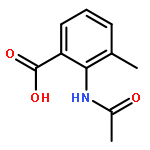
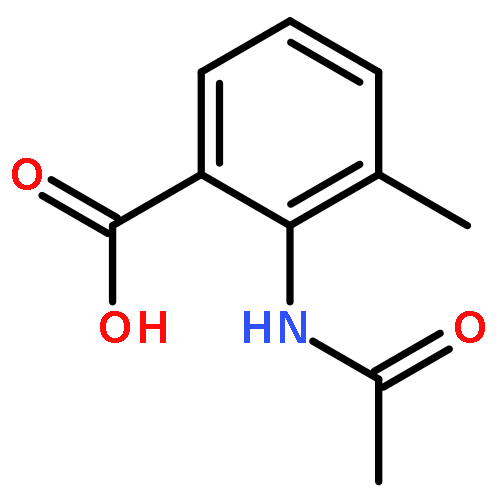
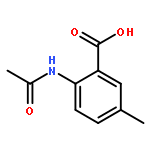
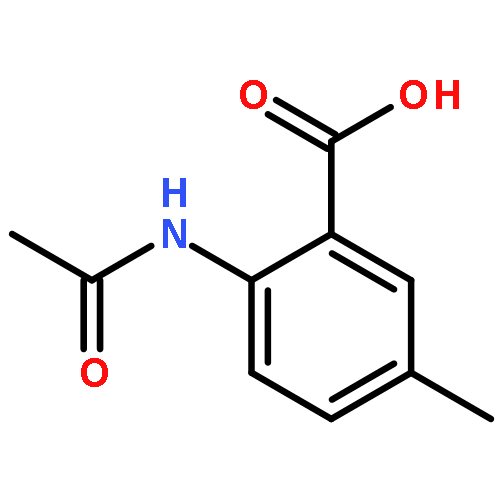
Co-reporter:Chen Zhang, Ling-Jun Feng, Yiyou Huang, Deyan Wu, Zhe Li, Qian Zhou, Yinuo Wu, and Hai-Bin Luo
Journal of Chemical Information and Modeling 2017 Volume 57(Issue 2) pp:
Publication Date(Web):January 5, 2017
DOI:10.1021/acs.jcim.6b00551
Phosphodiesterase-2A (PDE2A) is a potential therapeutic target for treatment of Alzheimer’s disease and pulmonary hypertension. However, most of the current PDE2A inhibitors have moderate selectivity over other PDEs. In the present study, we described the discovery of novel PDE2A inhibitors by structure-based virtual screening combining pharmacophore model screening, molecular docking, molecular dynamics simulations, and bioassay validation. Nine hits out of 30 molecules from the SPECS database (a hit rate of 30%) inhibited PDE2A with affinity less than 50 μM. Optimization of compound AQ-390/10779040 (IC50 = 4.6 μM) from the virtual screening, which holds a novel scaffold of benzo[cd]indol-2(1H)-one among PDE inhibitors, leads to discovery of a new compound LHB-8 with a significant improvement of inhibition (IC50 = 570 nM). The modeling studies demonstrated that LHB-8 formed an extra hydrogen bond with Asp808 and a hydrophobic interaction with Thr768, in addition to the common interactions with Gln859 and Phe862 of PDE2A. The novel scaffolds discovered in the present study can be used for rational design of PDE2A inhibitors with high affinity.
Co-reporter:Yiyou Huang, Xin Liu, Deyan Wu, Guihua Tang, ... Hai-Bin Luo
Biochemical Pharmacology 2017 Volume 130(Volume 130) pp:
Publication Date(Web):15 April 2017
DOI:10.1016/j.bcp.2017.01.016
Phosphodiesterase-4 (PDE4) is an important drug target for treatment of inflammation-related diseases. Till now, natural PDE4 inhibitors are rare and their co-crystal structures with PDE4 are hardly available. In the present study, selaginpulvilins K and L (1 and 2), two novel fluorene derivatives, were isolated from a traditional Chinese medicine Selaginella pulvinata and exhibited remarkable inhibition against phosphodiesterase-4D (PDE4D) at IC50 11 nM and 90 nM, respectively. Compound 1 also showed a good selectivity across PDE families with the selective fold ranging from 30 to 909. To understand the recognition mechanism of selaginpulvilins towards PDE4, the crystal structure of PDE4D bound with 1 was successfully determined by the X-ray diffraction method and presented an unusual binding mode in which the stretched skeleton of the inhibitor bound shallowly to the active site but had interactions with multi sub-pockets, such as Q, HC, M, and S, especially strong interaction with the metal region. Assisted with molecular modeling, the structure–activity relationship and the selectivity of selaginpulvilins were also well explored, which would facilitate the future rational inhibitor design or structural optimizations.Download high-res image (260KB)Download full-size image
Co-reporter:Yinuo Wu, Cheng Jiang, Deyan Wu, Qiong Gu, Zhang-Yi Luo and Hai-Bin Luo
Chemical Communications 2016 vol. 52(Issue 6) pp:1286-1289
Publication Date(Web):17 Nov 2015
DOI:10.1039/C5CC07890C
N,N-Dimethyloxamic acid can be successfully employed as a carboxylate precursor in the palladium-catalyzed direct C–H carboxylation of acetanilides. The reaction proceeds smoothly under mild conditions over a broad range of substrates with high functional group tolerance, affording substituted N-acyl anthranilic acids in moderate to high yields.
Co-reporter:Yinuo Wu, Lei Sun, Yunyun Chen, Qian Zhou, Jia-Wu Huang, Hui Miao, and Hai-Bin Luo
The Journal of Organic Chemistry 2016 Volume 81(Issue 3) pp:1244-1250
Publication Date(Web):January 8, 2016
DOI:10.1021/acs.joc.5b02535
A palladium-catalyzed oxidative C–H bond decarboxylative acylation of N-nitrosoanilines using α-oxocarboxylic acid as the acyl source is described. The catalyst Pd(OAc)2 and oxidant (NH4)2S2O8 enabled ortho-acylation of N-nitrosoanilines at room temperature, affording an array of N-nitroso-2-aminobenzophenones in moderate to excellent yields.
Co-reporter:Jingwei Zhou, Ruibo Wu and Hai-Bin Luo
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 44) pp:29483-29488
Publication Date(Web):08 Oct 2015
DOI:10.1039/C5CP05633K
SAHA (vorinostat, Merck) is a famous clinical drug for zinc-containing histone deacetylase (HDAC) targets against cancer and several other human disorders, whose inhibition mechanism (namely the protonation mechanism) upon binding to HDAC has been debated for more than ten years. It is very challenging to verify experimentally and is still controversial theoretically. The popular “Class-dependent” (namely “Tyr-dependent”) hypothesis is that the deprotonation of SAHA is mostly regulated by the conserved Tyr308 in class I HDAC while it is replaced by the His843 in class IIa HDAC. Herein, by elaborate QM(DFT)/MM MD simulations, we exclude the prevalent “Class-dependent” mechanism and advance a novel “Metal-dependent” mechanism, where the remote second metal site (K+ in most HDAC and Ca2+ in HDAC2) determines the protonation of SAHA. This proof-of-principle “Metal-dependent” mechanism opens up a new avenue to utilize the second metal site for isoform-selective inhibitor design.
