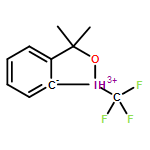
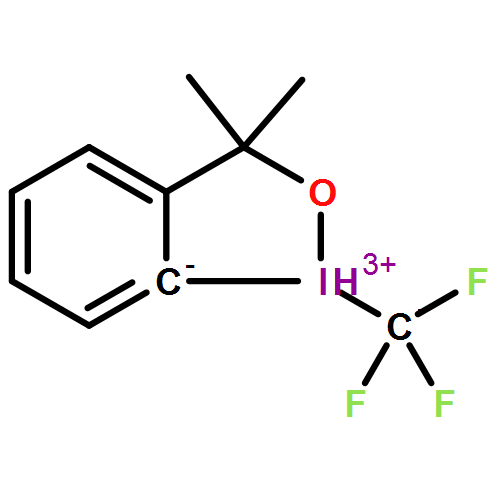
Co-reporter:Halua Pinto de Magalhães, Antonio Togni, and Hans Peter Lüthi
The Journal of Organic Chemistry November 17, 2017 Volume 82(Issue 22) pp:11799-11799
Publication Date(Web):October 7, 2017
DOI:10.1021/acs.joc.7b01716
Key for the observed reactivity of λ3-iodanes, powerful reagents for the selective transfer of functional groups to nucleophiles, are the properties of the 3-center-4-electron bond involving the iodine atom and the two linearly arranged ligands. This bond is also involved in the formation of the initial complex between the λ3-iodane and a nucleophile, which can be a solvent molecule or a reactant. The bonding in such complexes can be described by means of σ-hole interactions. In halogen compounds, σ-hole interaction was identified as a force in crystal packing or in the formation of supramolecular chains. More recently, σ-hole interactions were also shown to affect the reactivity of the iodine-based hypervalent reagents. Relative to their monovalent counterparts, where the σ-hole is located on the extension of the sigma-bond, in the hypervalent species our DFT calculations reveal the formation of a nonclassical σ-hole region with one or even two maxima. This observation is also made in fully relativistic calculations. The SAPT analysis shows that the σ-hole bond between the λ3-iodane and the nucleophile is not necessarily of purely electrostatic nature but may also contain a significant covalent component. This covalent component may facilitate chemical transformation of the compound by means of reductive elimination or other mechanisms and is therefore an indicator for its reactivity. Here, we also show that the shape, location, and strength of the σ-holes can be tuned by the choice of ligands and measures such as Brønsted activation of the iodane reagent. At the limit, the tuning transforms the nonclassical σ-hole regions into coordination sites, which allows us to control how a nucleophile will bind and react with the iodane.
Co-reporter:Halua Pinto de Magalhães, Hans Peter Lüthi and Patrick Bultinck
Physical Chemistry Chemical Physics 2016 vol. 18(Issue 2) pp:846-856
Publication Date(Web):20 Nov 2015
DOI:10.1039/C5CP05343A
Hypervalent iodine compounds, in particular λ3-iodanes, have become important reagents in organic synthesis for the electrophilic transfer of substituents to arenes and other nucleophiles. The structure and reactivity of these compounds are usually described based on a 3-center–4-electron bond model, involving the iodine central atom and its two trans substituents. The goal of this computational study is to explore Fermi correlation in view of a more advanced description of bonding in these compounds. For that matter, we apply the analysis of Domain Averaged Fermi Holes (DAFH). The DAFH analysis reveals a relationship between the occurrence of multicenter bonding and structural parameters which cannot be easily observed based on simple MO theory. Whereas for λ3-iodanes carrying electron-rich ligands pairing of electrons over three centers is indeed observed, compounds with electron-withdrawing substituents fall into a different category: the pairing of electrons is restricted to extend over two centers only, thus challenging the multicenter bonding picture in this case. Accordingly, a drastic reduction of the DAFH three center bond index is observed. The establishment of the multicenter bond in λ3-iodanes is driven by a pseudo Jahn–Teller (PJT) effect, whose extent is tightly coupled to the reactivity of the corresponding compound. The PJT stabilization scales with the degree of s–p hybridization of the central atom, which, in return, depends on the electron-withdrawing power of the ligands in the trans position. The response of the multicenter bond to the iodine “ligand field” can be expressed quantitatively in terms of DAFH bond indices. These show, for example, that the activation of the reacting hypervalent species by means of protonation results in a weaker 3-center–4-electron bond, thus making the reagent more reactive. In this work we explain a number of experimentally known facts concerning the reactivity of these compounds. We also show that the DAFH analysis offers a more complete understanding of hypervalency in λ3-iodanes, and that it is a tool to assist the search for novel reagents.
