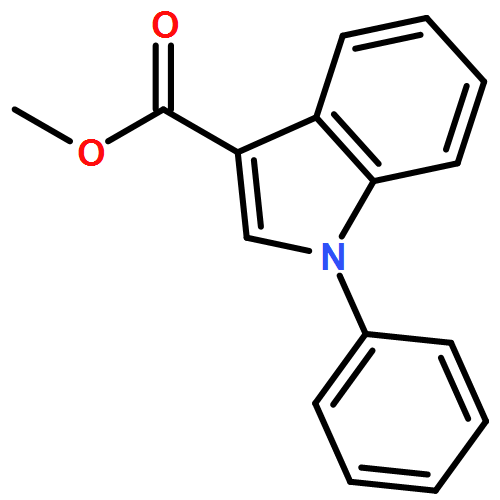
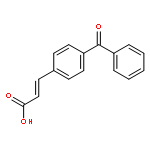
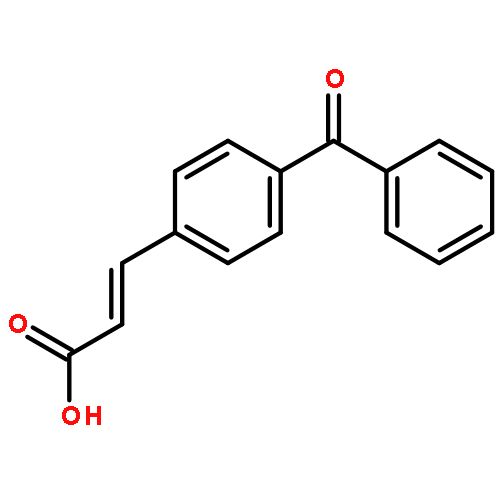
![Benzo[b]thiophene-2-carboxamide, N-hydroxy-](http://img.cochemist.com/ccimg/211200/211172-97-3.png)
![Benzo[b]thiophene-2-carboxamide, N-hydroxy-](http://img.cochemist.com/ccimg/211200/211172-97-3_b.png)
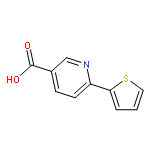
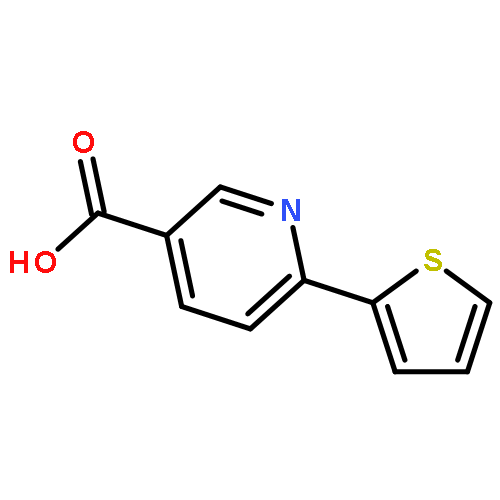
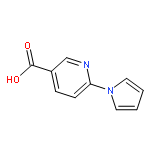
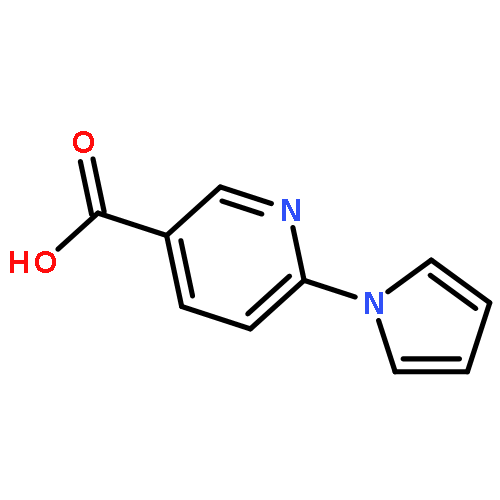


![Methyl 5-(trifluoromethyl)benzo[b]thiophene-2-carboxylate](http://img.cochemist.com/ccimg/146200/146137-92-0.png)
![Methyl 5-(trifluoromethyl)benzo[b]thiophene-2-carboxylate](http://img.cochemist.com/ccimg/146200/146137-92-0_b.png)
![2-Propenoic acid,3-[2-(trifluoromethyl)phenyl]-, (2E)-](http://img.cochemist.com/ccimg/98400/98386-81-3.png)