Co-reporter:Zhe Li, Xiao Lu, Ling-Jun Feng, Ying Gu, Xingshu Li, Yinuo Wu and Hai-Bin Luo
Molecular BioSystems 2015 vol. 11(Issue 1) pp:115-125
Publication Date(Web):10 Oct 2014
DOI:10.1039/C4MB00389F
Phosphodiesterase-9A (PDE9A) is a promising therapeutic target for the treatment of diabetes and Alzheimer's disease (AD). The Pfizer PDE9A inhibitor PF-04447943 has completed Phase II clinical trials in subjects with mild to moderate AD in 2013. However, most of the reported PDE9A inhibitors share the same scaffold as pyrazolopyrimidinone, which lacks structural diversity and is unfavorable for the development of novel PDE9A inhibitors. In the present study, a combinatorial method including pharmacophores, molecular docking, molecular dynamics simulations, binding free energy calculations, and bioassay was used to discover novel PDE9A inhibitors with new scaffolds rather than pyrazolopyrimidinones from the SPECS database containing about 200000 compounds. As a result, 15 hits out of 29 molecules (a hit rate of 52%) with five novel scaffolds were identified to be PDE9A inhibitors with inhibitory affinities no more than 50 μM to enrich the structural diversity, different from the pyrazolopyrimidinone-derived family. The high hit ratio of 52% for this virtual screening method indicated that the combinatorial method is a good compromise between computational cost and accuracy. Binding pattern analyses indicate that those hits with non-pyrazolopyrimidinone scaffolds can bind the same active site pocket of PDE9A as classical PDE9A inhibitors. In addition, structural modification of compound AG-690/40135604 (IC50 = 8.0 μM) led to a new one, 16, with an improved inhibitory affinity of 2.1 μM as expected. The five novel scaffolds discovered in the present study can be used for the rational design of PDE9A inhibitors with higher affinities.
Co-reporter:Yinuo Wu, Ling-Jun Feng, Xiao Lu, Fuk Yee Kwong and Hai-Bin Luo
Chemical Communications 2014 vol. 50(Issue 97) pp:15352-15354
Publication Date(Web):15 Oct 2014
DOI:10.1039/C4CC07440H
A palladium-catalyzed cascade cross-coupling of N-nitroso-anilines and toluene derivatives for the direct synthesis of N-alkyl-2-aminobenzophenones is described. N-nitroso groups in anilines can act as the traceless directing groups while toluene derivatives can serve as effective acyl precursors under mild reaction conditions.
Co-reporter:Yong-xian Shao ; Manna Huang ; Wenjun Cui ; Ling-Jun Feng ; Yinuo Wu ; Yinghong Cai ; Zhe Li ; Xinhai Zhu ; Peiqing Liu ; Yiqian Wan ; Hengming Ke ;Hai-Bin Luo
Journal of Medicinal Chemistry 2014 Volume 57(Issue 24) pp:10304-10313
Publication Date(Web):November 28, 2014
DOI:10.1021/jm500836h
Phosphodiesterase 9 (PDE9) inhibitors have been studied as potential therapeutics for treatment of diabetes and Alzheimer’s disease. Here we report a potent PDE9 inhibitor 3r that has an IC50 of 0.6 nM and >150-fold selectivity over other PDEs. The HepG2 cell-based assay shows that 3r inhibits the mRNA expression of phosphoenolpyruvate carboxykinase and glucose 6-phosphatase. These activities of 3r, together with the reasonable pharmacokinetic properties and no acute toxicity at 1200 mg/kg dosage, suggest its potential as a hypoglycemic agent. The crystal structure of PDE9-3r reveals significantly different conformation and hydrogen bonding pattern of 3r from those of previously published 28s. Both 3r and 28s form a hydrogen bond with Tyr424, a unique PDE9 residue (except for PDE8), but 3r shows an additional hydrogen bond with Ala452. This structure information might be useful for design of PDE9 inhibitors.
Co-reporter:Zhe Li, Yinuo Wu, Ling-Jun Feng, Ruibo Wu, and Hai-Bin Luo
Journal of Chemical Theory and Computation 2014 Volume 10(Issue 12) pp:5448-5457
Publication Date(Web):November 6, 2014
DOI:10.1021/ct500761d
Phosphodiesterases (PDEs) are the sole enzymes hydrolyzing the important second messengers cGMP and cAMP and have been identified as therapeutic targets for several diseases. The most successful examples are PDE5 inhibitors (i.e., sildenafil and tadalafil), which have been approved for the treatment of male erectile dysfunction and pulmonary hypertension. However, the side effects mostly due to nonselective inhibition toward other PDE isoforms, set back the clinical usage of PDE5 inhibitors. Until now, the exact catalytic mechanism of the substrate cGMP by PDE5 is still unclear. Herein, the first computational study on the catalytic hydrolysis mechanism of cGMP for PDE5 (catalytic domain) is performed by employing the state-of-the-art ab initio quantum mechanics/molecular mechanics (QM/MM) molecular dynamics (MD) simulations. Our simulations show a SN2 type reaction procedure via a highly dissociated transition state with a reaction barrier of 8.88 kcal/mol, which is quite different from the previously suggested hydrolysis mechanism of cAMP for PDE4. Furthermore, the subsequent ligand exchange and the release of the product GMP have also been investigated by binding energy analysis and MD simulations. It is deduced that ligand exchange would be the rate-determining step of the whole reaction, which is consistent with many previous experimental results. The obtained mechanistic insights should be valuable for not only the rational design of more specific inhibitors toward PDE5 but also understanding the general hydrolysis mechanism of cGMP-specific PDEs.
Co-reporter:Ting-Ting Lin ; Yi-You Huang ; Gui-Hua Tang ; Zhong-Bin Cheng ; Xin Liu ; Hai-Bin Luo ;Sheng Yin
Journal of Natural Products 2014 Volume 77(Issue 4) pp:955-962
Publication Date(Web):March 5, 2014
DOI:10.1021/np401040d
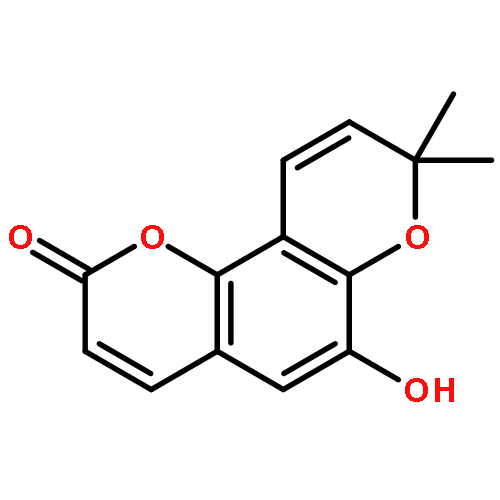
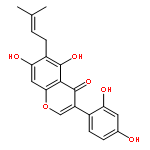
Bioassay-guided fractionation of the ethanolic extract of the roots of Toddalia asiatica led to the isolation of seven new prenylated coumarins (1–7) and 14 known analogues (8–21). The structures of 1–7 were elucidated by spectroscopic analysis, and their absolute configurations were determined by combined chemical methods and chiral separation analysis. Compounds 1–5, named toddalin A, 3‴-O-demethyltoddalin A, and toddalins B–D, represent an unusual group of phenylpropenoic acid-coupled prenylated coumarins. Compounds 1–21 and four modified analogues, 10a, 11a, 13a, and 17a, were screened by using tritium-labeled adenosine 3′,5′-cyclic monophosphate ([3H]-cAMP) as substrate for their inhibitory activity against phosphodiesterase-4 (PDE4), which is a drug target for the treatment of asthma and chronic obstructive pulmonary disease. Compounds 3, 8, 10, 10a, 11, 11a, 12, 13, 17, and 21 exhibited inhibition with IC50 values less than 10 μM. Toddacoumalone (8), the most active compound (IC50 = 0.14 μM), was more active than the positive control, rolipram (IC50 = 0.59 μM). In addition, the structure–activity relationship and possible inhibitory mechanism of the active compounds are also discussed.
