Co-reporter:John R. Bleeke, Meghan Stouffer, Nigam P. Rath
Journal of Organometallic Chemistry 2015 Volume 781() pp:11-22
Publication Date(Web):1 April 2015
DOI:10.1016/j.jorganchem.2015.01.006
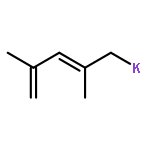
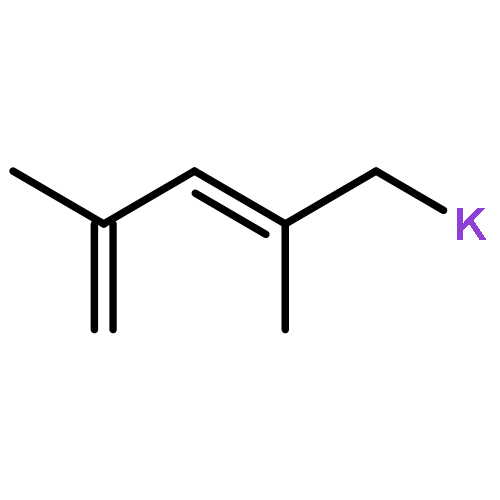
•The first examples of bis-(η3-azapentadienyl)RuL2 complexes have been synthesized and characterized.•The complexes adopt a variety of structural motifs, which differ in the orientation of the azapentadienyl ligands.•Dynamic interconversions between these structural types are observed.•The azapentadienyl nitrogens are the preferred sites of electrophilic attack.We report the synthesis, spectroscopy, structure, and reactivity of the first examples of bis-azapentadienyl–ruthenium complexes. The parent compound, [(1,2,3-η3)-(5-tert-butylazapentadienyl)]2Ru(PPh3)2 (1), is produced by reacting Cl2Ru(PPh3)3 with two equivalents of potassium tert-butylazapentadienide. Treatment of 1 with CNCMe3, P(OMe)3, PMe3, or PEt3 (L) in THF at room temperature yields single-ligand substitution products [(1,2,3-η3)-(5-tert-butylazapentadienyl)]2Ru(PPh3)(L) (2, L = CNCMe3; 3, L = P(OMe)3; 4, L = PMe3; 5, L = PEt3), and the double-ligand substitution product, [(1,2,3-η3)-(5-tert-butylazapentadienyl)]2Ru(PEt3)2 (6). Other double-ligand substitution products, [(1,2,3-η3)-(5-tert-butylazapentadienyl)]2Ru(L)2 (7, L = PMe3; 8, L = P(OMe)3), are obtained when 1 is treated with PMe3 or P(OMe)3 in THF at reflux. Compounds 7 and 8 exist in solution as equilibrium mixtures of two structural isomers. Electron-rich compounds 6 and 7 react with triflic acid to generate dicationic products,{[(1,2,3-η3)-(CH2CHCHCHN(H)(CMe3)]2Ru(L)2}2+(−O3SCF3)2 (9, L = PEt3; 10, L = PMe3), in which both azapentadienyl nitrogen atoms have been protonated. All of the compounds reported herein have been characterized by NMR spectroscopy, and the structures of 2, 3, 4, 6, and 9 have been confirmed by single-crystal X-ray diffraction.The first examples of bis-(η3-azapentadienyl)RuL2 complexes have been synthesized, and a variety of structural motifs have been observed by NMR and X-ray diffraction. Treatment of electron-rich members of this family with triflic acid leads to protonation at both azapentadienyl nitrogen centres.
Co-reporter:John R. Bleeke and Bryn L. Lutes, Nigam P. Rath
Organometallics 2009 Volume 28(Issue 15) pp:4577-4583
Publication Date(Web):July 14, 2009
DOI:10.1021/om9004912
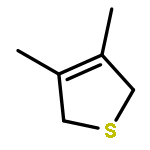
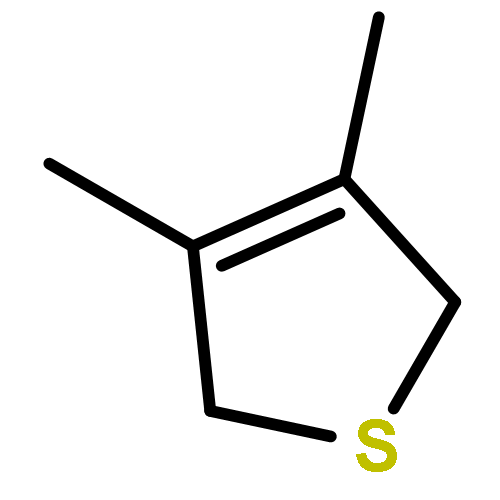
The first examples of thiapentadienyl-cobalt complexes have been synthesized and structurally characterized. Treatment of (Cl)Co(PMe3)3 with potassium thiapentadienide yields a dimetallic product, 1, which is held together by two bridging thiapentadienyl ligands, one of which possesses a cis internal double bond and the other a trans internal double bond. Treatment of 1 with carbon monoxide breaks apart the cluster, producing a mixture of (5-η1-cis-5-thiapentadienyl)Co(PMe3)2(CO)2 (cis-2) and its trans isomer (trans-2). cis-2 can be made independently by reacting (Cl)Co(PMe3)2(CO)2 with potassium thiapentadienide. In contrast, treatment of (Cl)Co(PMe3)3 with lithium 2,3-dimethyl-5-thiapentadienide generates a trimetallic product, 3. This species is held together by four bridging thiapentadienyl ligands, all possessing trans internal double bonds. In compound 3, the central cobalt is a Co(II) center, produced through a disproportionation reaction, while the two terminal cobalts are Co(I) centers. Treatment of 3 with carbon monoxide again breaks apart the cluster, producing (5-η1-trans-2,3-dimethyl-5-thiapentadienyl)Co(PMe3)2(CO)2 (trans-4). The cis isomer, cis-4, can be produced by treating (Cl)Co(PMe3)2(CO)2 with lithium 2,3-dimethyl-5-thiapentadienide. Compounds 1, cis-2, 3, and trans-4 have been characterized by single-crystal X-ray diffraction.
