Co-reporter:Hannes K. Buchholz, Rebecca K. Hylton, Jan Gerit Brandenburg, Andreas Seidel-Morgenstern, Heike Lorenz, Matthias Stein, and Sarah L. Price
Crystal Growth & Design September 6, 2017 Volume 17(Issue 9) pp:4676-4676
Publication Date(Web):July 18, 2017
DOI:10.1021/acs.cgd.7b00582
The separation of an enantiomer from a racemic mixture is of primary relevance to the pharmaceutical industry. The thermochemical properties of organic enantiopure and racemate crystals can be exploited to design an enantioselective crystallization process. The thermodynamic difference between the two crystal forms is accessible by two cycles which give the eutectic composition in solution. The “sublimation cycle” requires calculating the lattice energy and phonon frequencies of the crystal structures. Experimental results from heat capacity and other thermodynamic measurements of enantiopure and racemic crystals are compared with a variety of molecular and crystal structure-based calculations. This is done for three prototypes of pharmaceutical-like molecules with different degrees of molecular flexibility. Differences in crystal packing result in varying temperature-dependent heat capacities and affect the sublimation thermodynamics, relative solubility, and eutectic composition. Many simplifying assumptions about the thermodynamics and solubilities of the racemic and enantiopure crystals are critically evaluated. We show that calculations and experimental information using the sublimation cycle can guide the design of processes to resolve enantiomers by crystallization.
Co-reporter:Luca Iuzzolino, Anthony M. Reilly, Patrick McCabe, and Sarah L. Price
Journal of Chemical Theory and Computation October 10, 2017 Volume 13(Issue 10) pp:5163-5163
Publication Date(Web):September 11, 2017
DOI:10.1021/acs.jctc.7b00623
Determining the range of conformations that a flexible pharmaceutical-like molecule could plausibly adopt in a crystal structure is a key to successful crystal structure prediction (CSP) studies. We aim to use conformational information from the crystal structures in the Cambridge Structural Database (CSD) to facilitate this task. The conformations produced by the CSD Conformer Generator are reduced in number by considering the underlying rotamer distributions, an analysis of changes in molecular shape, and a minimal number of molecular ab initio calculations. This method is tested for five pharmaceutical-like molecules where an extensive CSP study has already been performed. The CSD informatics-derived set of crystal structure searches generates almost all the low-energy crystal structures previously found, including all experimental structures. The workflow effectively combines information on individual torsion angles and then eliminates the combinations that are too high in energy to be found in the solid state, reducing the resources needed to cover the solid-state conformational space of a molecule. This provides insights into how the low-energy solid-state and isolated-molecule conformations are related to the properties of the individual flexible torsion angles.
Co-reporter:Sarah L. Price, Doris E. Braun and Susan M. Reutzel-Edens
Chemical Communications 2016 vol. 52(Issue 44) pp:7065-7077
Publication Date(Web):04 Apr 2016
DOI:10.1039/C6CC00721J
Computational crystal structure prediction (CSP) methods can now be applied to the smaller pharmaceutical molecules currently in drug development. We review the recent uses of computed crystal energy landscapes for pharmaceuticals, concentrating on examples where they have been used in collaboration with industrial-style experimental solid form screening. There is a strong complementarity in aiding experiment to find and characterise practically important solid forms and understanding the nature of the solid form landscape.
Co-reporter:Rebecca K. Hylton; Graham J. Tizzard; Terence L. Threlfall; Amy L. Ellis; Simon J. Coles; Colin C. Seaton+; Eric Schulze; Heike Lorenz; Andreas Seidel-Morgenstern; Matthias Stein
Journal of the American Chemical Society 2015 Volume 137(Issue 34) pp:11095-11104
Publication Date(Web):August 5, 2015
DOI:10.1021/jacs.5b05938
Mandelic acids are prototypic chiral molecules where the sensitivity of crystallized forms (enantiopure/racemic compound/polymorphs) to both conditions and substituents provides a new insight into the factors that may allow chiral separation by crystallization. The determination of a significant number of single crystal structures allows the analysis of 13 enantiopure and 30 racemic crystal structures of 21 (F/Cl/Br/CH3/CH3O) substituted mandelic acid derivatives. There are some common phenyl packing motifs between some groups of racemic and enantiopure structures, although they show very different hydrogen-bonding motifs. The computed crystal energy landscape of 3-chloromandelic acid, which has at least two enantiopure and three racemic crystal polymorphs, reveals that there are many more possible structures, some of which are predicted to be thermodynamically more favorable as well as slightly denser than the known forms. Simulations of mandelic acid dimers in isolation, water, and toluene do not differentiate between racemic and enantiopure dimers and also suggest that the phenyl ring interactions play a major role in the crystallization mechanism. The observed crystallization behavior of mandelic acids does not correspond to any simple “crystal engineering rules” as there is a range of thermodynamically feasible structures with no distinction between the enantiopure and racemic forms. Nucleation and crystallization appear to be determined by the kinetics of crystal growth with a statistical bias, but the diversity of the mandelic acid crystallization behavior demonstrates that the factors that influence the kinetics of crystal nucleation and growth are not yet adequately understood.