Co-reporter:Zhong-Bin Cheng ; Xiao Lu ; Jing-Mei Bao ; Qing-Hua Han ; Zhen Dong ; Gui-Hua Tang ; Li-She Gan ; Hai-Bin Luo ;Sheng Yin
Journal of Natural Products 2014 Volume 77(Issue 12) pp:2651-2657
Publication Date(Web):December 12, 2014
DOI:10.1021/np500528u
(±)-Torreyunlignans A–D (1a/1b–4a/4b), four pairs of new 8–9′ linked neolignan enantiomers featuring a rare (E)-2-styryl-1,3-dioxane moiety, were isolated from the trunk of Torreya yunnanensis. The structures were determined by combined spectroscopic and chemical methods, and the absolute configurations were elucidated by ECD calculations. The compounds were screened by using tritium-labeled adenosine 3′,5′-cyclic monophosphate ([3H]-cGMP) as a substrate for inhibitory affinities against phosphodiesterase-9A (PDE9A), which is a potential target for the treatment of diabetes and Alzheimer’s disease. All of the enantiomers exhibited inhibition against PDE9A with IC50 values ranging from 5.6 to 15.0 μM. This is the first report of PDE9A inhibitors from nature.
Co-reporter:Jingwei Zhou, Hujun Xie, Zhihong Liu, Hai-Bin Luo, and Ruibo Wu
Journal of Chemical Information and Modeling 2014 Volume 54(Issue 11) pp:3162-3171
Publication Date(Web):October 31, 2014
DOI:10.1021/ci500513n
Discovery of the isoform-selective histone deacetylases (HDACs) inhibitors is of great medical importance and still a challenge. The comparison studies on the structure–function relationship of the conserved residues, which are located in the linker binding channel among class I HDACs (including 4 isoforms: HDAC1/2/3/8), have been carried out by using ab initio QM/MM MD simulations, a state-of-the-art approach to simulate metallo-enzymes. We found that the conserved tyrosine (Y303/308/286/306 in HDAC1/2/3/8, respectively) could modulate the zinc-inhibitor chelation among all class I HDACs with different regulatory mechanisms. For HDAC1/2/3 selective-inhibitor benzamide, the conserved tyrosine could modulate the coordinative ability of the central atom (Zn2+), while for pan-inhibitor SAHA, the conserved tyrosine could increase the chelating ability of the ligand (SAHA). Moreover, it is first found that the conserved tyrosine is correlated with the intertransformation of π–π stacking styles (parallel shift vs T-shaped) by the aromatic ring in benzamide and the two conserved phenylalanine residues of HDACs. In addition, the catalytic roles of the conserved tyrosine in stabilizing the transition state and intermediate are further revealed. These findings provide useful molecular basis knowledge for further isoform-selective inhibitor design among class I HDACs.
Co-reporter:Zhong-Bin Cheng ; Ya-Lin Deng ; Cheng-Qi Fan ; Qing-Hua Han ; Shu-Ling Lin ; Gui-Hua Tang ; Hai-Bin Luo ;Sheng Yin
Journal of Natural Products 2014 Volume 77(Issue 8) pp:1928-1936
Publication Date(Web):July 30, 2014
DOI:10.1021/np500394d
Ten new prostaglandin derivatives (PGs), sarcoehrendins A–J (1–10), together with five known analogues (11–15) were isolated from the soft coral Sarcophyton ehrenbergi. Compounds 4–8 represented the first examples of PGs featuring an 18-ketone group. The structures including the absolute configurations were elucidated on the basis of spectroscopic analysis and chemical evidence. All of the isolates and six synthetic analogues (3a, 3b, 4a, and 11a–11c) were screened for inhibitory activity against phosphodiesterase-4 (PDE4), which is a drug target for the treatment of asthma and chronic obstructive pulmonary disease. Compounds 2, 10, 11a, 11b, and 13–15 exhibited inhibition with IC50 values less than 10 μM, and compound 15 (IC50 = 1.4 μM) showed comparable activity to the positive control rolipram (IC50 = 0.60 μM). The active natural PGs (2, 10, and 13–15) represent the first examples of PDE4 inhibitors without an aromatic moiety, and a preliminary structure–activity relationship is also proposed.
Co-reporter:Zhe Li, Ying-Hong Cai, Yuen-Kit Cheng, Xiao Lu, Yong-Xian Shao, Xingshu Li, Ming Liu, Peiqing Liu, and Hai-Bin Luo
Journal of Chemical Information and Modeling 2013 Volume 53(Issue 4) pp:972-981
Publication Date(Web):March 21, 2013
DOI:10.1021/ci400063s
Phosphodiesterase-4D (PDE4D) has been proved to be a potential therapeutic target against strokes. In the present study, a procedure of integrating pharmacophore, molecular docking, molecular dynamics (MD) simulations, binding free energy calculations, and finally validation with bioassay was developed and described to search for novel PDE4D inhibitors from the SPECS database. Among the 29 compounds selected by our MD-augmented strategy, 15 hits were found with IC50 between 1.9 and 50 μM (a hit rate of 52%) and 6 potent hits showed IC50 less than 10 μM, which suggested that MD simulations can explore the intermolecular interactions of PDE4D–inhibitor complexes more precisely and thus significantly enhanced the hit rate of this screening. The effective and efficient integrated procedures described in this study could be readily applied to screening studies toward other drug targets.
Co-reporter:Yi-You Huang, Zhe Li, Ying-Hong Cai, Ling-Jun Feng, Yinuo Wu, Xingshu Li, and Hai-Bin Luo
Journal of Chemical Information and Modeling 2013 Volume 53(Issue 11) pp:3044-3053
Publication Date(Web):November 1, 2013
DOI:10.1021/ci400458z
Great attention has been paid to the clinical significance of phosphodiesterase 5 (PDE5) inhibitors, such as sildenafil, tadalafil, and vardenafil widely used for erectile dysfunction. However, sildenafil causes side effects on visual functions since it shows similar potencies to inhibit PDE5 and PDE6, whereas tadalafil gives a high selectivity of 1020-fold against PDE6. Till now, their molecular mechanisms of selectivity of PDE5 versus PDE6 have remained unknown in the absence of the crystal structure of PDE6. In order to elucidate its isoform-selective inhibitory mechanism, a 3D model of PDE6 was constructed by homology modeling, and its interaction patterns with tadalafil plus sildenafil were exploited by molecular docking, molecular dynamics (MD) simulations, and binding free energy calculations. The present work reveals that tadalafil exhibits a less negative predicted binding free energy of −35.21 kcal/mol with PDE6 compared with the value of −41.12 kcal/mol for PDE5, which suggests that tadalafil prefers PDE5 rather than PDE6 and confers a high selectivity for PDE5 versus PDE6. The binding free energy results for tadalafil were consistent with external bioassay studies (IC50 = 5100 and 5 nM toward PDE6 and PDE5, respectively). Two important residues from the Q2 pockets (Val782 and Leu804 in PDE5 and their corresponding Val738 and Met760 in PDE6) were further identified to account for the high selectivity of tadalafil for PDE5 versus PDE6. These findings have shed light on the continuous puzzle of why sildenafil (IC50 = 74 and 6 nM toward PDE6 and PDE5, respectively) causes visual disorders because of its poor selectivity but tadalafil does not. In addition, the homology model of PDE6 can be used to design more potent and selective second-generation PDE5 inhibitors with less inhibitory potency against PDE6.