![2-Propenoic acid,3-[2-(trifluoromethyl)phenyl]-, (2E)-](http://img.cochemist.com/ccimg/98400/98386-81-3_b.png)
![(2E)-3-[4-(Methylsulfonyl)phenyl]propenoic acid](http://img.cochemist.com/ccimg/88900/88899-85-8.png)
![(2E)-3-[4-(Methylsulfonyl)phenyl]propenoic acid](http://img.cochemist.com/ccimg/88900/88899-85-8_b.png)
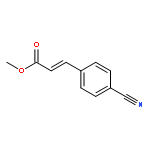

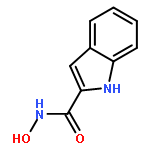
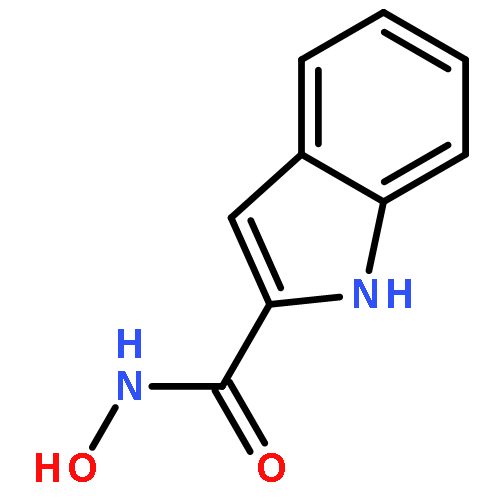
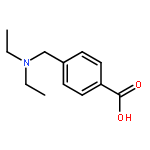
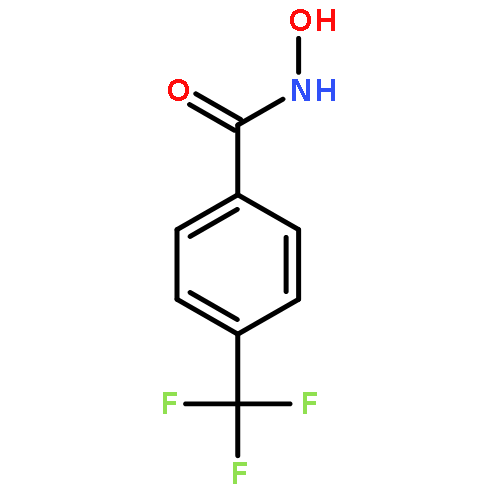
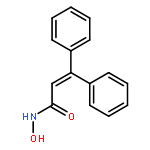
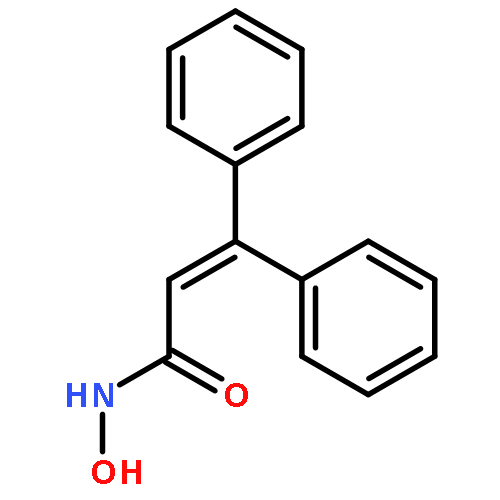
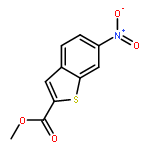

![Benzoic acid, 4-[(methylamino)carbonyl]-](http://img.cochemist.com/ccimg/23800/23754-45-2.png)
![Benzoic acid, 4-[(methylamino)carbonyl]-](http://img.cochemist.com/ccimg/23800/23754-45-2_b.png)