Co-reporter:Ogaga G. Uzoh, Peter T. A. Galek and Sarah L. Price
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 12) pp:7936-7948
Publication Date(Web):2015/02/16
DOI:10.1039/C4CP05525J
In traditional molecular mechanics force fields, intramolecular non-bonded interactions are modelled as intermolecular interactions, and the form of the torsion potential is based on the conformational profiles of small organic molecules. We investigate how a separate model for the intramolecular forces in pharmaceuticals could be more realistic by analysing the low barrier to rotation of the phenyl ring in the fenamates (substituted N-phenyl-aminobenzoic acids), that results in a wide range of observed angles in the numerous fenamate crystal structures. Although the conformational energy changes by significantly less than 10 kJ mol−1 for a complete rotation of the phenyl ring for fenamic acid, the barrier is only small because of small correlated changes in the other bond and torsion angles. The maxima for conformations where the two aromatic rings approach coplanarity arise from steric repulsion, but the maxima when the two rings are approximately perpendicular arise from a combination of an electronic effect and intramolecular dispersion. Representing the ab initio conformational energy profiles as a cosine series alone is ineffective; however, combining a cos2ξ term to represent the electronic barrier with an intramolecular atom–atom exp-6 term for all atom pairs separated by three or more bonds (1–4 interactions) provides a very effective representation. Thus we propose a new, physically motivated, generic analytical model of conformational energy, which could be combined with an intermolecular model to form more accurate force-fields for modelling the condensed phases of pharmaceutical-like organic molecules.
Co-reporter:Nizar Issa, Sarah A. Barnett, Sharmarke Mohamed, Doris E. Braun, Royston C. B. Copley, Derek A. Tocher and Sarah L. Price
CrystEngComm 2012 vol. 14(Issue 7) pp:2454-2464
Publication Date(Web):25 Jan 2012
DOI:10.1039/C2CE06325E
The ability of the pharmaceutically acceptable cocrystallising agents, succinic acid and 4-aminobenzoic acid, to form cocrystals with ten small organic molecules with hydrogen bonding acceptors but no donors, was investigated by grinding, hot-stage microscopy and solution based crystallisation experiments. The reproducible results obtained by different methods showed that only six cocrystals formed. The crystal structures of the four novel cocrystals, succinic acid·2,2′-bipyridine (1:1, P21/c, I), succinic acid·diphenylcyclopropenone (1:2, P21/c, II), 4-aminobenzoic acid·antipyrine (1:1, P21, III) and 4-aminobenzoic acid·phenazine (1:2, P, IV), are reported. The computed crystal energy landscapes of the cocrystals and their components show why succinic acid·1,4-dicyanobenzene did not form a cocrystal as well as predicting the observed structure of succinic acid·2,2′-bipyridine as the most stable. The most stable hypothetical structures of a 1:1 succinic acid·1,4-dicyanobenzene cocrystal are closely related to those of the components. The results demonstrate that cocrystal formation requires both hydrogen bonding and close packing, and so markedly non-planar pharmaceuticals will be quite specific in the steric and hydrogen bonding disposition requirement of coformers.
Co-reporter:Ogaga G. Uzoh, Aurora J. Cruz-Cabeza, and Sarah L. Price
Crystal Growth & Design 2012 Volume 12(Issue 8) pp:4230-4239
Publication Date(Web):July 11, 2012
DOI:10.1021/cg3007348
The concept of a polymorphophore was investigated by contrasting the crystal energy landscapes of monomorphic fenamic acid (2-(phenylamino)-benzoic acid, FA) and one of its highly polymorphic derivatives, tolfenamic acid (2-[(3-chloro-2-methylphenyl)amino]-benzoic acid, TA). The crystal energy landscapes of both molecules show that the benzoic acid R22(8) dimer motif is found in all low energy crystal structures, but conformational flexibility of the phenyl rings leads to a wide range of crystal structures with different packings of this dimer. Many of the observed fenamate crystal structures can overlay a significant proportion of the coordination environment with other observed or calculated structures, but the substituents of the phenyl group affect the ordering of the related low energy crystal structures. The crystal energy landscape of tolfenamic acid has several crystal structures, including the observed polymorphs, tightly clustered around the global minimum, whereas the corresponding cluster contains only the observed and a closely related structure for fenamic acid. Hence, the fenamate fragment is potentially permissive of a large number of structures because of the conformational flexibility, but the substituents determine whether a specific fenamate will be polymorphic. Thus, a polymorphophore promotes but does not guarantee polymorphism.
Co-reporter:Doris E. Braun, Panagiotis G. Karamertzanis and Sarah L. Price
Chemical Communications 2011 vol. 47(Issue 19) pp:5443-5445
Publication Date(Web):08 Apr 2011
DOI:10.1039/C1CC10762C
A study of two dihydroxybenzoic acid isomers shows that computational methods can be used to predict hydrate formation, the compound∶water ratio and hydrate crystal structures. The calculations also help identify a novel hydrate found in the solid form screening that validates this study.
Co-reporter:Doris E. Braun, Miguel Ardid-Candel, Emiliana D’Oria, Panagiotis G. Karamertzanis, Jean-Baptiste Arlin, Alastair J. Florence, Alan G. Jones, and Sarah L. Price
Crystal Growth & Design 2011 Volume 11(Issue 12) pp:5659-5669
Publication Date(Web):November 2, 2011
DOI:10.1021/cg201203u
Following the computational prediction that (RS)-naproxen would be more stable than the therapeutically used and more studied homochiral (S)-naproxen, we performed an interdisciplinary study contrasting the two compounds. The crystal structure of the racemic compound was solved from powder X-ray diffraction data (Pbca) and showed no packing similarity with the homochiral structure (P21). The binary melting point phase diagram was constructed to confirm the nature of the racemic species, and differential scanning calorimetric and solubility measurements were used to estimate the enthalpy difference between the crystals (ΔHR+S→RScry) to be −1.5 ± 0.3 kJ·mol–1 at T ∼ 156 °C and −2.4 ± 1.0 kJ·mol–1 in the range 10–40 °C. A comparison of the different approximations involved in estimating ΔHR+S→RScry implied that the difference in the lattice energies overestimated the stability of the (RS) crystal. The naproxen lattice energy landscape confirmed that all the practically important crystal structures have been found and characterized and provided insights into the crystal growth problems of the racemic form. This highlights the complementarity of computational modeling in investigating chiral crystallization.