Co-reporter:Peng Zhao, Shang-Ke Chen, Ying-Hong Cai, Xiao Lu, Zhe Li, Yuen-Kit Cheng, Cuixian Zhang, Xiaopeng Hu, Xixin He, Hai-Bin Luo
Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics 2013 Volume 1834(Issue 10) pp:2089-2096
Publication Date(Web):October 2013
DOI:10.1016/j.bbapap.2013.07.004
•Pterostilbene represents a different binding pattern from other two compounds.•Hydrophobic/aromatic forces play the major contributions to the binding of ligands.•A linear correlation was derived between the predicted ΔGbind and experimental ΔGbind′.The phosphodiesterase-4 (PDE4) enzyme is a promising therapeutic target for several diseases. Our previous studies found resveratrol and moracin M to be natural PDE4 inhibitors. In the present study, three natural resveratrol analogs [pterostilbene, (E)-2′,3,5′,5-tetrahydroxystilbene (THSB), and oxyresveratrol] are structurally related to resveratrol and moracin M, but their inhibition and mechanism against PDE4 are still unclear. A combined method consisting of molecular docking, molecular dynamics (MD) simulations, binding free energy, and bioassay was performed to better understand their inhibitory mechanism. The binding pattern of pterostilbene demonstrates that it involves hydrophobic/aromatic interactions with Phe340 and Phe372, and forms hydrogen bond(s) with His160 and Gln369 in the active site pocket. The present work also reveals that oxyresveratrol and THSB can bind to PDE4D and exhibits less negative predicted binding free energies than pterostilbene, which was qualitatively validated by bioassay (IC50 = 96.6, 36.1, and 27.0 μM, respectively). Additionally, a linear correlation (R2 = 0.953) is achieved for five PDE4D/ligand complexes between the predicted binding free energies and the experimental counterparts approximately estimated from their IC50 values (≈RT ln IC50). Our results imply that hydrophobic/aromatic forces are the primary factors in explaining the mechanism of inhibition by the three products. Results of the study help to understand the inhibitory mechanism of the three natural products, and thus help the discovery of novel PDE4 inhibitors from resveratrol, moracin M, and other natural products.
Co-reporter:Yan-Ping Li, Xiang Weng, Fang-Xian Ning, Jie-Bin Ou, Jin-Qiang Hou, Hai-Bin Luo, Ding Li, Zhi-Shu Huang, Shi-Liang Huang, Lian-Quan Gu
Journal of Molecular Graphics and Modelling 2013 Volume 41() pp:61-67
Publication Date(Web):April 2013
DOI:10.1016/j.jmgm.2013.02.003
In the present study, a series of novel azaoxoisoaporphine derivatives were reported and their inhibitory activities toward acetylcholinesterase (AChE), butyrylcholinesterase (BuChE), and Aβ aggregation were evaluated. The new compounds remained high inhibitory potency on Aβ aggregation, with inhibitory activity from 29.42% to 89.63% at a concentration of 10 μM, but had no action on AChE or BuChE, which was very different from our previously reported oxoaporphine and oxoisoaporphine derivatives. By 3D-QSAR studies, we constructed a reliable CoMFA model (q2 = 0.856 and r2 = 0.986) based on the inhibitory activities toward AChE and discovered key information on structure and anti-AChE activities among the azaoxoisoaporphine, oxoaporphine, and oxoisoaporphine derivatives. The model was further confirmed by the test-set validation (q2 = 0.873, r2 = 0.937, and slope k = 0.902) and Y-randomization examination. The statistically significant and physically meaningful 3D-QSAR/CoMFA model provided better insight into understanding the inhibitory behaviors of those chemicals, which may provide useful information for the rational molecular design of azaoxoisoaporphine derivatives anti-AChE and anti-AD agents.Graphical abstractHighlights► This paper analyzed a new series of azaoxoisoaporphine derivatives. ► The derivatives showed high inhibition on Aβ aggregation but no ChE inhibition. ► A statistically significant and physically meaningful 3D-QSAR model was derived. ► The molecular shape, size, and charge were crucial for AChE inhibition.
Co-reporter:Fei Meng ; Jing Hou ; Yong-Xian Shao ; Pei-Ying Wu ; Manna Huang ; Xinhai Zhu ; Yonghong Cai ; Zhe Li ; Jie Xu ; Peiqing Liu ; Hai-Bin Luo ; Yiqian Wan ;Hengming Ke
Journal of Medicinal Chemistry 2012 Volume 55(Issue 19) pp:8549-8558
Publication Date(Web):September 17, 2012
DOI:10.1021/jm301189c
A new series of phosphodiesterase-9 (PDE9) inhibitors that contain a scaffold of 6-amino-pyrazolopyrimidinone have been discovered by a combination of structure-based design and computational docking. This procedure significantly saved the load of chemical synthesis and is an effective method for the discovery of inhibitors. The best compound 28 has an IC50 of 21 nM and 3.3 μM, respectively, for PDE9 and PDE5 and about 3 orders of magnitude of selectivity against other PDE families. The crystal structure of the PDE9 catalytic domain in complex with 28 has been determined and shows a hydrogen bond between 28 and Tyr424. This hydrogen bond may account for the 860-fold selectivity of 28 against PDE1B, in comparison with about 30-fold selectivity of BAY73-6691. Thus, our studies suggest that Tyr424, a unique residue of PDE8 and PDE9, is a potential target for improvement of selectivity of PDE9 inhibitors.
Co-reporter:Xue-Hua Zheng;Yong-Xian Shao;Zhe Li;Ming Liu;Xianzhang Bu;Hai-Bin Luo;Xiaopeng Hu
Journal of Separation Science 2012 Volume 35( Issue 4) pp:505-512
Publication Date(Web):
DOI:10.1002/jssc.201100903
Abstract
The natural product curcumin is widely used in Asian countries for the treatment of several diseases. However, the clinical potential of curcumins remains limited due to their relatively poor bioavailability and no experimental data about their lipophilicity for bioavailability prediction. To evaluate the retention and lipophilicity of curcumin and its 31 newly synthesized analogues, they were subjected to 3D quantitative structure–retention relationship studies by RP-HPLC. Superior than the classical four-variant quantitative structure–retention relationship model (conventional r2=0.734), the 3D comparative molecular similarity index analysis model with combined steric, electrostatic, and H-bond donor fields, resulted in a robust structure–retention correlation (cross-validated q2=0.613 and r2=0.979). The statistical analyses indicate that the electrostatic and H-bond donor fields have a primary influence on the chromatographic retention of analytes. The predictive power and robustness of the derived comparative molecular similarity index analysis model was further confirmed by the test-set validation (q2=0.702, r2=0.905, and the slope K=1.016) and Y-randomization examination. Statistically significant and physically meaningful 3D-quantitative structure–retention relationship provided better insight into understanding the retention behaviors of curcumin and its analogues, and their separation mechanism in a given RP-HPLC system.