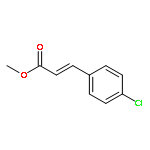
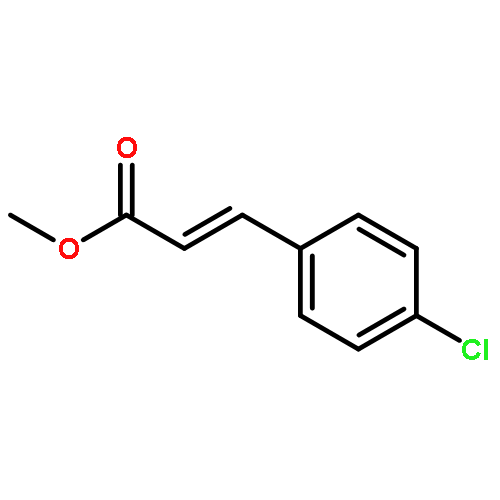
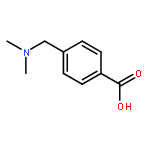
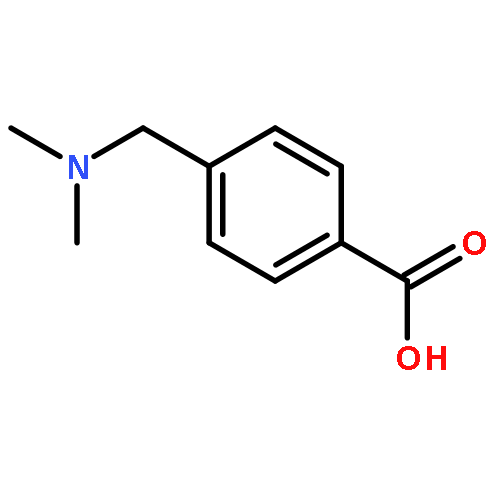
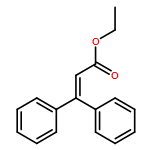
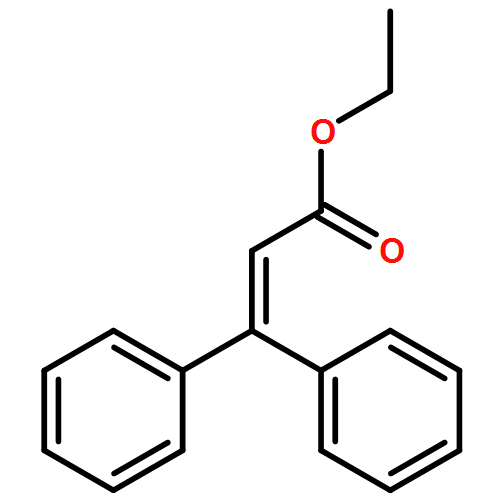

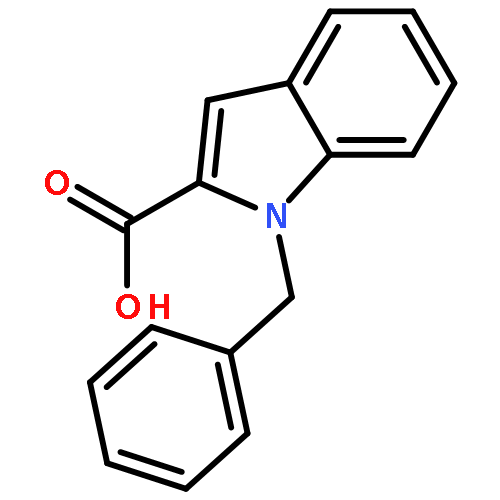
![NSC-1125476, Tetrahydro-5-[(2-hydroxy-1-naphthalenyl)methyl]-6-phenyl-2-thioxo-4(1H)-Pyrimidinone, 5-(2-Hydroxynaphthalen-1-ylmethyl)-6-phenyl-2-thioxo-2,3-dihydro-1H-pyrimidin-4-one, 5-[(2-hydroxy-1-naphthyl)methyl]-2-mercapto-6-phenyl-4(3H)-Pyrimidinone](http://img.cochemist.com/ccimg/14600/14513-15-6.png)
![NSC-1125476, Tetrahydro-5-[(2-hydroxy-1-naphthalenyl)methyl]-6-phenyl-2-thioxo-4(1H)-Pyrimidinone, 5-(2-Hydroxynaphthalen-1-ylmethyl)-6-phenyl-2-thioxo-2,3-dihydro-1H-pyrimidin-4-one, 