Co-reporter:Emiliana D’Oria, Panagiotis G. Karamertzanis and Sarah L. Price
Crystal Growth & Design 2010 Volume 10(Issue 4) pp:1749-1756
Publication Date(Web):February 26, 2010
DOI:10.1021/cg9014306
We used crystal structure prediction methods to generate racemic and homochiral crystal structures of benzo(c)phenanthrene, 3,4-dehydroproline anhydride, and 2,6-dimethylglycoluril, which are all known to spontaneously resolve. The known homochiral crystal structures were found at or near the global minimum in lattice energy; however, in all three cases there were hypothetical racemic crystal structures within a few kJ mol−1 in energy. The comparison of hypothetical racemic structures with the known homochiral crystal structures showed structural similarities, despite the symmetry differences, suggesting that most molecules are very unlikely to crystallize in a chiral crystal structure that is markedly more stable than any racemic crystal. Thus the experimentally observed asymmetry in the thermodynamic favorability of racemic and homochiral crystal structures is not due to experimental bias; that is, any thermodynamic drive for spontaneous resolution is genuinely small. Hence, whereas the formation of a racemic crystal can have a significant enthalpic stability advantage over all possible homochiral crystal structures and be more readily predicted, spontaneous resolution cannot be predicted without careful consideration of entropic effects and accurate computational models.
Co-reporter:Matthew Habgood and Sarah L. Price
Crystal Growth & Design 2010 Volume 10(Issue 7) pp:3263
Publication Date(Web):June 1, 2010
DOI:10.1021/cg100405s
Caffeine cocrystallizes with each of the monohydroxybenzoic acids (2-, 3-, and 4-hydroxybenzoic acid (HBA)) but with very different behavior.(1) The computed crystal energy landscapes for 1:1 systems of caffeine with 2HBA, two conformers of 3HBA, and 4HBA, presented in this paper, help rationalize this diversity. The known cocrystal structures of caffeine/2HBA and caffeine/3HBA are found and calculated to be stable relative to their separate components. The instability of any possible cocrystal between caffeine and the more stable conformer of 3HBA, relative to the experimental structure, is confirmed, as is the ability of caffeine and 4HBA to form cocrystals. Comparison of all the energy landscapes, and in particular the distributions of different hydrogen bonding motifs, provides a rationalization of the variation in crystallization behavior between the isomers and conformers in this superficially similar homologous series. In particular, an explanation is suggested for the formation of three-independent-molecule cocrystals in the case of 4HBA but not 2- or 3HBA.
Co-reporter:Sarah L. Price, Maurice Leslie, Gareth W. A. Welch, Matthew Habgood, Louise S. Price, Panagiotis G. Karamertzanis and Graeme M. Day
Physical Chemistry Chemical Physics 2010 vol. 12(Issue 30) pp:8478-8490
Publication Date(Web):07 Jul 2010
DOI:10.1039/C004164E
Crystal structure prediction for organic molecules requires both the fast assessment of thousands to millions of crystal structures and the greatest possible accuracy in their relative energies. We describe a crystal lattice simulation program, DMACRYS, emphasizing the features that make it suitable for use in crystal structure prediction for pharmaceutical molecules using accurate anisotropic atom–atom model intermolecular potentials based on the theory of intermolecular forces. DMACRYS can optimize the lattice energy of a crystal, calculate the second derivative properties, and reduce the symmetry of the spacegroup to move away from a transition state. The calculated terahertz frequency k = 0 rigid-body lattice modes and elastic tensor can be used to estimate free energies. The program uses a distributed multipole electrostatic model (Qat, t = 00,…,44s) for the electrostatic fields, and can use anisotropic atom–atom repulsion models, damped isotropic dispersion up to R−10, as well as a range of empirically fitted isotropic exp-6 atom–atom models with different definitions of atomic types. A new feature is that an accurate model for the induction energy contribution to the lattice energy has been implemented that uses atomic anisotropic dipole polarizability models (αat, t = (10,10)…(11c,11s)) to evaluate the changes in the molecular charge density induced by the electrostatic field within the crystal. It is demonstrated, using the four polymorphs of the pharmaceutical carbamazepine C15H12N2O, that whilst reproducing crystal structures is relatively easy, calculating the polymorphic energy differences to the accuracy of a few kJ mol−1 required for applications is very demanding of assumptions made in the modelling. Thus DMACRYS enables the comparison of both known and hypothetical crystal structures as an aid to the development of pharmaceuticals and other speciality organic materials, and provides a tool to develop the modelling of the intermolecular forces involved in molecular recognition processes.