Co-reporter:Shang-Ke Chen, Peng Zhao, Yong-Xian Shao, Zhe Li, Cuixian Zhang, Peiqing Liu, Xixin He, Hai-Bin Luo, Xiaopeng Hu
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 9) pp:3261-3264
Publication Date(Web):1 May 2012
DOI:10.1016/j.bmcl.2012.03.026
Phosphodiesterase-4 (PDE4) has been identified to be a promising target for treatment of asthma. Moracin M extracted from Chinese herbal drug ‘Sang-Bai-Pi’ (Morus alba L.) was studied for the inhibitory affinity towards PDE4. It inhibited PDE4D2, PDE4B2, PDE5A1, and PDE9A2 with the IC50 values of 2.9, 4.5, >40, and >100 μM, respectively. Our molecular docking and 8 ns molecular dynamics (MD) simulations demonstrated that moracin M forms three hydrogen bonds with Gln369, Asn321, and Asp318 in the active site and stacks against Phe372. In addition, comparative kinetics analysis of its analog moracin C was carried out to qualitatively validate their inhibitory potency as predicted by the binding free energy calculations after MD simulations.
Co-reporter:Yong-Xian Shao;Peng Zhao;Zhe Li;Ming Liu;Peiqing Liu
European Biophysics Journal 2012 Volume 41( Issue 3) pp:297-306
Publication Date(Web):2012 March
DOI:10.1007/s00249-011-0785-1
In our previous kinetics studies the natural products oroxylin and wogonin were shown to have strong biological affinity for, and inhibitory effects against, human cytochrome P450 1A2, with IC50 values of 579 and 248 nM, respectively; this might lead to the occurrence of drug–drug interactions when co-administered clinically. However, their inhibitory mechanisms against 1A2 remain elusive. In this study, molecular docking and molecular dynamics simulations were performed to better understand the molecular basis of their inhibitory mechanisms towards 1A2. Structural analysis revealed that oroxylin has a different binding pattern from wogonin and another very strongly binding inhibitor α-naphthoflavone (ANF, IC50 = 49 nM). The O7 atom of oroxylin forms hydrogen bonds with the OD1/OD2 atoms of Asp313, which is not observed in the 1A2–wogonin complex. Because of energetically unfavorable repulsions with the methoxy group at the 6 position of the oroxylin ring, significant conformational changes were observed for the sidechain of Thr118 in the MD simulated model. As a result, the larger and much more open binding-site architecture of the 1A2–oroxylin complex may account for its weaker inhibitory effect relative to the 1A2–ANF complex. Energy analysis indicated that oroxylin has a less negative predicted binding free energy of −19.8 kcal/mol than wogonin (−21.1 kcal/mol), which is consistent with our experimental assays. Additionally, our energy results suggest that van der Waals/hydrophobic and hydrogen-bonding interactions are important in the inhibitory mechanisms of oroxylin whereas the former is the underlying force responsible for strong inhibition by ANF and wogonin.
Co-reporter:Jiansong Fang, Dane Huang, Wenxia Zhao, Hu Ge, Hai-Bin Luo, and Jun Xu
Journal of Chemical Information and Modeling 2011 Volume 51(Issue 6) pp:1431-1438
Publication Date(Web):May 26, 2011
DOI:10.1021/ci2001154
Glycogen synthase kinase 3β (GSK-3β) is a potential therapeutic target for cancer, type-2 diabetes, and Alzheimer’s disease. This paper proposes a new lead identification protocol that predicts new GSK-3β ATP competitive inhibitors with topologically diverse scaffolds. First, three-dimensional quantitative structure–activity relationship (3D QSAR) models were built and validated. These models are based upon known GSK-3β inhibitors, benzofuran-3-yl-(indol-3-yl) maleimides, by means of comparative molecular field analysis (CoMFA) and comparative molecular similarity indices analysis (CoMSIA). Second, 28 826 maleimide derivatives were selected from the PubChem database. After filtration via Lipinski’s rules, 10 429 maleimide derivatives were left. Third, the FlexX-dock program was employed to virtually screen the 10 429 compounds against GSK-3β. This resulted in 617 virtual hits. Fourth, the 3D QSAR models predicted that from the 617 virtual hits, 93 compounds would have GSK-3β inhibition values of less than 15 nM. Finally, from the 93 predicted active hits, 23 compounds were confirmed as GSK-3β inhibitors from literatures; their GSK-3β inhibition ranged from 1.3 to 480 nM. Therefore, the hits rate of our virtual screening protocol is greater than 25%. The protocol combines ligand- and structure-based approaches and therefore validates both approaches and is capable of identifying new hits with topologically diverse scaffolds.
Co-reporter:Ming Liu, Minggui Yuan, Zhe Li, Yuen-Kit Cheng, Hai-Bin Luo, Xiaopeng Hu
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 14) pp:4243-4247
Publication Date(Web):15 July 2011
DOI:10.1016/j.bmcl.2011.05.095
In the present work, a combined study of kinetic analysis, molecular docking, and molecular dynamics simulations on indomethacin and its analogues is performed to better understand their inhibitory mechanisms towards human glyoxalase I (GLOI). A remarkable correlation (R2 = 0.974) was observed for six inhibitors including indomethacin between their experimental inhibitory affinities and predicted binding free energy parameter (ΔGbind,pred). This suggests that ΔGbind,pred of a GLOI/inhibitor complex can be efficiently used to interpolate the experimental inhibitory affinity of a ligand of similar nature in the GLOI enzyme system. Energetic analyses revealed that electrostatic contribution plays an important role in their inhibitory mechanisms, which reflects the significant contribution of the coordination bond between zinc and ligands. The present work highlights that indomethacin is a promising lead as GLOI inhibitors for further development since it may bind all subsites in the active site pocket of GLOI and stabilize the flexible loop (152–159).
Co-reporter:Ming Liu, Minggui Yuan, Minxian Luo, Xianzhang Bu, Hai-Bin Luo, Xiaopeng Hu
Biophysical Chemistry 2010 Volume 147(1–2) pp:28-34
Publication Date(Web):March 2010
DOI:10.1016/j.bpc.2009.12.007
Glyoxalase I (GLOI) is a key metalloenzyme in glycolytic pathway by detoxifying reactive α-ketoaldehydes such as methylglyoxal. Recent studies demonstrate that the nature product curcumin is an efficient inhibitor of GLOI, but its binding mechanism towards GLOI is still unclear. In the present study, molecular docking and molecular dynamics (MD) simulations were performed to better understand the inhibitory mechanism of curcumin towards GLOI. The enol form of curcumin coordinates with the catalytic zinc ion of GLOI and forms a strong hydrogen bond with Glu 172, whereas its keto tautomer displays unfavorable electrostatic interactions with Glu 172 and Glu 99. The calculated binding free energies suggest that GLOI prefers the primary enol form (ΔG = − 30.38 kcal/mol) to the keto tautomer (ΔG = − 24.16 kcal/mol). The present work also reveals that bisdemethoxycurcumin binds to GLOI in a similar manner as curcumin and exhibits a slightly less negative predicted binding free energy, which is further validated by our comparative kinetics analysis (Ki = 18.2 and 10.3 μM for bisdemethoxycurcumin and curcumin, respectively). Results of the study can provide an insight into the development of novel and more effective GLOI inhibitors.