5-[(2-hydroxy-1-naphthyl)methyl]-2-mercapto-6-phenyl-4(3H)-Pyrimidinone](http://img.cochemist.com/ccimg/14600/14513-15-6_b.png)
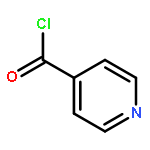
![1H-Benzo[f]chromen-3(2H)-one](http://img.cochemist.com/ccimg/5700/5690-03-9.png)
![1H-Benzo[f]chromen-3(2H)-one](http://img.cochemist.com/ccimg/5700/5690-03-9_b.png)


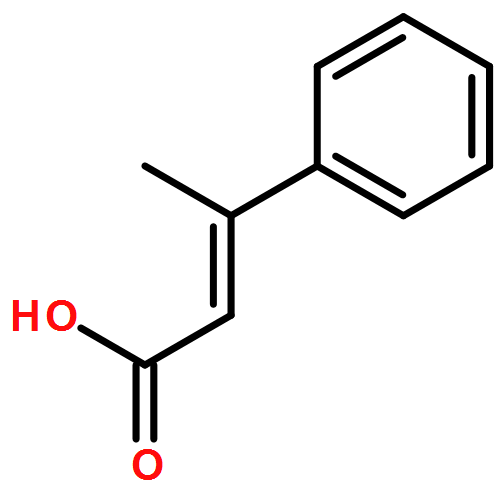
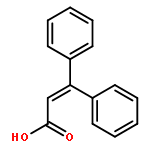
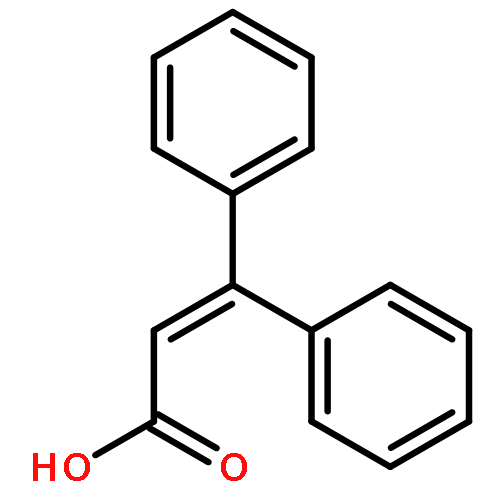




![1,3,5-Naphthalenetrisulfonicacid,8,8'-[carbonylbis[imino-3,1-phenylenecarbonylimino(4-methyl-3,1-phenylene)carbonylimino]]bis-](http://img.cochemist.com/ccimg/200/145-63-1.png)
![1,3,5-Naphthalenetrisulfonicacid,8,8'-[carbonylbis[imino-3,1-phenylenecarbonylimino(4-methyl-3,1-phenylene)carbonylimino]]bis-](http://img.cochemist.com/ccimg/200/145-63-1_b.png)