Co-reporter:Katarzyna Grubel, Amy L. Fuller, Bonnie M. Chambers, Atta M. Arif and Lisa M. Berreau
Inorganic Chemistry February 1, 2010 Volume 49(Issue 3) pp:1071-1081
Publication Date(Web):December 29, 2009
DOI:10.1021/ic901981y
A mononuclear Ni(II) complex having an acireductone type ligand, and supported by the bnpapa (N,N-bis((6-neopentylamino-2-pyridyl)methyl)-N-((2-pyridyl)methyl)amine) ligand, [(bnpapa)Ni(PhC(O)C(OH)C(O)Ph)]ClO4 (14), has been prepared and characterized by elemental analysis, 1H NMR, FTIR, and UV−vis. To gain insight into the 1H NMR features of 14, the air stable analogue complexes [(bnpapa)Ni(CH3C(O)CHC(O)CH3)]ClO4 (16) and [(bnpapa)Ni(ONHC(O)CH3)]ClO4 (17) were prepared and characterized by X-ray crystallography, 1H NMR, FTIR, UV−vis, mass spectrometry, and solution conductivity measurements. Compounds 16 and 17 are 1:1 electrolyte species in CH3CN. 1H and 2H NMR studies of 14, 16, and 17 and deuterated analogues revealed that the complexes having six-membered chelate rings for the exogenous ligand (14 and 16) do not have a plane of symmetry within the solvated cation and thus exhibit more complicated 1H NMR spectra. Compound 17, as well as other simple Ni(II) complexes of the bnpapa ligand (e.g., [(bnpapa)Ni(ClO4)(CH3CN)]ClO4 (18) and [(bnpapaNi)2(μ-Cl)2](ClO4)2 (19)), exhibit 1H NMR spectra consistent with the presence of a plane of symmetry within the cation. Treatment of [(bnpapa)Ni(PhC(O)C(OH)C(O)Ph)]ClO4 (14) with O2 results in aliphatic carbon−carbon bond cleavage within the acireductone-type ligand and the formation of [(bnpapa)Ni(O2CPh)]ClO4 (9), benzoic acid, benzil, and CO. Use of 18O2 in the reaction gives high levels of incorporation (>80%) of one labeled oxygen atom into 9 and benzoic acid. The product mixture and level of 18O incorporation in this reaction is different than that exhibited by the analogue supported the hydrophobic 6-Ph2TPA ligand, [(6-Ph2TPA)Ni(PhC(O)C(OH)C(O)Ph)]ClO4 (2). We propose that this difference is due to variations in the reactivity of bnpapa- and 6-Ph2TPA-ligated Ni(II) complexes with triketone and/or peroxide species produced in the reaction pathway.
Co-reporter:Marina Popova;Tatiana Soboleva;Atta M. Arif
RSC Advances (2011-Present) 2017 vol. 7(Issue 36) pp:21997-22007
Publication Date(Web):2017/04/19
DOI:10.1039/C7RA02653F
The properties of the extended flavonol 3-hydroxy-2-phenyl-benzo[g]chromen-4-one (2a) in DMSO : aqueous buffer solutions at pH = 7.4, including in the presence of metal ions, surfactants and serum albumin proteins, have been examined. Absorption and emission spectral studies of 2a in 1 : 1 DMSO : PBS buffer (pH = 7.4) indicate that a mixture of neutral and anionic forms of the flavonol are present. Notably, in 1 : 1 DMSO : TRIS buffer (pH = 7.4) only the neutral form of the flavonol is present. These results indicate that the nature of the buffer influences the acid/base equilibrium properties of 2a. Introduction of a Zn(II) complex of 2a− to a 1 : 1 DMSO : aqueous buffer (TRIS or PBS, pH = 7.4) solution produces absorption and emission spectral features consistent with the presence of a mixture of neutral 2a along with Zn(II)-coordinated or free 2a−. The nature of the anionic species present depends on the buffer composition. PBS buffered solutions (pH = 7.4) containing the surfactants CTAB or SDS enable 2a to be solubilized at a much lower percentage of DMSO (3.3–4.0%). Solutions containing the cationic surfactant CTAB include a mixture of 2a and 2a− whereas only the neutral flavonol is present in SDS-containing buffered solution. Compound 2a is also solubilized in TRIS buffer solutions at low cocentrations of DMSO (3.3%, pH = 7.4) in the presence of serum albumin proteins. Stern–Volmer analysis of the quenching of the inherent protein fluorescence indicates static binding of 2a to the proteins. The binding constant for this interaction is lower than that found for naturally-occurring flavonols (quercetin or morin) or 3-hydroxyflavone. Compound 2a binds to Site I of bovine and human serum albumin proteins as indicated by competition studies with warfarin and ibuprofen, as well as by docking investigations. The quantum yield for CO release from 2a (λirr = 419 nm) under aqueous conditions ranges from 0.0006(3) when the compound is bound to bovine serum albumin to 0.017(1) when present as a zinc complex in a 1 : 1 DMSO : H2O solution. Overall, the results of these studies demonstrate that 2a is a predictable visible light-induced CO release compound under a variety of aqueous conditions, including in the presence of proteins.