Co-reporter:Lin He, Fan He, Huichang Bi, Jiankang Li, Su Zeng, Hai-Bin Luo, Min Huang
Bioorganic & Medicinal Chemistry Letters 2010 Volume 20(Issue 20) pp:6008-6012
Publication Date(Web):15 October 2010
DOI:10.1016/j.bmcl.2010.08.072
Our kinetics studies demonstrated that the nature product chrysin exhibited a high inhibitory affinity of 54 nM towards human cytochrome P450 1A2 and was comparable to α-naphthoflavone (49 nM), whereas it represented a moderate affinity of 5225 nM against human cytochrome P450 2C9. However, it remains unclear how this inhibitor selectively binds 1A2. To better understand the isoform selectivity of chrysin, molecular docking and molecular dynamics simulations were performed. Chrysin formed a strong H-bond with Asp313 of 1A2. The stacking interactions with Phe226 also contributed to its tight binding to 1A2. The larger and much more open active site architectures of 2C9 may explain the weaker inhibitory affinity of chrysin towards 2C9. The predicted binding free energies suggest that chrysin preferred 1A2 (ΔGbind, pred = −23.11 kcal/mol) to 2C9 (−20.41 kcal/mol). Additionally, the present work revealed that 7-hydroxy-flavone bound to 1A2 in a similar pattern as chrysin and represented a slightly less negative predicted binding free energy, which was further validated by our kinetics analysis (IC50 = 240 nM). Results of the study can provide insight for designing novel isoform-selective 1A2 inhibitors.Both experimental and computational methods have been performed to explore the inhibitory mechanism of chrysin (IC50 = 54 nM) towards human cytochrome P450 1A2.
Co-reporter:Ming Liu, Lin He, Xiaopeng Hu, Peiqing Liu, Hai-Bin Luo
Bioorganic & Medicinal Chemistry Letters 2010 Volume 20(Issue 23) pp:7004-7010
Publication Date(Web):1 December 2010
DOI:10.1016/j.bmcl.2010.09.116
The nociceptin/orphanin FQ receptor (NOP) has been implicated in a wide range of biological functions, including pain, anxiety, depression and drug abuse. Especially, its agonists have a great potential to be developed into anxiolytics. However, the crystal structure of NOP is still not available. In the present work, both structure-based and ligand-based modeling methods have been used to achieve a comprehensive understanding on 67 N-substituted spiropiperidine analogues as NOP agonists. The comparative molecular-field analysis method was performed to formulate a reasonable 3D-QSAR model (cross-validated coefficient q2 = 0.819 and conventional r2 = 0.950), whose robustness and predictability were further verified by leave-eight-out, Y-randomization, and external test-set validations. The excellent performance of CoMFA to the affinity differences among these compounds was attributed to the contributions of electrostatic/hydrogen-bonding and steric/hydrophobic interactions, which was supported by the Surflex-Dock and CDOCKER molecular-docking simulations based on the 3D model of NOP built by the homology modeling method. The CoMFA contour maps and the molecular docking simulations were integrated to propose a binding mode for the spiropiperidine analogues at the binding site of NOP.We studied 3D-QSAR and molecular mechanism of spiropiperidine analogues targeting nociceptin/orphanin FQ receptor (NOP). Molecular docking simulations reveal salt bridge, hydrogen-bonding, and hydrophobic interactions stabilize the most active agonist P67 in the binding site pocket of NOP.
Co-reporter:Xuehua Zheng, Yinuo Wu, Deyan Wu, Xinhua Wang, Chao Zhang, Xiaolei Guo, Hai-Bin Luo
Bioorganic & Medicinal Chemistry Letters (1 December 2016) Volume 26(Issue 23) pp:5631-5638
Publication Date(Web):1 December 2016
DOI:10.1016/j.bmcl.2016.10.073
Co-reporter:Xuehua Zheng, Yinuo Wu, Deyan Wu, Xinhua Wang, Chao Zhang, Xiaolei Guo, Hai-Bin Luo
Bioorganic & Medicinal Chemistry Letters (1 December 2016) Volume 26(Issue 23) pp:5631-5638
Publication Date(Web):1 December 2016
DOI:10.1016/j.bmcl.2016.10.073
AKR1C3 is a promising drug target for castration-resistant prostate cancer (CRPC). Here, 3D-QSAR analysis were performed on 3-(3,4-dihydroisoquinolin-2(1H)-ylsulfonyl)benzoic acids to correlate their chemical structures with their observed AKR1C3 inhibitory activity. Three structural alignment methods employing various conformers were used to scrutinize the effect of conformation selection on the predictive accuracy of QSAR models. Using docked conformation, the best CoMFA and CoMSIA models were developed and validated with a training set of 61 molecules and a test set of 7 molecules. Detailed analysis of contour maps provided helpful structural insights to rational design of AKR1C3 inhibitors with enhanced potency.
Co-reporter:Yinuo Wu, Cheng Jiang, Deyan Wu, Qiong Gu, Zhang-Yi Luo and Hai-Bin Luo
Chemical Communications 2016 - vol. 52(Issue 6) pp:NaN1289-1289
Publication Date(Web):2015/11/17
DOI:10.1039/C5CC07890C
N,N-Dimethyloxamic acid can be successfully employed as a carboxylate precursor in the palladium-catalyzed direct C–H carboxylation of acetanilides. The reaction proceeds smoothly under mild conditions over a broad range of substrates with high functional group tolerance, affording substituted N-acyl anthranilic acids in moderate to high yields.
Co-reporter:Jingwei Zhou, Ruibo Wu and Hai-Bin Luo
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 44) pp:NaN29488-29488
Publication Date(Web):2015/10/08
DOI:10.1039/C5CP05633K
SAHA (vorinostat, Merck) is a famous clinical drug for zinc-containing histone deacetylase (HDAC) targets against cancer and several other human disorders, whose inhibition mechanism (namely the protonation mechanism) upon binding to HDAC has been debated for more than ten years. It is very challenging to verify experimentally and is still controversial theoretically. The popular “Class-dependent” (namely “Tyr-dependent”) hypothesis is that the deprotonation of SAHA is mostly regulated by the conserved Tyr308 in class I HDAC while it is replaced by the His843 in class IIa HDAC. Herein, by elaborate QM(DFT)/MM MD simulations, we exclude the prevalent “Class-dependent” mechanism and advance a novel “Metal-dependent” mechanism, where the remote second metal site (K+ in most HDAC and Ca2+ in HDAC2) determines the protonation of SAHA. This proof-of-principle “Metal-dependent” mechanism opens up a new avenue to utilize the second metal site for isoform-selective inhibitor design.
Co-reporter:Yinuo Wu, Ling-Jun Feng, Xiao Lu, Fuk Yee Kwong and Hai-Bin Luo
Chemical Communications 2014 - vol. 50(Issue 97) pp:NaN15354-15354
Publication Date(Web):2014/10/15
DOI:10.1039/C4CC07440H
A palladium-catalyzed cascade cross-coupling of N-nitroso-anilines and toluene derivatives for the direct synthesis of N-alkyl-2-aminobenzophenones is described. N-nitroso groups in anilines can act as the traceless directing groups while toluene derivatives can serve as effective acyl precursors under mild reaction conditions.