Co-reporter:Halua Pinto de Magalhães, Hans Peter Lüthi, and Antonio Togni
The Journal of Organic Chemistry 2014 Volume 79(Issue 17) pp:8374-8382
Publication Date(Web):August 11, 2014
DOI:10.1021/jo501714f
The functionalization of arenes via diaryliodonium salts has gained considerable attention in synthesis, as these compounds react under mild conditions. Mechanistic studies have shown that the formation of corresponding λ3-iodane intermediates takes a key role, as they determine the course and selectivity of the reaction. Bridged diaryliodonium salts, featuring a heterocyclic moiety involving the iodine atom, were shown to exhibit a distinctly different reactivity, leading to different products. These products are not just the result of reductive elimination reactions but may also arise via radical mechanisms. Our quantum chemical calculations reveal that the λ3-iodane intermediate is also the “gateway” for reactions that are observed only for strained bridged systems. At the same time, we find a remarkable affinity of the hypervalent region to planarity for all reaction mechanisms. This also explains the correlation between the size of the bridge connecting the aryl groups and the reaction products observed. Furthermore, the energetics of these competing reactions are examined by analysis of the mechanisms. Finally, using model compounds, some of the basic features governing the reactivity of λ3-iodanes are discussed.
Co-reporter:Halua Pinto de Magalhães, Hans Peter Lüthi, and Antonio Togni
Organic Letters 2012 Volume 14(Issue 15) pp:3830-3833
Publication Date(Web):July 19, 2012
DOI:10.1021/ol3014039
This computational study investigates the factors governing the selectivity of the reductive eliminations from rapidly equilibrating isomeric λ3-iodanes derived from a diaryl iodonium salt and a nucleophile. The chemoselectivity is mainly determined by the partial charge at the ipso-carbon atom involved in the 3-center–4-electron bond.
Co-reporter:Stefano Borini, Peter A. Limacher and Hans Peter Lüthi
The Journal of Physical Chemistry A 2010 Volume 114(Issue 5) pp:2221-2229
Publication Date(Web):January 19, 2010
DOI:10.1021/jp908439x
As part of a systematic study of the impact of donor−acceptor substitution on the structure and properties of π-conjugated compounds, we present a theoretical investigation of the all-trans polyacetylene backbone, end-capped with moieties of different donor or acceptor natures and different strengths, focusing on the effects induced by these substituents on bond lengths and shape of the conjugated chain. Optimized geometries for polyacetylene containing 15 and 20 double bonds have been computed by means of density functional theory with the Coulomb-attenuating B3LYP (CAM-B3LYP) functional. We show that the simultaneous presence of two substituents has a cooperative effect on the lengths of single and double bonds. We also show that, depending on the substitution pattern, distortion toward bow- or S-shaped structures occurs. Two new geometric parameters are defined in order to evaluate the mode and intensity of this distortion. Cubic Bézier curves have been used as simple geometric models for the chain bending behavior.
Co-reporter:Halua Pinto de Magalhães, Hans Peter Lüthi and Patrick Bultinck
Physical Chemistry Chemical Physics 2016 - vol. 18(Issue 2) pp:NaN856-856
Publication Date(Web):2015/11/20
DOI:10.1039/C5CP05343A
Hypervalent iodine compounds, in particular λ3-iodanes, have become important reagents in organic synthesis for the electrophilic transfer of substituents to arenes and other nucleophiles. The structure and reactivity of these compounds are usually described based on a 3-center–4-electron bond model, involving the iodine central atom and its two trans substituents. The goal of this computational study is to explore Fermi correlation in view of a more advanced description of bonding in these compounds. For that matter, we apply the analysis of Domain Averaged Fermi Holes (DAFH). The DAFH analysis reveals a relationship between the occurrence of multicenter bonding and structural parameters which cannot be easily observed based on simple MO theory. Whereas for λ3-iodanes carrying electron-rich ligands pairing of electrons over three centers is indeed observed, compounds with electron-withdrawing substituents fall into a different category: the pairing of electrons is restricted to extend over two centers only, thus challenging the multicenter bonding picture in this case. Accordingly, a drastic reduction of the DAFH three center bond index is observed. The establishment of the multicenter bond in λ3-iodanes is driven by a pseudo Jahn–Teller (PJT) effect, whose extent is tightly coupled to the reactivity of the corresponding compound. The PJT stabilization scales with the degree of s–p hybridization of the central atom, which, in return, depends on the electron-withdrawing power of the ligands in the trans position. The response of the multicenter bond to the iodine “ligand field” can be expressed quantitatively in terms of DAFH bond indices. These show, for example, that the activation of the reacting hypervalent species by means of protonation results in a weaker 3-center–4-electron bond, thus making the reagent more reactive. In this work we explain a number of experimentally known facts concerning the reactivity of these compounds. We also show that the DAFH analysis offers a more complete understanding of hypervalency in λ3-iodanes, and that it is a tool to assist the search for novel reagents.