Co-reporter:Sushma L. Saraf, Anna Miłaczewska, Tomasz Borowski, Christopher D. James, David L. Tierney, Marina Popova, Atta M. Arif, and Lisa M. Berreau
Inorganic Chemistry 2016 Volume 55(Issue 14) pp:6916-6928
Publication Date(Web):July 5, 2016
DOI:10.1021/acs.inorgchem.6b00456
Aliphatic oxidative carbon–carbon bond cleavage reactions involving Cu(II) catalysts and O2 as the terminal oxidant are of significant current interest. However, little is currently known regarding how the nature of the Cu(II) catalyst, including the anions present, influence the reaction with O2. In previous work, we found that exposure of the Cu(II) chlorodiketonate complex [(6-Ph2TPA)Cu(PhC(O)CClC(O)Ph)]ClO4 (1) to O2 results in oxidative aliphatic carbon–carbon bond cleavage within the diketonate unit, leading to the formation of benzoic acid, benzoic anhydride, benzil, and 1,3-diphenylpropanedione as organic products. Kinetic studies of this reaction revealed a slow induction phase followed by a rapid decay of the absorption features of 1. Notably, the induction phase is not present when the reaction is performed in the presence of a catalytic amount of chloride anion. In the studies presented herein, a combination of spectroscopic (UV–vis, EPR) and density functional theory (DFT) methods have been used to examine the chloride and benzoate ion binding properties of 1 under anaerobic conditions. These studies provide evidence that each anion coordinates in an axial position of the Cu(II) center. DFT studies reveal that the presence of the anion in the Cu(II) coordination sphere decreases the barrier for O2 activation and the formation of a Cu(II)–peroxo species. Notably, the chloride anion more effectively lowers the barrier associated with O–O bond cleavage. Thus, the nature of the anion plays an important role in determining the rate of reaction of the diketonate complex with O2. The same type of anion effects were observed in the O2 reactivity of the simple Cu(II)–bipyridine complex [(bpy)Cu(PhC(O)C(Cl)C(O)Ph)ClO4] (3).
Co-reporter:Stacey N. Anderson;Jason M. Richards;Hector J. Esquer; Abby D. Benninghoff;Dr. Atta M. Arif; Lisa M. Berreau
ChemistryOpen 2015 Volume 4( Issue 5) pp:590-594
Publication Date(Web):
DOI:10.1002/open.201500167
Abstract
Molecules that can be used to deliver a controlled amount of carbon monoxide (CO) have the potential to facilitate investigations into the roles of this gaseous molecule in biology and advance therapeutic treatments. This has led to the development of light-induced CO-releasing molecules (photoCORMs). A goal in this field of research is the development of molecules that exhibit a combination of controlled CO release, favorable biological properties (e.g., low toxicity and trackability in cells), and structural tunability to affect CO release. Herein, we report a new biologically-inspired organic photoCORM motif that exhibits several features that are desirable in a next-generation photoCORM. We show that 3-hydroxyflavone-based compounds are easily synthesized and modified to impart changes in absorption features and quantum yield for CO release, exhibit low toxicity, are trackable in cells, and can exhibit both O2-dependent and -independent CO release reactivity.
Co-reporter:Caleb J. Allpress ; Anna Miłaczewska ; Tomasz Borowski ; Jami R. Bennett ; David L. Tierney ; Atta M. Arif ||
Journal of the American Chemical Society 2014 Volume 136(Issue 22) pp:7821-7824
Publication Date(Web):May 14, 2014
DOI:10.1021/ja502577b
A mononuclear Cu(II) chlorodiketonate complex was prepared, characterized, and found to undergo oxidative aliphatic carbon–carbon bond cleavage within the diketonate unit upon exposure to O2 at ambient temperature. Mechanistic studies provide evidence for a dioxygenase-type C–C bond cleavage reaction pathway involving trione and hypochlorite intermediates. Significantly, the presence of a catalytic amount of chloride ion accelerates the oxygen activation step via the formation of a Cu–Cl species, which facilitates monodentate diketonate formation and lowers the barrier for O2 activation. The observed reactivity and chloride catalysis is relevant to Cu(II) halide-catalyzed reactions in which diketonates are oxidatively cleaved using O2 as the terminal oxidant. The results of this study suggest that anion coordination can play a significant role in influencing copper-mediated oxygen activation in such systems.
Co-reporter:Caleb J. Allpress
European Journal of Inorganic Chemistry 2014 Volume 2014( Issue 27) pp:4642-4649
Publication Date(Web):
DOI:10.1002/ejic.201402254
Abstract
A mononuclear NiII complex bearing the monoanion of 1-acetoxy-3-phenylpropane-2,3-dione (4) as a ligand has been prepared {[(6-Ph2TPA)Ni{PhC(O)C(O)CHOC(O)CH3)]ClO4, 5; 6-Ph2TPA = N,N-bis[(6-phenyl-2-pyridyl)methyl]-N-(2-pyridylmethyl)amine}. This complex was characterized by 1H NMR, UV/Vis and IR spectroscopy, mass spectrometry, and elemental analysis. Exposure of solutions of 5 to O2 did not result in any reaction over the course of hours. Deprotection of 5 by the addition of NaOCH3 in methanol generated a NiII species (6) that contains a coordinated dianionic C(1)-H acireductone. Exposure of 6 to O2 led to regioselective oxidative cleavage reactivity akin to that found for the NiII-containing acireductone dioxygenase enzyme. The strategy outlined herein is the first synthetic approach that enables examination of the oxidative reactivity of a synthetic NiII species containing a dianionic C(1)-H acireductone ligand.
Co-reporter:Sushma L. Saraf, Trevor J. Fish, Abby D. Benninghoff, Ashley A. Buelt, Rhett C. Smith, and Lisa M. Berreau
Organometallics 2014 Volume 33(Issue 22) pp:6341-6351
Publication Date(Web):November 4, 2014
DOI:10.1021/om5006337
Photoactivation is a promising approach for modulating the biological activity of RuII compounds. In this work, RuII flavonolato compounds, [Ru(η6-p-cymene)(L)(3-Hfl)]OTf (8; 3-Hfl = monoanion of 3-hydroxyflavone; L = solvent) and [Ru(η6-p-cymene)Cl(3-Hfl-X)] (3a–3c; 3-Hfl-X = p-H, -Cl, or -F on the flavonolato phenyl substituent), have been evaluated for photoinduced reactivity within the flavonolato unit upon irradiation with UV (300 nm) or visible (419 nm) light under aerobic conditions. For each compound, irradiation in CH3CN was found to result in the loss of the p-cymene ligand and the formation of products resulting from oxidative ring opening of the flavonolato ligand in a dioxygenase-type reaction. This reaction also results in the release of carbon monoxide. The RuII products generated in these reactions are [RuII(solvent)(carboxylato)]+ and [Ru(CO)(solvent)(carboxylato)]+ (carboxylato = O-benzoylsalicylato or benzoato) species, as determined by ESI-MS. The amount of free CO generated depends on the wavelength of irradiation, with 300 nm light giving a higher amount of free CO. Evaluation of the photoinduced reactivity of 8 in DMSO/H2O (10:90) at 300 nm showed similar reactivity to that found in organic solvent, although the reaction occurs more slowly. The products of the photoreactions of 8 and 3a at 419 nm are nontoxic toward human T-lymphocyte (Jurkat) and non-small-cell lung carcinoma (A549) cells. This lack of toxicity versus the starting compounds is likely due to differences in interactions of the [RuII(solvent)(carboxylato)]+ and [RuII(CO)(solvent)(carboxylato)]+ species with biomolecules (e.g., serum proteins), thus resulting in reduced bioavailabilty. 1H NMR studies provide evidence that the photoreaction products coordinate to biologically relevant donors such as histidine and 5′-GMP in d6-DMSO/D2O (10:90) and exhibit reactivity with these small molecules that is distinctly different from that exhibited by the starting compounds. Overall, the photoreactivity of 8 and 3a–3c may represent an approach toward altering the biological chemistry of these compounds.