Co-reporter:Sarah (Sally) L. Price
Accounts of Chemical Research 2009 Volume 42(Issue 1) pp:117
Publication Date(Web):October 17, 2008
DOI:10.1021/ar800147t
The phenomenon of polymorphism, the ability of a molecule to adopt more than one crystal structure, is a well-established property of crystalline solids. The possible variations in physical properties between polymorphs make the reliable reproduction of a crystalline form essential for all research using organic materials, as well as quality control in manufacture. Thus, the last two decades have seen both an increase in interest in polymorphism and the availability of the computer power needed to make the computational prediction of organic crystal structures a practical possibility. In the past decade, researchers have made considerable improvements in the theoretical basis for calculating the sets of structures that are within the energy range of possible polymorphism, called crystal energy landscapes. It is common to find that a molecule has a wide variety of ways of packing with lattice energy within a few kilojoules per mole of the most stable structure. However, as we develop methods to search for and characterize “all” solid forms, it is also now usual for polymorphs and solvates to be found. Thus, the computed crystal energy landscape reflects and to an increasing extent “predicts” the emerging complexity of the solid state observed for many organic molecules. This Account will discuss the ways in which the calculation of the crystal energy landscape of a molecule can be used as a complementary technique to solid form screening for polymorphs. Current methods can predict the known crystal structure, even under “blind test” conditions, but such successes are generally restricted to those structures that are the most stable over a wide range of thermodynamic conditions. The other low-energy structures can be alternative polymorphs, which have sometimes been found in later experimental studies. Examining the computed structures reveals the various compromises between close packing, hydrogen bonding, and π−π stacking that can result in energetically feasible structures. Indeed, we have observed that systems with many almost equi-energetic structures that contain a common interchangeable motif correlate with a tendency to disorder and problems with control of the crystallization product. Thus, contrasting the computed crystal energy landscape with the known crystal structures of a given molecule provides a valuable complement to solid form screening, and the examination of the low-energy structures often leads to a rationalization of the forms found.
Co-reporter:Nizar Issa, Panagiotis G. Karamertzanis, Gareth W. A. Welch and Sarah L. Price
Crystal Growth & Design 2009 Volume 9(Issue 1) pp:442-453
Publication Date(Web):November 24, 2008
DOI:10.1021/cg800685z
A cocrystal is only expected to form if it is thermodynamically more stable than the crystals of its components. To test whether this can be predicted with a current computational methodology, we compare the lattice energies of 12 cocrystals of 4-aminobenzoic acid, 8 of succinic acid and 6 of caffeine, with the sums of the lattice energies of their components. These three molecules were chosen for their potential use in pharmaceutical cocrystals and because they had sufficient determinations of cocrystals and corresponding partner crystal structures in the Cambridge Structural Database. The lattice energies were evaluated using anisotropic intermolecular atom−atom potentials, with the electrostatic model and the intramolecular energy penalty for changes in specified torsion angles derived from ab initio calculations on the isolated molecules. The majority of the cocrystals are calculated to be more stable than their components, but the energy difference is only large in a few of the cases where the partner molecule cannot hydrogen bond to itself. More typically, the cocrystal stabilization is comparable to polymorphic energy differences and some of the specifically identified errors in the computational modeling. The cocrystals will be more stable relative to the observed disordered structures of caffeine and the kinetically preferred polymorph of 4-aminobenzoic acid, highlighting kinetic factors that may be involved in cocrystal formation. Overall, it appears that cocrystal formation should generally be predictable by comparing the relative stability of the most stable cocrystal and its pure components found on the computed crystal energy landscapes, but this is often very demanding of the accuracy of the method used to calculate the crystal energy.
Co-reporter:Royston C. B. Copley, Sarah A. Barnett, Panagiotis G. Karamertzanis, Kenneth D. M. Harris, Benson M. Kariuki, Mingcan Xu, E. Anne Nickels, Robert W. Lancaster and Sarah L. Price
Crystal Growth & Design 2008 Volume 8(Issue 9) pp:3474-3481
Publication Date(Web):August 6, 2008
DOI:10.1021/cg800517h
Detailed analysis of X-ray diffraction data from four single crystals of eniluracil, prepared under different crystallization conditions, confirms a picture in which the crystals exhibit different degrees of disorder, which is also suggested by the computed low energy crystal structures. Since several of these crystal structures that effectively differ by an interchange of the oxygen and hydrogen atoms on C(4) and C(6) are essentially equi-energetic, growth errors that may be difficult to reverse are practically inevitable. The structural variations observed for the crystals of eniluracil studied are more appropriately described in terms of variable degrees of disorder rather than polymorphism. Analysis of the computed crystal energy landscape for interchangeable hydrogen-bonded (or other strong) motifs is, therefore, shown to be a valuable complement to X-ray diffraction and solid-state NMR for understanding and characterizing disorder in organic solid state systems. In the case of eniluracil, this detailed picture probably accounts for the challenges in devising a robust production process for this anticancer agent in the 1990s. The specific nature of the disorder accounts for different structures being obtained from powder X-ray diffraction data of different samples, and the possibility of publishable single crystal X-ray refinements also being interpreted as polymorphism rather than disorder.
Co-reporter:Sarah L. Price
Physical Chemistry Chemical Physics 2008 vol. 10(Issue 15) pp:1996-2009
Publication Date(Web):19 Feb 2008
DOI:10.1039/B719351C
Many organic molecules are emerging as having many crystalline forms, including polymorphs and solvates, as more techniques are being used to generate and characterise the organic solid state. The fundamental scientific and industrial interest in controlling crystallisation is inspiring the development of computational methods of predicting which crystal structures are thermodynamically feasible. Sometimes, computing this crystal energy landscape will reveal that a molecule has one way of packing with itself that is sufficiently more favourable than any other so only this crystal structure will be observed. More frequently, there will be many energy minima that are energetically feasible, showing approximately equi-energetic compromises between the various intermolecular interactions allowed by the conformational flexibility. Such cases generally lead to multiple solid forms. At the moment, we usually calculate the lattice energy landscape, an approximation to the real crystal energy landscape at 0 K. Despite its limitations, many studies show that this is a valuable complement to solid form screening, which helps in discovering new structures as well as rationalising the solid forms that are found in experimental searches. The range of factors that can determine which of the thermodynamically feasible crystal structures are observed polymorphs, shows the many further challenges in developing crystal energy landscapes as a tool for control of the organic solid state.