![[5-METHYL-2-(METHYLAMINO)PHENYL]-PHENYLMETHANONE](http://img.cochemist.com/ccimg/39200/39106-78-0.png)
![[5-METHYL-2-(METHYLAMINO)PHENYL]-PHENYLMETHANONE](http://img.cochemist.com/ccimg/39200/39106-78-0_b.png)
![SODIUM (2S)-2-[(2,2-DICHLOROACETYL)AMINO]-3-HYDROXY-PROPANOATE](http://img.cochemist.com/ccimg/34500/34425-25-7.png)
![SODIUM (2S)-2-[(2,2-DICHLOROACETYL)AMINO]-3-HYDROXY-PROPANOATE](http://img.cochemist.com/ccimg/34500/34425-25-7_b.png)
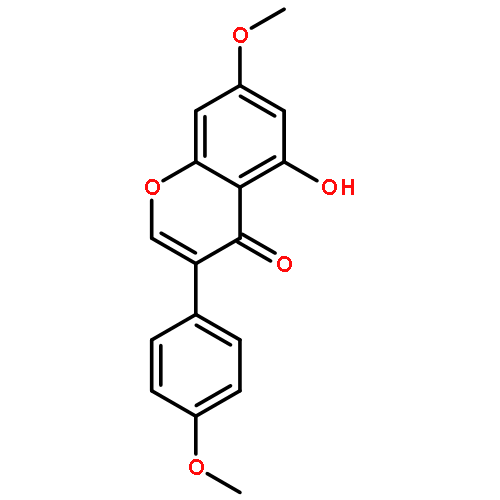
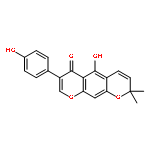









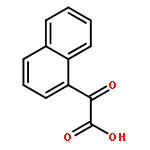
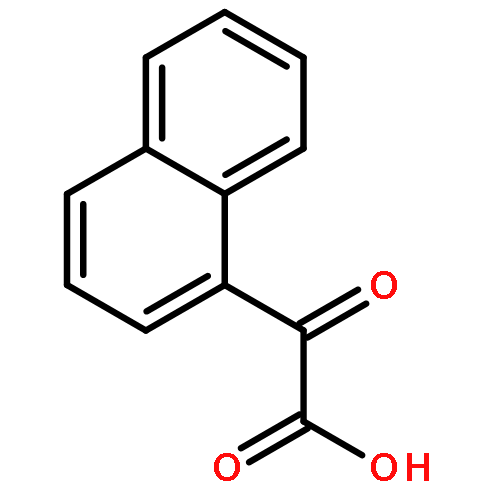


![Methanone, phenyl[2-(phenylamino)phenyl]-](http://img.cochemist.com/ccimg/23700/23699-69-6.png)
![Methanone, phenyl[2-(phenylamino)phenyl]-](http://img.cochemist.com/ccimg/23700/23699-69-6_b.png)












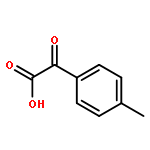
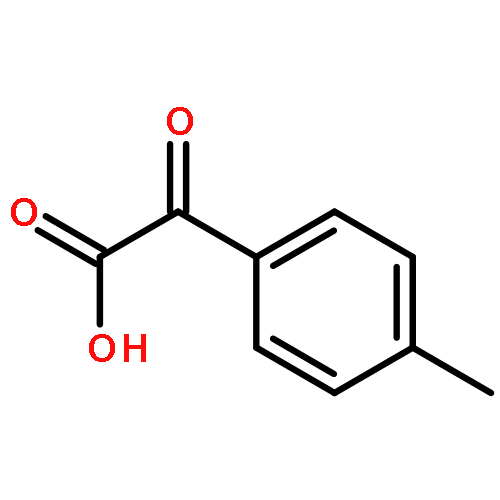
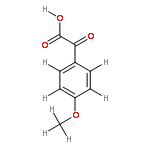
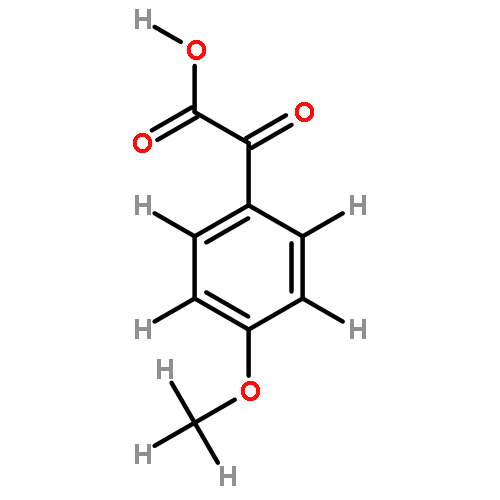
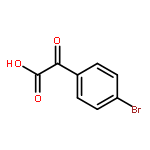
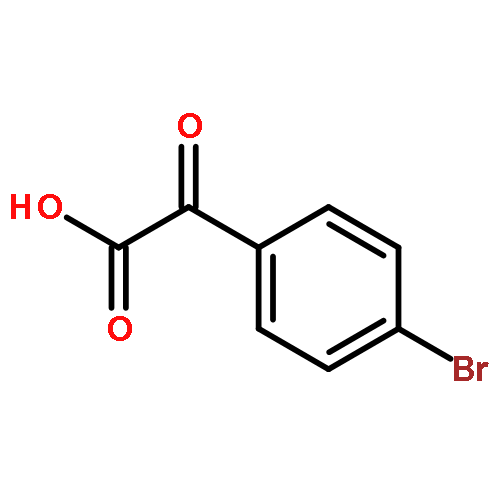


![2H,8H-Benzo[1,2-b:3,4-b']dipyran-2-one,6-methoxy-8,8-dimethyl-](http://img.cochemist.com/ccimg/6100/6054-10-0.png)
![2H,8H-Benzo[1,2-b:3,4-b']dipyran-2-one,6-methoxy-8,8-dimethyl-](http://img.cochemist.com/ccimg/6100/6054-10-0_b.png)
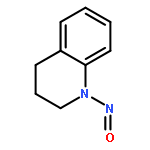
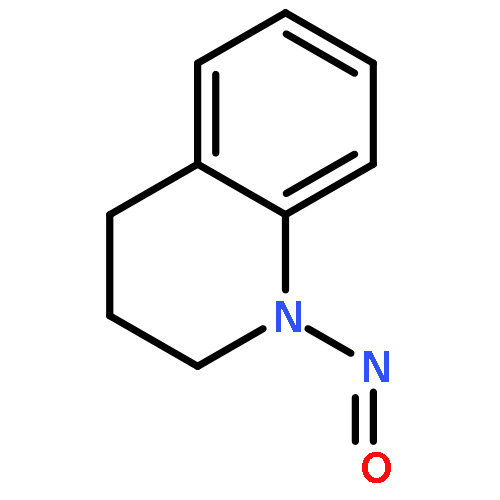
![Methanone, (4-chlorophenyl)[2-(methylamino)phenyl]-](http://img.cochemist.com/ccimg/5000/4937-54-6.png)
![Methanone, (4-chlorophenyl)[2-(methylamino)phenyl]-](http://img.cochemist.com/ccimg/5000/4937-54-6_b.png)








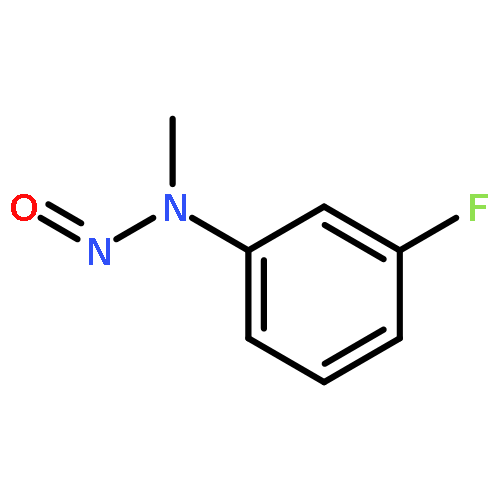
![Methanone, [2-(ethylamino)phenyl]phenyl-](http://img.cochemist.com/ccimg/1900/1859-77-4.png)
![Methanone, [2-(ethylamino)phenyl]phenyl-](http://img.cochemist.com/ccimg/1900/1859-77-4_b.png)
methanone](http://img.cochemist.com/ccimg/1900/1859-76-3.png)
methanone](http://img.cochemist.com/ccimg/1900/1859-76-3_b.png)




![(1AR,4R,4AR,7S,7AS,7BR)-1,1,4,7-TETRAMETHYLDECAHYDRO-1H-CYCLOPROP<WBR />A[E]AZULENE-4,7-DIOL](http://img.cochemist.com/ccimg/800/763-10-0.png)