Co-reporter:Caleb J. Allpress, Lisa M. Berreau
Coordination Chemistry Reviews 2013 Volume 257(21–22) pp:3005-3029
Publication Date(Web):November 2013
DOI:10.1016/j.ccr.2013.06.001
•Metalloenzyme-catalyzed aliphatic carbon–carbon bond cleavage reactions recently identified.•Substrates vary in terms of their O2 reactivity in the absence of enzyme.•Reaction pathways depend on the nature of the substrate; metal versus substrate reactivity with O2.•Regioselectivity of carbon–carbon bond cleavage influenced by nature of metal ion.•Model systems providing insight into structure/reactivity relationships.Over the past decade, several metalloenzymes have been characterized which catalyze dioxygenase-type aliphatic carbon–carbon bond cleavage reactions. The substrates for these enzymes vary from species that are stable with respect to O2 under ambient conditions, to examples that in anionic form exhibit O2 reactivity in the absence of enzyme. Described herein are advances from studies of the enzymes themselves and model systems. These combined investigations provide insight into novel mechanistic pathways leading to aliphatic carbon–carbon bond cleavage and/or the factors that influence regioselectivity in the oxidative carbon–carbon bond cleavage reactions.
Co-reporter:Eric S. Elton, Tingting Zhang, Rajeev Prabhakar, Atta M. Arif, and Lisa M. Berreau
Inorganic Chemistry 2013 Volume 52(Issue 19) pp:11480-11492
Publication Date(Web):September 25, 2013
DOI:10.1021/ic401782x
Two new Pb(II) complexes of the amide-appended nitrogen/sulfur epppa (N-((2-ethylthio)ethyl)-N-((6-pivaloylamido-2-pyridyl)methyl)-N-((2-pyridyl)methyl)amine) chelate ligand, [(epppa)Pb(NO3)2] (4-NO3) and [(epppa)Pb(ClO4)2] (4-ClO4), were prepared and characterized. In the solid state, 4-NO3 exhibits κ5-epppa chelate ligand coordination as well as the coordination of two bidentate nitrate ions. In acetonitrile, 4-NO3 is a 1:1 electrolyte with a coordinated NO3–, whereas 4-ClO4 is a 1:2 electrolyte. Treatment of 4-ClO4 with 1 equiv Me4NOH·5H2O in CH3CN:CH3OH (3:5) results in amide methanolysis in a reaction that is akin to that previously reported for the Zn(II) analogue [(epppa)Zn](ClO4)2 (3-ClO4). 1H NMR kinetic studies of the amide methanolysis reactions of 4-ClO4 and 3-ClO4 as a function of temperature revealed free energies of activation of 21.3 and 24.5 kcal/mol, respectively. The amide methanolysis reactions of 4-ClO4 and 3-ClO4 differ in terms of the effect of the concentration of methanol (saturation kinetics for 4-ClO4; second-order behavior for 3-ClO4), the observation of a small solvent kinetic isotope effect (SKIE) only for the reaction of the Zn(II)-containing 3-ClO4, and the properties of an initial intermediate isolated from each reaction upon treatment with Me4NOH·5H2O. These experimental results, combined with computational studies of the amide methanolysis reaction pathways of 4-ClO4 and 3-ClO4, indicate that the Zn(II)-containing 3-ClO4 initially undergoes amide deprotonation upon treatment with Me4NOH·5H2O. Subsequent amide protonation from coordinated methanol yields a structure containing a coordinated neutral amide and methoxide anion from which amide cleavage can then proceed. The rate-determining step in this pathway is either amide protonation or protonation of the leaving group. The Pb(II)-containing 4-ClO4 instead directly forms a neutral amide-containing, epppa-ligated Pb(II)–OH/Pb(II)-OCH3 equilibrium mixture upon treatment with Me4NOH·5H2O in methanol. The rate-determining step in the amide methanolysis pathway of 4-ClO4 is nucleophilic attack of the Pb(II)-OCH3 moiety on the coordinated amide. Overall, it is the larger size of the Pb(II) center and the availability of coordination positions that enable direct formation of a Pb(II)–OH/Pb(II)-OCH3 mixture versus the initial amide deprotonation identified in the reaction of the Zn(II)-containing 3-ClO4.