Co-reporter:Sarah A. Barnett, Ashley T. Hulme, Nizar Issa, Thomas C. Lewis, Louise S. Price, Derek A. Tocher and Sarah L. Price
New Journal of Chemistry 2008 vol. 32(Issue 10) pp:1761-1775
Publication Date(Web):12 Aug 2008
DOI:10.1039/B806763E
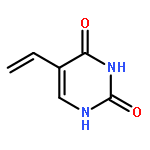
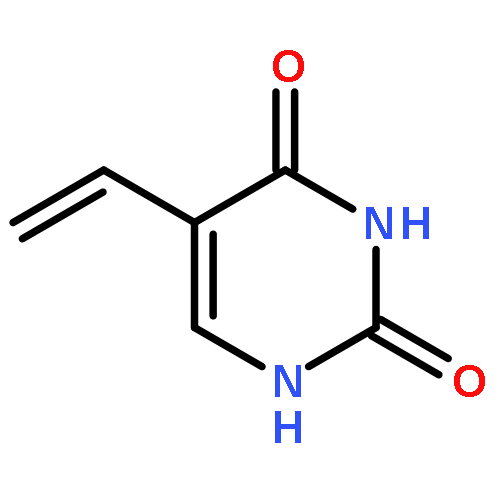
A search of the Cambridge Structural Database for crystal structures of 5-substituted uracils shows that, although there is a recurrent motif with symmetric hydrogen bonding and interdigitation of the 5-substituent R, a range of other hydrogen bonded ribbons, sheets and three-dimensional motifs are possible. In order to try and rationalize this, we have performed a combination of experimental studies and computational searches for low energy structures for the 12 simple 5-substituted uracils with R = H, CH3, CH2CH3, CHCH2, CN, OH, NH2, NO2, F, Cl, Br and I. Crystallization experiments on these compounds yielded the first single crystal X-ray determinations of 5-ethyluracil and 5-cyanouracil, as well as low temperature redeterminations of the disordered structures of 5-chlorouracil and 5-bromouracil. The lattice energies were calculated for the known crystal structures and compared with the computed lattice energy landscape for each molecule (except R = Br and I). Although the symmetric ribbon motif often dominates the computed crystal energy landscape, all of the molecules show a variety of different hydrogen bonding structures within a small energy range (5 kJ mol−1) of the global minimum and exhibit quite a diverse range of energetically competitive motifs. Thus, the range of crystallization outcomes, from polymorphism and other multiple forms, to the difficulty in growing single crystals (R = CHCH2 and NH2) probably reflects the sensitivity of the various hydrogen bonding motifs to the substituent and limited range of crystallization conditions that can be applied.
Co-reporter:Robert W. Lancaster;Panagiotis G. Karamertzanis;Ashley T. Hulme;Derek A. Tocher;Thomas C. Lewis
Journal of Pharmaceutical Sciences 2007 Volume 96(Issue 12) pp:3419-3431
Publication Date(Web):9 JUL 2007
DOI:10.1002/jps.20983
Progesterone has been known to be polymorphic for over 70 years, and crystallization conditions for the production of both experimentally characterized polymorphs have been repeatedly reported in the literature up to 1975. Nevertheless, our attempts to produce crystals of the metastable form 2 suitable for single crystal X-ray diffraction failed until the structurally related molecule pregnenolone was introduced as an additive into the crystallization solution. Accurate low temperature crystal structures were obtained for forms 1 and 2, pregnenolone and a newly discovered pregnenolone–progesterone co-crystal, which appeared concomitantly with progesterone forms 1 and 2. Computational work based on the experimental crystal structures and those generated by a search for low energy structures showed that the crystallization of enantiomerically pure progesterone results in a more strained conformation compared with the racemate due to the rotation of the acetyl and 21-methyl groups. The role of impurities or additives in influencing crystallization outcome is discussed. © 2007 Wiley-Liss, Inc. and the American Pharmacists Association J Pharm Sci 96: 3419–3431, 2007
Co-reporter:Robert W. Lancaster, Panagiotis G. Karamertzanis, Ashley T. Hulme, Derek A. Tocher, Douglas F. Covey and Sarah L. Price
Chemical Communications 2006 (Issue 47) pp:4921-4923
Publication Date(Web):16 Oct 2006
DOI:10.1039/B611599C
A computational prediction that mixing the synthetic mirror image of progesterone with its natural form would produce a specific racemic crystal structure was validated.
Co-reporter:Royston C. B. Copley, Lucie S. Deprez, Thomas C. Lewis and Sarah L. Price
CrystEngComm 2005 vol. 7(Issue 69) pp:421-428
Publication Date(Web):28 Jun 2005
DOI:10.1039/B504756K
A manual crystallization screen was performed on 3-oxauracil and 5-hydroxyuracil (isobarbituric acid), culminating in the first determination of their crystal structures. Concurrently but independently, the low energy crystal structures of these molecules were computed by a search for minima in the lattice energy. The crystal structure of 3-oxauracil corresponded to the global minimum in the lattice energy, with an unusually large energy gap of 4 kJ mol−1 between the observed and other hypothetical crystal structures. Therefore, this structure was easily predicted despite some inadequacies in the computational model. The combination of the experimental and computational search suggests that this is the most thermodynamically stable anhydrous crystal structure of 3-oxauracil and it seems unlikely that it will have any readily produced polymorphs. The experimental crystal structure of 5-hydroxyuracil was also found as a low energy crystal structure in the search but a few other hypothetical structures with different hydrogen bonding motifs were predicted to be thermodynamically competitive. It is therefore possible that other polymorphs might be found for 5-hydroxyuracil, although they were not found in this crystallization screen. These successful crystal structure predictions illustrate that the confidence with which crystal structures and polymorphism can be predicted varies between structurally similar molecules.