Co-reporter:John R. Bleeke, Todsapon Thananatthanachon and Nigam P. Rath
Organometallics 2008 Volume 27(Issue 11) pp:2436-2446
Publication Date(Web):May 10, 2008
DOI:10.1021/om800041b
A series of η1-silapentadienyl−iridium complexes have been synthesized by reacting (η2-cyclooctene)(X)Ir(PMe3)3 (X = Cl or Me) with butadienyldimethylsilanes, and the reactivity of these species has been investigated. Treatment of (η2-cyclooctene)(Cl)Ir(PMe3)3 wi th E- or (Z-butadienyl)dimethylsilane produces, via Si−H bond activation, (η1-E-dimethylsilapentadienyl)(H)(Cl)Ir(PMe3)3, 1E. Similarly, treatment of (η2-cyclooctene)(Me)Ir(PMe3)3 with E- or (Z-butadienyl)dimethylsilane produces η1-E- or (η1-Z-dimethylsilapentadienyl)(H)(Me)Ir(PMe3)3, 2E or 2Z. Upon heating in benzene at 100 °C (under pressure), 2E decomposes, but 2Z loses methane and coordinates the terminal double bond of the silapentadienyl ligand, producing (η1,η2-dimethylsilapentadienyl)Ir(PMe3)3, 3. Treatment of (η2-cyclooctene)(Cl)Ir(PMe3)3 with (E-2,3-dimethylbutadienyl)dimethylsilane produces (η1-E-2,3,5,5-tetramethylsilapentadienyl)(H)(Cl)Ir(PMe3)3, 4E. In acetone, this species isomerizes via 2,3-dimethylbutadienyl migration from silicon to iridium, leading ultimately to production of (η1-E-2,3-dimethylbutadienyl)(H)(SiMe2Cl)Ir(PMe3)3, 5. Treatment of (η2-cyclooctene)(Cl)Ir(PMe3)3 with the Z isomer of (2,3-dimethylbutadienyl)dimethylsilane produces transient (η1-Z-2,3,5,5-tetramethylsilapentadienyl)(H)(Cl)Ir(PMe3)3, 4Z, which also rearranges. In this case, hydride first migrates from iridium to C2 of the silapentadienyl ligand, and then the resulting butenyl group migrates from silicon to iridium, forming an iridacyclopentene product, 6. The reactions of (η2-cyclooctene)(Me)Ir(PMe3)3 with E- or (Z-2,3-dimethylbutadienyl)dimethylsilane generate η1-E- or (η1-Z-2,3,5,5-tetramethylsilapentadienyl)(H)(Me)Ir(PMe3)3, 7E or 7Z. Heating these compounds in toluene or benzene (under pressure) leads to methane loss, followed by C−H bond activation. In the case of 7E, a silapentadienyl methyl group is activated, producing an iridasilacyclopentene product, 8. In the case of 7Z, a C−H bond on the end of the silapentadienyl chain is activated, producing the first example of an iridasilacyclohexadiene, 9. X-ray crystal structures of (η2-cyclooctene)(Me)Ir(PMe3)3, 5-acetone, 6, and 7E are reported here, while structures of 1E and 3 were reported in a prior communication.1
Co-reporter:John R. Bleeke, Phawit Putprasert and Todsapon Thananatthanachon, Nigam P. Rath
Organometallics 2008 Volume 27(Issue 22) pp:5744-5747
Publication Date(Web):October 15, 2008
DOI:10.1021/om8008712
Treatment of aromatic nitriles with methyllithium produces N-lithiated imine reagents, which, when reacted with (η2-cyclooctene)(Cl)Ir(PMe3)3, generate fused iridaazacycles via ortho-metalation. Monoprotonation of these iridaazacycles produces fused iridapyrrole derivatives, while diprotonation leads to several different pathways.
Co-reporter:John R. Bleeke
Accounts of Chemical Research 2007 Volume 40(Issue 10) pp:1035
Publication Date(Web):June 27, 2007
DOI:10.1021/ar700071p
The first example of a family of related aromatic metallacycles has been synthesized, and the physical and chemical properties of its individual members have been investigated. The metallacyclic rings in these compounds are, in general, constructed from pentadienyl- or heteropentadienyliridium precursors via C–H bond activation processes. The molecules display the structural and spectroscopic features of aromaticity, including ring planarity, π-bond delocalization, and diamagnetic ring current effects. The reactivity profile of the molecules is complex, reflecting the simultaneous presence of an aromatic ring and a reactive metal center.
.jpg)


![Phosphine, bis[2-(diethylphosphino)ethyl]phenyl-](http://img.cochemist.com/ccimg/102800/102747-21-7.png)
![Phosphine, bis[2-(diethylphosphino)ethyl]phenyl-](http://img.cochemist.com/ccimg/102800/102747-21-7_b.png)





![Phosphine,[2-[(dimethylphosphino)methyl]-2-methyl-1,3-propanediyl]bis[dimethyl-](http://img.cochemist.com/ccimg/77700/77609-83-7.png)
![Phosphine,[2-[(dimethylphosphino)methyl]-2-methyl-1,3-propanediyl]bis[dimethyl-](http://img.cochemist.com/ccimg/77700/77609-83-7_b.png)