![(1AR,4R,4AR,7S,7AS,7BR)-1,1,4,7-TETRAMETHYLDECAHYDRO-1H-CYCLOPROP<WBR />A[E]AZULENE-4,7-DIOL](http://img.cochemist.com/ccimg/800/763-10-0_b.png)




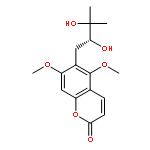
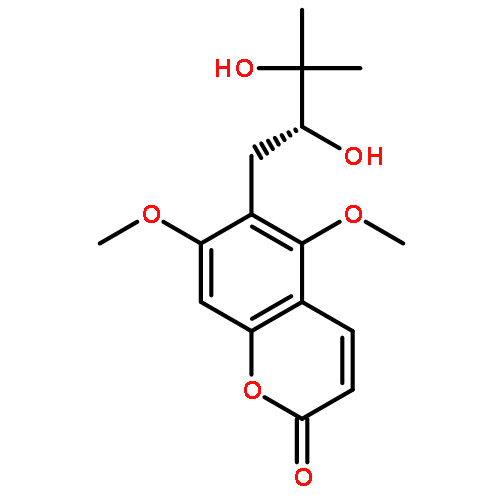
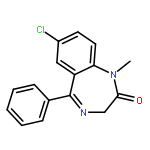
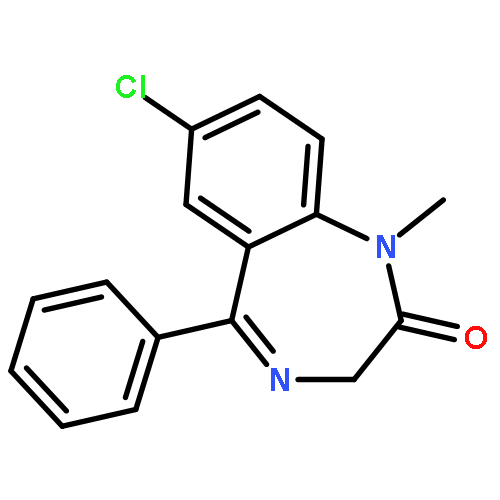






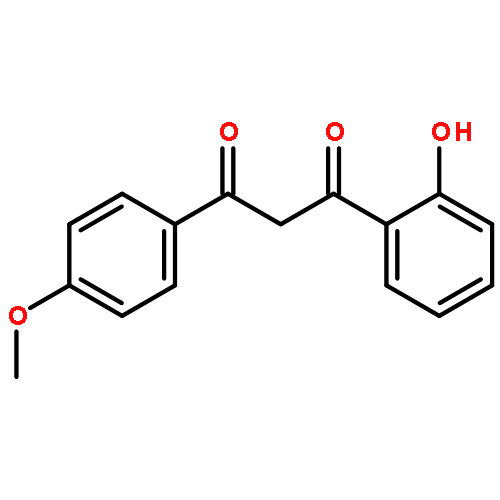
![4H-Pyrazolo[3,4-d]pyrimidin-4-one,1-(2-chlorophenyl)-1,5-dihydro-6-[(2R)-3,3,3-trifluoro-2-methylpropyl]-](http://img.cochemist.com/ccimg/794600/794568-92-6.png)
![4H-Pyrazolo[3,4-d]pyrimidin-4-one,1-(2-chlorophenyl)-1,5-dihydro-6-[(2R)-3,3,3-trifluoro-2-methylpropyl]-](http://img.cochemist.com/ccimg/794600/794568-92-6_b.png)






























![Benzo[b]thiophene-2-carboxamide,](http://img.cochemist.com/ccimg/1082800/1082743-32-5.png)
![Benzo[b]thiophene-2-carboxamide,](http://img.cochemist.com/ccimg/1082800/1082743-32-5_b.png)
![(R,S)-4-[(4'-hydroxy-3-((4-hydroxyphenyl)ethynyl)biphenyl-2-yl)(4-hydroxyphenyl)methylene]-2,5-cyclohexadien-1-one](http://img.cochemist.com/ccimg/1062200/1062124-28-0.png)
![(R,S)-4-[(4'-hydroxy-3-((4-hydroxyphenyl)ethynyl)biphenyl-2-yl)(4-hydroxyphenyl)methylene]-2,5-cyclohexadien-1-one](http://img.cochemist.com/ccimg/1062200/1062124-28-0_b.png)
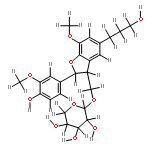
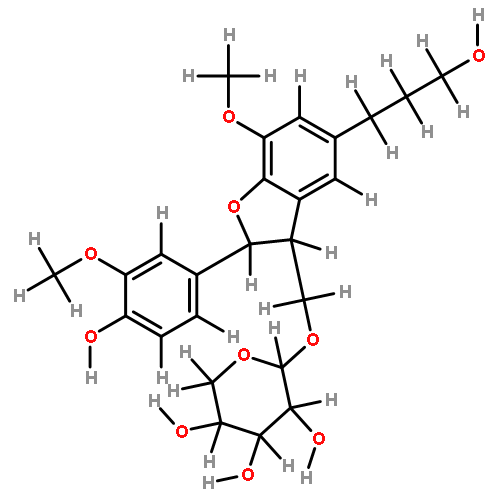

![4H-1-Benzopyran-4-one,3-[2,4-dihydroxy-3-(3-methyl-2-buten-1-yl)phenyl]-7-hydroxy-](http://img.cochemist.com/ccimg/166600/166547-20-2.png)
![4H-1-Benzopyran-4-one,3-[2,4-dihydroxy-3-(3-methyl-2-buten-1-yl)phenyl]-7-hydroxy-](http://img.cochemist.com/ccimg/166600/166547-20-2_b.png)






![Methanone, (2-fluorophenyl)[2-(methylamino)phenyl]-](http://img.cochemist.com/ccimg/106900/106849-49-4.png)
![Methanone, (2-fluorophenyl)[2-(methylamino)phenyl]-](http://img.cochemist.com/ccimg/106900/106849-49-4_b.png)





![METHANONE, [2-(BUTYLAMINO)PHENYL]PHENYL-](http://img.cochemist.com/ccimg/72400/72342-55-3.png)
![METHANONE, [2-(BUTYLAMINO)PHENYL]PHENYL-](http://img.cochemist.com/ccimg/72400/72342-55-3_b.png)