Co-reporter:T. C. Lewis, D. A. Tocher, G. M. Day and S. L. Price
CrystEngComm 2003 vol. 5(Issue 2) pp:3-9
Publication Date(Web):14 Jan 2003
DOI:10.1039/B211784C
A computational search to predict the crystal structure of parabanic acid produced the known P21/c crystal structure as the global minimum in the lattice energy. However, there are many hypothetical structures only 2–6 kJ mol−1 less stable than the known form, which are within the energy range of possible polymorphism and have reasonable mechanical properties and relative growth rates. The harmonic intermolecular frequencies and the attachment energy estimate of relative growth rates suggest that the known polymorph is thermodynamically and kinetically favoured, but the possibility of other polymorphs cannot be excluded. A simultaneous experimental search for new polymorphs found crystals with a new morphology and X-ray powder pattern when a solution of parabanic acid in methanol was left to evaporate. Eventually, the structure was shown by single crystal X-ray diffraction to be that of oxo-ureido-acetic acid methyl ester. Thus, under the conditions of recrystallisation from methanol, parabanic acid had undergone a previously unreported ring-opening reaction, and had not crystallised as a new polymorph as had seemed likely prior to single crystal characterisation. The combination of the experimental and theoretical studies indicates that new polymorphs of parabanic acid are unlikely to be found readily.
Co-reporter:A. T. Anghel, G. M. Day and S. L. Price
CrystEngComm 2002 vol. 4(Issue 62) pp:348-355
Publication Date(Web):18 Jul 2002
DOI:10.1039/B202084J
The oldest crystal structure of pyridine is unusually complex, with four molecules in the asymmetric unit cell of Pna21 symmetry. In an attempt to understand why pyridine crystallises with 16 molecules in the unit cell, we have considered its thermodynamic stability relative to hypothetical pyridine structures. These were generated by a search for minima in the lattice energy of pyridine amongst the more common space groups, using the crystal structure prediction procedure MOLPAK followed by lattice energy minimisation using a distributed multipole-based intermolecular potential. We find over two dozen distinct crystal structures in the energy gap of less than 6 kJ mol−1 between the corresponding models for the observed and most stable (hypothetical) structure. Adding harmonic phonon estimates of the intermolecular zero point energy and entropy at the melting point of pyridine slightly improves the relative stability of the observed Z
=
16 structure. Several of these hypothetical structures can be eliminated as only just mechanically stable, or because the growth rate of the crystal is estimated to be very slow by the attachment energy model. Nevertheless, there are still over a dozen structures that appear competitive with the known structure as polymorphs of pyridine. Following these predictions, an intense experimental search has found a new polymorph of perdeutero-pyridine (form II), which was not found in the search. This structure is also predicted to be metastable with a similar energy to form I. Although there is some evidence for kinetic factors favouring the observed structures, the metastable Z
= 16 structure and the new form II remain a challenge for our understanding of crystallisation.
Co-reporter:Theresa Beyer, Thomas Lewis and Sarah L. Price
CrystEngComm 2001 vol. 3(Issue 44) pp:178-212
Publication Date(Web):
DOI:10.1039/B108135G
A survey of the molecules which have been used in crystal structure prediction studies is presented. The results of these studies have been analysed in terms of whether the experimentally observed crystal structures are found at or near the global minimum in the lattice energy. The results suggest that whilst some crystal structures can be predicted just on the basis of lattice energy searches, there is yet insufficient experience to judge for which molecules this energetic criterion is sufficient, within the limitations of current force-field accuracy. The molecules chosen to test crystal structure prediction methods appear to be biased away from the types that would be expected to be readily predictable and suitable for crystal engineering. The survey highlights the need for more theoretical and experimental collaboration to understand what determines whether a molecule's crystal structure will be so favourable that other polymorphs are unlikely.
Co-reporter:Ola Engkvist, Sarah L. Price and Anthony J. Stone
Physical Chemistry Chemical Physics 2000 vol. 2(Issue 13) pp:3017-3027
Publication Date(Web):31 May 2000
DOI:10.1039/A910352J
A new method of probing surface–surface interactions and calculating attachment energies for morphology predictions, based on the interactions between an infinite surface and a thin finite slice (a nano-crystallite), has been implemented in the ORIENT program package. This, together with existing capabilities for studying 2D periodic surface adlayers, or isolated molecular clusters on a surface, enables a wide range of complementary calculations to be performed to study crystallization phenomena of organic molecules with accurate anisotropic atom–atom intermolecular potentials, including distributed-multipole electrostatic models. Properties pertinent to the morphology and agglomeration of urea crystals are reported, including surface relaxation, attachment
energies and surface energies, solvent and solute binding energies, and the inter-surface interaction energy. We correctly predict the two major forms {110} and {001} of vapour-grown urea crystals, including an observed aspect ratio. The polar cap facets of the crystals probably arise from the unusually large relaxation of
a polar {111} surface which provides a further kinetic barrier to growth. A comparison of the binding energies of water and urea molecules to the different surfaces shows that the growth of the {110} surfaces will be particularly impeded by the presence of water. This rationalizes the increased morphological dominance of this face in crystals grown from solution. The interfacial energy between the dominant (110) and (001) crystal faces
has also been calculated, and was found to be only about 20% smaller than the interaction between (110) surfaces.