Co-reporter:Katarzyna Grubel, Sushma L. Saraf, Stacey N. Anderson, Brynna J. Laughlin, Rhett C. Smith, Atta M. Arif, Lisa M. Berreau
Inorganica Chimica Acta 2013 Volume 407() pp:91-97
Publication Date(Web):1 October 2013
DOI:10.1016/j.ica.2013.07.029
Highlights•First structural characterization of a Pb(II) flavonolate complex.•Pb(II) flavonolate exhibits unique structural and spectroscopic features versus Group 12 analogs.•Irradiation with UV-light induces stoiochiometric or catalytic dioxygenase-type CO release.The synthesis, characterization, and photoinduced CO-release reactivity of [(6-Ph2TPA)Pb(3-Hfl)]ClO4 (1; 6-Ph2TPA = N,N-bis((6-phenyl-2-pyridyl)methyl)-N-((2-pyridyl)methyl)amine; 3-Hfl = anion of 3-hydroxyflavone) is reported and compared with Group 12 analogues ([(6-Ph2TPA)M(3-Hfl)]ClO4 (M = Hg(II) (2), Cd(II) (3), Zn(II) (4)). The Pb(II) complex exhibits unique features within this group of complexes in terms of its structural and spectroscopic features involving the coordinated flavonolate ligand. Similar to the Group 12 compounds, irradiation of 1 at 300 nm in an O2-containing environment results in the quantitative release of CO and the formation of a Pb(II) O-benzoylsalicylate (O-bs, depside) complex via a photoinduced dioxygenase-type reaction. Comparison of the quantum yield for the reaction of the Pb(II) flavonolate complex (Φ = 0.21(6)) versus the reactions of the structurally-related Group 12 metal complexes (6-Ph2TPA)M(3-Hfl)]ClO4 (M = Hg(II) (2, Φ = 0.31(2)), Cd(II) (3, Φ = 0.28(2)), Zn(II) (4), Φ = 0. 09(1)) revealed that flavonolate complexes of a heavy metal ion (Cd(II), Hg(II), Pb(II)) exhibit a higher reaction quantum yield than the Zn(II) derivative. Both Pb(II) and Zn(II) flavonolate complexes were found to be catalysts for the oxidative photoinduced degradation of 3-hydroxyflavone. The combined results of these investigations suggest that metal contaminants typically present in soil, including toxic heavy metal ions, might facilitate the oxidative decomposition of plant-derived flavonols via photoinduced reactions.Graphical abstractThe structural, spectroscopic, and photoinduced CO-release reactivity properties of a novel Pb(II) flavonolate complex are reported and compared to Group 12 analogs. The results suggest that divalent metal ions that are typically found in contaminated soils may promote the degradation of plant-derived flavonols via photoinduced oxidative pathways.
Co-reporter:Caleb J. Allpress ; Katarzyna Grubel ; Ewa Szajna-Fuller ; Atta M. Arif
Journal of the American Chemical Society 2012 Volume 135(Issue 2) pp:659-668
Publication Date(Web):December 7, 2012
DOI:10.1021/ja3038189
Mononuclear Fe(II) complexes ([(6-Ph2TPA)Fe(PhC(O)C(R)C(O)Ph)]X (3-X: R = OH, X = ClO4 or OTf; 4: R = H, X = ClO4)) supported by the 6-Ph2TPA chelate ligand (6-Ph2TPA = N,N-bis((6-phenyl-2-pyridyl)methyl)-N-(2-pyridylmethyl)amine) and containing a β-diketonate ligand bound via a six-membered chelate ring have been synthesized. The complexes have all been characterized by 1H NMR, UV–vis, and infrared spectroscopy and variably by elemental analysis, mass spectrometry, and X-ray crystallography. Treatment of dry CH3CN solutions of 3-OTf with O2 leads to oxidative cleavage of the C(1)–C(2) and C(2)–C(3) bonds of the acireductone via a dioxygenase reaction, leading to formation of carbon monoxide and 2 equiv of benzoic acid as well as two other products not derived from dioxygenase reactivity: 2-oxo-2-phenylethylbenzoate and benzil. Treatment of CH3CN/H2O solutions of 3-X with O2 leads to the formation of an additional product, benzoylformic acid, indicative of the operation of a new reaction pathway in which only the C(1)–C(2) bond is cleaved. Mechanistic studies show that the change in regioselectivity is due to the hydration of a vicinal triketone intermediate in the presence of both an iron center and water. This is the first structural and functional model of relevance to iron-containing acireductone dioxygenase (Fe-ARD′), an enzyme in the methionine salvage pathway that catalyzes the regiospecific oxidation of 1,2-dihydroxy-3-oxo-(S)-methylthiopentene to form 2-oxo-4-methylthiobutyrate. Importantly, this model system is found to control the regioselectivity of aliphatic carbon–carbon bond cleavage by changes involving an intermediate in the reaction pathway, rather than by the binding mode of the substrate, as had been proposed in studies of acireductone enzymes.
Co-reporter:Katarzyna Grubel;Amy R. Marts;Samuel M. Greer;David L. Tierney;Caleb J. Allpress;Stacey N. Anderson;Brynna J. Laughlin;Rhett C. Smith;Atta M. Arif
European Journal of Inorganic Chemistry 2012 Volume 2012( Issue 29) pp:4750-4757
Publication Date(Web):
DOI:10.1002/ejic.201200212
Abstract
Irradiation of 3-hydroxyflavonolato (3-Hfl) complexes of MnII, CoII, NiII and CuII (1–4) at 300 nm under aerobic conditions results in dioxygenase-type reactivity and the formation of the corresponding divalent metal O-benzoylsalicylato (O-bs) complexes 8–11 and CO. The latter were characterized by using multiple methods, including elemental analysis, X-ray crystallography, NMR and/or EPR spectroscopy, mass spectrometry and IR spectroscopy. Compounds 1–4 serve as catalysts for the photoinduced reactivity of 3-hydroxyflavonol (3-HflH) to produce O-benzoylsalicylic acid as the major product. Spectroscopic studies (UV/Vis and 1H NMR) show that each O-benzoylsalicylato complex 8–11 reacts with one equiv. of 3-hydroxyflavonol to regenerate 1–4 and enable turnover reactivity. Unlike what is observed for free 3-HflH, photoinduced reactions involving 1–4 and excess flavonol show only minor amounts of flavonol isomerization reactivity. These results indicate that the presence of a metal ion, whether under stoichiometric or catalytic conditions, facilitates the photoinduced degradation of 3-HflH to produce a carboxylic acid and CO as products.
Co-reporter:Katarzyna Grubel, Brynna J. Laughlin, Thora R. Maltais, Rhett C. Smith, Atta M. Arif and Lisa M. Berreau
Chemical Communications 2011 vol. 47(Issue 37) pp:10431-10433
Publication Date(Web):15 Aug 2011
DOI:10.1039/C1CC13961D
Exposure of 3-hydroxyflavonolate complexes of the group 12 metals to UV light under aerobic conditions results in oxidative carbon–carbon bond cleavage and CO release. This reactivity is novel in that it occurs under mild reaction conditions and suggests that light-induced CO-release reactivity involving metal flavonolate species may be possible in biological systems.
Co-reporter:Lisa M. Berreau ; Tomasz Borowski ; Katarzyna Grubel ; Caleb J. Allpress ; Jeffrey P. Wikstrom ; Meaghan E. Germain ; Elena V. Rybak-Akimova ;David L. Tierney
Inorganic Chemistry 2011 Volume 50(Issue 3) pp:1047-1057
Publication Date(Web):January 11, 2011
DOI:10.1021/ic1017888
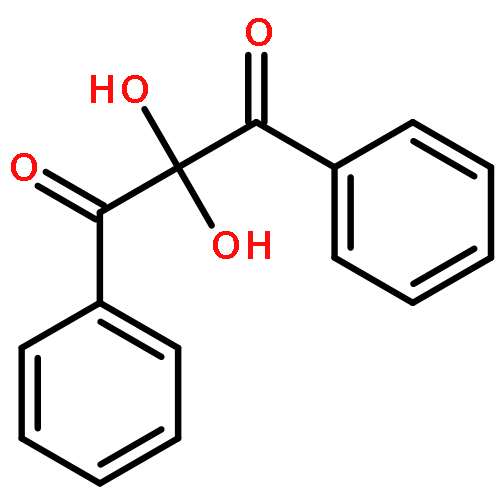
The mononuclear nickel(II) enolate complex [(6-Ph2TPA)Ni(PhC(O)C(OH)C(O)Ph]ClO4 (I) was the first reactive model complex for the enzyme/substrate (ES) adduct in nickel(II)-containing acireductone dioxygenases (ARDs) to be reported. In this contribution, the mechanism of its O2-dependent aliphatic carbon−carbon bond cleavage reactivity was further investigated. Stopped-flow kinetic studies revealed that the reaction of I with O2 is second-order overall and is ∼80 times slower at 25 °C than the reaction involving the enolate salt [Me4N][PhC(O)C(OH)C(O)Ph]. Computational studies of the reaction of the anion [PhC(O)C(OH)C(O)Ph]− with O2 support a hydroperoxide mechanism wherein the first step is a redox process that results in the formation of 1,3-diphenylpropanetrione and HOO−. Independent experiments indicate that the reaction between 1,3-diphenylpropanetrione and HOO− results in oxidative aliphatic carbon−carbon bond cleavage and the formation of benzoic acid, benzoate, and CO:CO2 (∼12:1). Experiments in the presence of a nickel(II) complex gave a similar product distribution, albeit benzil [PhC(O)C(O)Ph] is also formed, and the CO:CO2 ratio is ∼1.5:1. The results for the nickel(II)-containing reaction match those found for the reaction of I with O2 and provide support for a trione/HOO− pathway for aliphatic carbon−carbon bond cleavage. Overall, I is a reasonable structural model for the ES adduct formed in the active site of NiIIARD. However, the presence of phenyl appendages at both C(1) and C(3) in the [PhC(O)C(OH)C(O)Ph]− anion results in a reaction pathway for O2-dependent aliphatic carbon−carbon bond cleavage (via a trione intermediate) that differs from that accessible to C(1)−H acireductone species. This study, as the first detailed investigation of the O2 reactivity of a nickel(II) enolate complex of relevance to NiIIARD, provides insight toward understanding the chemical factors involved in the O2 reactivity of metal acireductone species.
Co-reporter:Katarzyna Grubel, Gajendrasingh K. Ingle, Amy L. Fuller, Atta M. Arif and Lisa M. Berreau
Dalton Transactions 2011 vol. 40(Issue 40) pp:10609-10620
Publication Date(Web):16 Aug 2011
DOI:10.1039/C1DT10587F
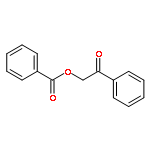
Reaction conditions were evaluated for the preparation of [(6-PhTPA)Ni(PhC(O)C(OH)C(O)Ph)]ClO4 (3) and [(6-Ph2TPA)Co(PhC(O)C(OH)C(O)Ph)]ClO4 (7), two complexes of structural relevance to the enzyme/substrate (ES) adduct in Ni(II)- and Co(II)-containing forms of acireductone dioxygenase. The presence of water in reactions directed at the preparation of 3 and 7 was found to result in isomerization of the enolate precursor 2-hydroxy-1,3-diphenylpropane-1,3-dione to give the ester 2-oxo-2-phenylethylbenzoate. Performing synthetic procedures under dryer conditions reduced the amount of ester production and enabled the isolation of 3 in high yield. This complex was comprehensively characterized, including by X-ray crystallography. Using similar conditions for the 6-Ph2TPACo-containing system, the amount of ester generated was only modestly affected, but the formation of a benzoate complex ([(6-Ph2TPA)Co(O2CPh)]ClO4, 10) resulting from ester hydrolysis was prevented. The best preparation of 7 was found to involve dry conditions and short reaction times. The approach outlined herein toward determining appropriate reaction conditions for the preparation of 3 and 7 involved the preparation and characterization of several air-stable (6-PhTPA)Ni- and (6-Ph2TPA)Co-containing analog complexes having enolate, solvent, and benzoate ligands. These complexes were used as paramagnetic 1H NMR standards for evaluation of reaction mixtures containing 3 and 7.