Co-reporter:Chitrani Medhi;John B. O. Mitchell;Alethea B. Tabor
Biopolymers 1999 Volume 52(Issue 2) pp:
Publication Date(Web):11 JUL 2000
DOI:10.1002/1097-0282(1999)52:2<84::AID-BIP2>3.0.CO;2-S
The factors that determine the binding of a chromophore between the base pairs in DNA intercalation complexes are dissected. The electrostatic potential in the intercalation plane is calculated using an accurate ab initio based distributed multipole electrostatic model for a range of intercalation sites, involving different sequences of base pairs and relative twist angles. There will be a significant electrostatic contribution to the binding energy for chromophores with a predominantly positive electrostatic potential, but this varies significantly with sequence, and somewhat with twist angle. The usefulness of these potential maps for understanding the binding of intercalators is explored by calculating the electrostatic binding energy for 9-aminoacridine, ethidium, and daunomycin in a variety of model binding sites. The electrostatic forces play a major role in the positioning of an intercalating 9-aminoacridine and a significant stabilizing role in the binding of ethidium in its sterically constrained position, but the intercalation of daunomycin is determined by the side-chain binding. Sequence preferences are likely to be determined by a complex and subtle mixture of effects, with electrostatics being just one component. The electrostatic binding energy is also unlikely to be a major determinant of the twist angle, as its variation with angle is modest for most intercalation sites. Overall, the electrostatic potential maps give guidance on how positively charged chromophores can be chemically adapted by heteroatomic substitution to optimise their binding. © 2000 John Wiley & Sons, Inc. Biopoly 52: 84–93, 1999
Co-reporter:Sarah L. Price, Susan M. Reutzel-Edens
Drug Discovery Today (June 2016) Volume 21(Issue 6) pp:912-923
Publication Date(Web):1 June 2016
DOI:10.1016/j.drudis.2016.01.014
•Crystal structure prediction studies have been carried out with the pharmaceutical industry.•Crystal energy landscapes can help to define solid form landscapes.•Crystal structure prediction studies guide experiments to find new polymorphs.•The value of crystal structure prediction extends beyond right-sizing solid form screens.•Realistic crystal energy landscapes of smaller pharmaceuticals are now within reach.Solid-form screening to identify all solid forms of an active pharmaceutical ingredient (API) has become increasingly important in ensuring the quality by design of pharmaceutical products and their manufacturing processes. However, despite considerable enlargement of the range of techniques that have been shown capable of producing novel solid forms, it is possible that practically important forms might not be found in the short timescales currently allowed for solid-form screening. Here, we report on the state-of-the-art use of computed crystal energy landscapes to complement pharmaceutical solid-form screening. We illustrate how crystal energy landscapes can help establish molecular-level understanding of the crystallization behavior of APIs and enhance the ability of solid-form screening to facilitate pharmaceutical development.
Co-reporter:Sarah L. Price
Physical Chemistry Chemical Physics 2008 - vol. 10(Issue 15) pp:NaN2009-2009
Publication Date(Web):2008/02/19
DOI:10.1039/B719351C
Many organic molecules are emerging as having many crystalline forms, including polymorphs and solvates, as more techniques are being used to generate and characterise the organic solid state. The fundamental scientific and industrial interest in controlling crystallisation is inspiring the development of computational methods of predicting which crystal structures are thermodynamically feasible. Sometimes, computing this crystal energy landscape will reveal that a molecule has one way of packing with itself that is sufficiently more favourable than any other so only this crystal structure will be observed. More frequently, there will be many energy minima that are energetically feasible, showing approximately equi-energetic compromises between the various intermolecular interactions allowed by the conformational flexibility. Such cases generally lead to multiple solid forms. At the moment, we usually calculate the lattice energy landscape, an approximation to the real crystal energy landscape at 0 K. Despite its limitations, many studies show that this is a valuable complement to solid form screening, which helps in discovering new structures as well as rationalising the solid forms that are found in experimental searches. The range of factors that can determine which of the thermodynamically feasible crystal structures are observed polymorphs, shows the many further challenges in developing crystal energy landscapes as a tool for control of the organic solid state.
Co-reporter:Sarah L. Price, Doris E. Braun and Susan M. Reutzel-Edens
Chemical Communications 2016 - vol. 52(Issue 44) pp:NaN7077-7077
Publication Date(Web):2016/04/04
DOI:10.1039/C6CC00721J
Computational crystal structure prediction (CSP) methods can now be applied to the smaller pharmaceutical molecules currently in drug development. We review the recent uses of computed crystal energy landscapes for pharmaceuticals, concentrating on examples where they have been used in collaboration with industrial-style experimental solid form screening. There is a strong complementarity in aiding experiment to find and characterise practically important solid forms and understanding the nature of the solid form landscape.
Co-reporter:Doris E. Braun, Panagiotis G. Karamertzanis and Sarah L. Price
Chemical Communications 2011 - vol. 47(Issue 19) pp:NaN5445-5445
Publication Date(Web):2011/04/08
DOI:10.1039/C1CC10762C
A study of two dihydroxybenzoic acid isomers shows that computational methods can be used to predict hydrate formation, the compound∶water ratio and hydrate crystal structures. The calculations also help identify a novel hydrate found in the solid form screening that validates this study.