Co-reporter:Caleb J. Allpress;Dr. Atta M. Arif;Dylan T. Houghton;Dr. Lisa M. Berreau
Chemistry - A European Journal 2011 Volume 17( Issue 52) pp:14962-14973
Publication Date(Web):
DOI:10.1002/chem.201101962
Abstract
Three mononuclear NiII complexes containing a 2-chloro-1,3-diketonate ligand and supported by the 6-Ph2TPA chelate, as well as analogues that lack the 2-chloro substituent on the β-diketonate ligand, have been prepared and characterized. Upon irradiation at 350 nm under aerobic conditions, complexes containing the 2-chloro-substituted ligands undergo reactions to generate products resulting from oxidative cleavage, α-cleavage, and radical-derived reactions involving the 2-chloro-1,3-diketonate ligand. Mechanistic studies suggest that the oxidative cleavage reactivity, which leads to the production of carboxylic acids, is a result of the formation of superoxide, which occurs through reaction of reduced nickel complexes with O2. The presence of the 2-chloro substituent was found to be a prerequisite for oxidative carbon–carbon bond-cleavage reactivity, as complexes lacking this functional group did not undergo these reactions following prolonged irradiation. The approach toward investigating the oxidative reactivity of metal β-diketonate species outlined herein has yielded results of relevance to the proposed mechanistic pathways of metalloenzyme-catalyzed β-diketonate oxidative cleavage reactions.
Co-reporter:Katarzyna Grubel ; Katarzyna Rudzka ; Atta M. Arif ; Katie L. Klotz ; Jason A. Halfen
Inorganic Chemistry 2010 Volume 49(Issue 1) pp:82-96
Publication Date(Web):December 2, 2009
DOI:10.1021/ic901405h
A series of divalent metal flavonolate complexes of the general formula [(6-Ph2TPA)M(3-Hfl)]X (1−5-X; X = OTf− or ClO4−; 6-Ph2TPA = N,N-bis((6-phenyl-2-pyridyl)methyl)-N-((2-pyridyl)methyl)amine; M = Mn(II), Co(II), Ni(II), Cu(II), Zn(II); 3-Hfl = 3-hydroxyflavonolate) were prepared and characterized by X-ray crystallography, elemental analysis, FTIR, UV−vis, 1H NMR or EPR, and cyclic voltammetry. All of the complexes have a bidentate coordinated flavonolate ligand. The difference in M−O distances (ΔM−O) involving this ligand varies through the series, with the asymmetry of flavonolate coordination increasing in the order Mn(II) ∼ Ni(II) < Cu(II) < Zn(II) < Co(II). The hypsochromic shift of the absorption band I (π→π*) of the coordinated flavonolate ligand in 1−5-OTf (relative to that in free anion) increases in the order Ni(II) < Mn(II) < Cu(II) < Zn(II), Co(II). Previously reported 3-Hfl complexes of divalent metals fit well with this ordering. 1H NMR studies indicate that the 3-Hfl complexes of Co(II), Ni(II), and Zn(II) exhibit a pseudo-octahedral geometry in solution. EPR studies suggest that the Mn(II) complex 1-OTf may form binuclear structures in solution. The mononuclear Cu(II) complex 4-OTf has a distorted square pyramidal geometry. The oxidation potential of the flavonolate ligand depends on the metal ion present and/or the solution structure of the complex, with the Mn(II) complex 1-OTf exhibiting the lowest potential, followed by the pseudo-octahedral Ni(II) and Zn(II) 3-Hfl complexes, and the distorted square pyramidal Cu(II) complex 4-OTf. The Mn(II) complex [(6-Ph2TPA)Mn(3-Hfl)]OTf (1-OTf) is unique in the series in undergoing ligand exchange reactions in the presence of M(ClO4)2·6H2O (M = Co, Ni, Zn) in CD3CN to produce [(6-Ph2TPA)M(CD3CN)n](X)2, [Mn(3-Hfl)2·0.5H2O], and MnX2 (X = OTf− or ClO4−). Under similar conditions, the 3-Hfl complexes of Co(II), Ni(II), and Cu(II) undergo flavonolate ligand exchange to produce [(6-Ph2TPA)M(CD3CN)n](X)2 (M = Co, Ni, Cu; n = 1 or 2) and [Zn(3-Hfl)2·2H2O]. An Fe(II) complex of 3-Hfl, [(6-Ph2TPA)Fe(3-Hfl)]ClO4 (8), was isolated and characterized by elemental analysis, FTIR, UV−vis, 1H NMR, cyclic voltammetry, and a magnetic moment measurement. This complex reacts with O2 to produce the diiron(III) μ-oxo compound [(6-Ph2TPAFe(3Hfl))2(μ-O)](ClO4)2 (6).
Co-reporter:Katarzyna Rudzka, Katarzyna Grubel, Atta M. Arif and Lisa M. Berreau
Inorganic Chemistry 2010 Volume 49(Issue 17) pp:7623-7625
Publication Date(Web):August 6, 2010
DOI:10.1021/ic100775m
A nickel(II) enediolate cluster (2) forms upon treatment of [(6-Ph2TPA)Ni(PhC(O)C(OH)C(O)Ph)]ClO4 (1) with Me4NOH·5H2O in CH3CN. Crystallographic studies of 2 revealed a hexanuclear structure of S6 symmetry with a formula of {[Ni(PhC(O)C(O)C(O)Ph)(CH3OH)]·1.33CH3OH}6. Because isolation of bulk amounts of 2 from the reaction involving 1 proved impossible, a solvation analogue of 2 (labeled 5) was generated from admixture of Ni(ClO4)2·6H2O, 2-hydroxy-1,3-diphenylpropane-1,3-dione, and Me4NOH·5H2O in CH3OH/CH3CN. Complex 5 is formulated as {[Ni(PhC(O)C(O)C(O)Ph)(H2O)]·H2O·0.25CH3CN}6 based on elemental analysis, a molecular weight determination, UV−vis, and a magnetic moment measurement. Treatment of 5 with O2 and 6-Ph2TPA (6 equiv) results in the formation of CO and [(6-Ph2TPA)Ni(O2CPh)2(H2O)] (3), the latter of which was isolated in 69% yield. The level of 18O incorporation in this reaction matches that for a reaction wherein 2 is generated from 1. These results provide evidence that a nickel(II) enediolate cluster is the O2 reactive species in a previously reported model reaction for nickel(II)-containing acireductone dioxygenase.
Co-reporter:James J. Danford ; Piotr Dobrowolski
Inorganic Chemistry 2009 Volume 48(Issue 23) pp:11352-11361
Publication Date(Web):October 14, 2009
DOI:10.1021/ic901890d
Glyoxalase II enzymes catalyze the hydrolysis of a thioester substrate and have been found to coordinate a variety of dimetal combinations, including Fe(III)Zn(II), within the enzyme active site. Of relevance to these enzymes, the thioester hydrolysis reactivity of the Fe(III)Zn(II) compound [(BPBPMP)Fe(III)Zn(II)(μ-OAc)2]ClO4 (1) was evaluated in CH3CN/H2O (50:50; buffered) at 26.5 °C. Thioester hydrolysis in the absence and presence of 1 was monitored using 2H NMR by following the loss of the thioester −SCD3 signal. Two products are generated in the reaction involving the metal complex, D3CSSCD3 and CD3SH. Kinetic studies of this reaction as a function of pH revealed maximum rate above the pKa of a Zn−OH2 moiety of [(BPBPMP)Fe(III)(OH)(μ-OH)Zn(II)(OH2)]+, which forms from 1 in CH3CN/H2O (50:50). UV−vis and electron paramagnetic resonance (EPR) studies of a single turnover thioester hydrolysis reaction in the presence of 1 equiv of 1 at pH = 9.0 suggest that the thioester does not initially interact with the Fe(III) center, but that changes occur at this site over the course of the reaction. The formation of a Fe(III)−SCD3 moiety is proposed based on the observed D3CSSCD3 formation, which likely results from redox activity involving a iron(III) thiolate species. A mechanism for thioester hydrolysis is proposed involving initial coordination of the deprotonated α-hydroxy thioester to the zinc center followed by nucleophilic attack by a terminal Fe(III)−OH moiety and thiolate leaving group stabilization by the Fe(III) center. Overall, this study outlines a novel approach of using an aliphatic thioester substrate and 2H NMR to provide mechanistic insight into thioester hydrolysis involving an Fe(III)Zn(II) complex of relevance to glyoxalase II.
Co-reporter:Katarzyna Rudzka ; Atta M. Arif
Inorganic Chemistry 2008 Volume 47(Issue 23) pp:10832-10840
Publication Date(Web):October 28, 2008
DOI:10.1021/ic800947z
Using a new N4-donor chelate ligand having a mixture of hydrophobic phenyl and hydrogen-bond-donor appendages, a trinuclear nickel(II) complex of the doubly deprotonated form of 2-hydroxy-1,3-diphenylpropane-1,3-dione was isolated, characterized (X-ray crystallography, elemental analysis, UV−vis, 1H NMR, FTIR, and magnetic moment measurement), and evaluated for O2 reactivity. This complex, [(6-NA-6-Ph2TPANi)2(μ-PhC(O)C(O)C(O)Ph)2Ni](ClO4)2 (4), has two terminal pseudooctahedral NiII centers supported by the tetradentate chelate ligand and a central square-planar NiII ion ligated by oxygen atoms of two bridging enediolate ligands. In CH3CN, 4 exhibits a deep orange/brown color and λmax = 463 nm (ε = 16 000 M−1cm−1). The room temperature magnetic moment of 4, determined by Evans method, is μeff = 5.3(2) μB. This is consistent with the presence of two noninteracting high-spin NiII centers, a diamagnetic central NiII ion, and an overall quintet ground state. Exposure of a CH3CN solution of 4 to O2 results in the rapid loss of the orange/brown color to give a green solution. The products identified from this reaction are [(κ3-6-NA-6-Ph2TPA)Ni(O2Ph)(H2O)]ClO4 (5), benzil [PhC(O)C(O)Ph], and CO. Identification of 5 was achieved via its independent synthesis and a comparison of its 1H NMR and mass spectral features with those of the 6-NA-6-Ph2TPA-containing product generated upon reaction of 4 with O2. The independently prepared sample of 5 was characterized by X-ray crystallography, elemental analysis, UV−vis, mass spectrometry, and FTIR. The O2 reactivity of 4 has relevance to the active-site chemistry of NiII-containing acireductone dioxygenase (NiIIARD).
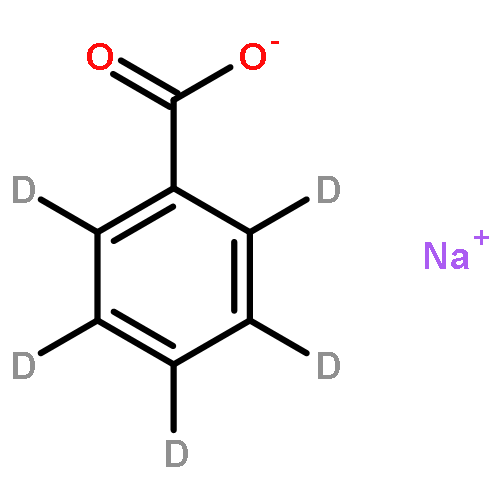
Co-reporter:Katarzyna Grubel ; Amy L. Fuller ; Bonnie M. Chambers ; Atta M. Arif
Inorganic Chemistry () pp:
Publication Date(Web):December 29, 2009
DOI:10.1021/ic901981y
A mononuclear Ni(II) complex having an acireductone type ligand, and supported by the bnpapa (N,N-bis((6-neopentylamino-2-pyridyl)methyl)-N-((2-pyridyl)methyl)amine) ligand, [(bnpapa)Ni(PhC(O)C(OH)C(O)Ph)]ClO4 (14), has been prepared and characterized by elemental analysis, 1H NMR, FTIR, and UV−vis. To gain insight into the 1H NMR features of 14, the air stable analogue complexes [(bnpapa)Ni(CH3C(O)CHC(O)CH3)]ClO4 (16) and [(bnpapa)Ni(ONHC(O)CH3)]ClO4 (17) were prepared and characterized by X-ray crystallography, 1H NMR, FTIR, UV−vis, mass spectrometry, and solution conductivity measurements. Compounds 16 and 17 are 1:1 electrolyte species in CH3CN. 1H and 2H NMR studies of 14, 16, and 17 and deuterated analogues revealed that the complexes having six-membered chelate rings for the exogenous ligand (14 and 16) do not have a plane of symmetry within the solvated cation and thus exhibit more complicated 1H NMR spectra. Compound 17, as well as other simple Ni(II) complexes of the bnpapa ligand (e.g., [(bnpapa)Ni(ClO4)(CH3CN)]ClO4 (18) and [(bnpapaNi)2(μ-Cl)2](ClO4)2 (19)), exhibit 1H NMR spectra consistent with the presence of a plane of symmetry within the cation. Treatment of [(bnpapa)Ni(PhC(O)C(OH)C(O)Ph)]ClO4 (14) with O2 results in aliphatic carbon−carbon bond cleavage within the acireductone-type ligand and the formation of [(bnpapa)Ni(O2CPh)]ClO4 (9), benzoic acid, benzil, and CO. Use of 18O2 in the reaction gives high levels of incorporation (>80%) of one labeled oxygen atom into 9 and benzoic acid. The product mixture and level of 18O incorporation in this reaction is different than that exhibited by the analogue supported the hydrophobic 6-Ph2TPA ligand, [(6-Ph2TPA)Ni(PhC(O)C(OH)C(O)Ph)]ClO4 (2). We propose that this difference is due to variations in the reactivity of bnpapa- and 6-Ph2TPA-ligated Ni(II) complexes with triketone and/or peroxide species produced in the reaction pathway.