Co-reporter:Sarah L. Price, Maurice Leslie, Gareth W. A. Welch, Matthew Habgood, Louise S. Price, Panagiotis G. Karamertzanis and Graeme M. Day
Physical Chemistry Chemical Physics 2010 - vol. 12(Issue 30) pp:NaN8490-8490
Publication Date(Web):2010/07/07
DOI:10.1039/C004164E
Crystal structure prediction for organic molecules requires both the fast assessment of thousands to millions of crystal structures and the greatest possible accuracy in their relative energies. We describe a crystal lattice simulation program, DMACRYS, emphasizing the features that make it suitable for use in crystal structure prediction for pharmaceutical molecules using accurate anisotropic atom–atom model intermolecular potentials based on the theory of intermolecular forces. DMACRYS can optimize the lattice energy of a crystal, calculate the second derivative properties, and reduce the symmetry of the spacegroup to move away from a transition state. The calculated terahertz frequency k = 0 rigid-body lattice modes and elastic tensor can be used to estimate free energies. The program uses a distributed multipole electrostatic model (Qat, t = 00,…,44s) for the electrostatic fields, and can use anisotropic atom–atom repulsion models, damped isotropic dispersion up to R−10, as well as a range of empirically fitted isotropic exp-6 atom–atom models with different definitions of atomic types. A new feature is that an accurate model for the induction energy contribution to the lattice energy has been implemented that uses atomic anisotropic dipole polarizability models (αat, t = (10,10)…(11c,11s)) to evaluate the changes in the molecular charge density induced by the electrostatic field within the crystal. It is demonstrated, using the four polymorphs of the pharmaceutical carbamazepine C15H12N2O, that whilst reproducing crystal structures is relatively easy, calculating the polymorphic energy differences to the accuracy of a few kJ mol−1 required for applications is very demanding of assumptions made in the modelling. Thus DMACRYS enables the comparison of both known and hypothetical crystal structures as an aid to the development of pharmaceuticals and other speciality organic materials, and provides a tool to develop the modelling of the intermolecular forces involved in molecular recognition processes.
Co-reporter:Ogaga G. Uzoh, Peter T. A. Galek and Sarah L. Price
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 12) pp:NaN7948-7948
Publication Date(Web):2015/02/16
DOI:10.1039/C4CP05525J
In traditional molecular mechanics force fields, intramolecular non-bonded interactions are modelled as intermolecular interactions, and the form of the torsion potential is based on the conformational profiles of small organic molecules. We investigate how a separate model for the intramolecular forces in pharmaceuticals could be more realistic by analysing the low barrier to rotation of the phenyl ring in the fenamates (substituted N-phenyl-aminobenzoic acids), that results in a wide range of observed angles in the numerous fenamate crystal structures. Although the conformational energy changes by significantly less than 10 kJ mol−1 for a complete rotation of the phenyl ring for fenamic acid, the barrier is only small because of small correlated changes in the other bond and torsion angles. The maxima for conformations where the two aromatic rings approach coplanarity arise from steric repulsion, but the maxima when the two rings are approximately perpendicular arise from a combination of an electronic effect and intramolecular dispersion. Representing the ab initio conformational energy profiles as a cosine series alone is ineffective; however, combining a cos2ξ term to represent the electronic barrier with an intramolecular atom–atom exp-6 term for all atom pairs separated by three or more bonds (1–4 interactions) provides a very effective representation. Thus we propose a new, physically motivated, generic analytical model of conformational energy, which could be combined with an intermolecular model to form more accurate force-fields for modelling the condensed phases of pharmaceutical-like organic molecules.


![1,3,2-Benzodioxaborole, 2-[(1E)-2-phenylethenyl]-](http://img.cochemist.com/ccimg/6800/6783-04-6.png)
![1,3,2-Benzodioxaborole, 2-[(1E)-2-phenylethenyl]-](http://img.cochemist.com/ccimg/6800/6783-04-6_b.png)


![3-Azabicyclo[3.3.1]nonane-2,4-dione](http://img.cochemist.com/ccimg/4400/4355-17-3.png)
![3-Azabicyclo[3.3.1]nonane-2,4-dione](http://img.cochemist.com/ccimg/4400/4355-17-3_b.png)








![4-Thiazolidinone,5-[[3,5-bis(1,1-dimethylethyl)-4-hydroxyphenyl]methyl]-](http://img.cochemist.com/ccimg/108000/107902-67-0.png)
![4-Thiazolidinone,5-[[3,5-bis(1,1-dimethylethyl)-4-hydroxyphenyl]methyl]-](http://img.cochemist.com/ccimg/108000/107902-67-0_b.png)

![Imidazo[2,1-b]quinazolin-2(3H)-one, 6-chloro-1,5-dihydro-3-methyl-](http://img.cochemist.com/ccimg/105700/105622-85-3.png)
![Imidazo[2,1-b]quinazolin-2(3H)-one, 6-chloro-1,5-dihydro-3-methyl-](http://img.cochemist.com/ccimg/105700/105622-85-3_b.png)













![(2-ISOPROPYL-3-INDOLIZINYL)(4-{3-[(2-METHYL-2-PROPANYL)AMINO]PROP<WBR />OXY}PHENYL)METHANONE](http://img.cochemist.com/ccimg/3600/3546-21-2.png)
![(2-ISOPROPYL-3-INDOLIZINYL)(4-{3-[(2-METHYL-2-PROPANYL)AMINO]PROP<WBR />OXY}PHENYL)METHANONE](http://img.cochemist.com/ccimg/3600/3546-21-2_b.png)





![Benzo[d]isothiazole-3(2H)-thione 1,1-dioxide](http://img.cochemist.com/ccimg/27200/27148-03-4.png)
![Benzo[d]isothiazole-3(2H)-thione 1,1-dioxide](http://img.cochemist.com/ccimg/27200/27148-03-4_b.png)






![1,3,5,7-Tetraazatricyclo[3.3.1.13,7]decane-2,2,4,4,6,6,8,8,9,9,10,10-d12(9CI)](http://img.cochemist.com/ccimg/23400/23304-08-7.png)
![1,3,5,7-Tetraazatricyclo[3.3.1.13,7]decane-2,2,4,4,6,6,8,8,9,9,10,10-d12(9CI)](http://img.cochemist.com/ccimg/23400/23304-08-7_b.png)