Co-reporter:Katarzyna Grubel, Brynna J. Laughlin, Thora R. Maltais, Rhett C. Smith, Atta M. Arif and Lisa M. Berreau
Chemical Communications 2011 - vol. 47(Issue 37) pp:NaN10433-10433
Publication Date(Web):2011/08/15
DOI:10.1039/C1CC13961D
Exposure of 3-hydroxyflavonolate complexes of the group 12 metals to UV light under aerobic conditions results in oxidative carbon–carbon bond cleavage and CO release. This reactivity is novel in that it occurs under mild reaction conditions and suggests that light-induced CO-release reactivity involving metal flavonolate species may be possible in biological systems.
Co-reporter:Stacey N. Anderson, Michael T. Larson and Lisa M. Berreau
Dalton Transactions 2016 - vol. 45(Issue 37) pp:NaN14580-14580
Publication Date(Web):2016/08/23
DOI:10.1039/C6DT01709F
Two types of zinc flavonolato complexes ([(6-Ph2TPA)Zn(flavonolato)]ClO4 and Zn(flavonolato)2) of four extended flavonols have been prepared, characterized, and evaluated for visible light-induced CO release reactivity. Zinc coordination of each flavonolato anion results in a red-shift of the lowest energy absorption feature and in some cases enhanced molar absorptivity relative to the free flavonol. The zinc-coordinated flavonolato ligands undergo visible light-induced CO release with enhanced reaction quantum yields relative to the neutral flavonols. Most notable is the discovery that both types of zinc flavonolato derivatives undergo similar visible light-induced CO release reactivity in solution and in the solid state. A solid film of a Zn(flavonolato)2 derivative was evaluated as an in situ CO release agent for aerobic oxidative palladium-catalyzed alkoxycarbonylation to produce esters in ethanol. The CO release product was found to undergo ester alcolysis under the conditions of the carbonylation reaction.
Co-reporter:Katarzyna Grubel, Gajendrasingh K. Ingle, Amy L. Fuller, Atta M. Arif and Lisa M. Berreau
Dalton Transactions 2011 - vol. 40(Issue 40) pp:NaN10620-10620
Publication Date(Web):2011/08/16
DOI:10.1039/C1DT10587F
Reaction conditions were evaluated for the preparation of [(6-PhTPA)Ni(PhC(O)C(OH)C(O)Ph)]ClO4 (3) and [(6-Ph2TPA)Co(PhC(O)C(OH)C(O)Ph)]ClO4 (7), two complexes of structural relevance to the enzyme/substrate (ES) adduct in Ni(II)- and Co(II)-containing forms of acireductone dioxygenase. The presence of water in reactions directed at the preparation of 3 and 7 was found to result in isomerization of the enolate precursor 2-hydroxy-1,3-diphenylpropane-1,3-dione to give the ester 2-oxo-2-phenylethylbenzoate. Performing synthetic procedures under dryer conditions reduced the amount of ester production and enabled the isolation of 3 in high yield. This complex was comprehensively characterized, including by X-ray crystallography. Using similar conditions for the 6-Ph2TPACo-containing system, the amount of ester generated was only modestly affected, but the formation of a benzoate complex ([(6-Ph2TPA)Co(O2CPh)]ClO4, 10) resulting from ester hydrolysis was prevented. The best preparation of 7 was found to involve dry conditions and short reaction times. The approach outlined herein toward determining appropriate reaction conditions for the preparation of 3 and 7 involved the preparation and characterization of several air-stable (6-PhTPA)Ni- and (6-Ph2TPA)Co-containing analog complexes having enolate, solvent, and benzoate ligands. These complexes were used as paramagnetic 1H NMR standards for evaluation of reaction mixtures containing 3 and 7.

amino]methyl]-2-pyridinyl]-2,2-dimethyl-](http://img.cochemist.com/ccimg/929700/929602-56-2.png)
amino]methyl]-2-pyridinyl]-2,2-dimethyl-](http://img.cochemist.com/ccimg/929700/929602-56-2_b.png)









![Phenol, 2-[[(2-pyridinylmethyl)amino]methyl]-](http://img.cochemist.com/ccimg/63700/63671-68-1.png)
![Phenol, 2-[[(2-pyridinylmethyl)amino]methyl]-](http://img.cochemist.com/ccimg/63700/63671-68-1_b.png)