Co-reporter:Brian R. Kusche, Adrienne E. Smith, Michele A. McGuirl and Nigel D. Priestley
Journal of the American Chemical Society December 2, 2009 Volume 131(Issue 47) pp:17155-17165
Publication Date(Web):November 10, 2009
DOI:10.1021/ja9050235
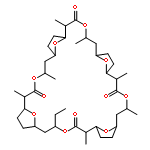
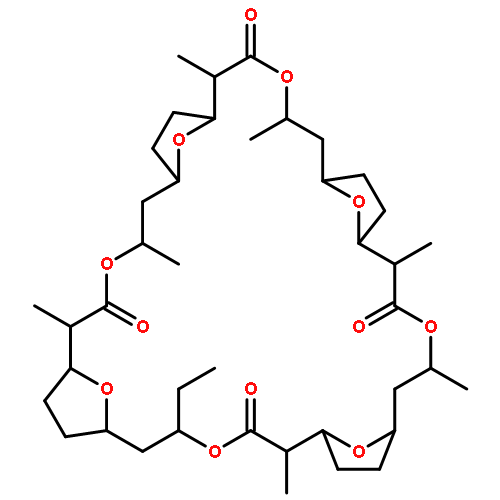
Nonactin is a polyketide antibiotic produced by Streptomyces griseus ETH A7796 and is an ionophore that is selective for K+ ions. It is a cyclic tetraester generated from two monomers of (+)-nonactic acid and two of (−)-nonactic acid, arranged (+)-(−)-(+)-(−) so that nonactin has S4 symmetry and is achiral. To understand why achiral nonactin is the naturally generated diastereoisomer, we generated two alternate diastereoisomers of nonactin, one prepared solely from (+)-nonactic acid and one prepared solely from (−)-nonactic acid, referred to here as ‘all-(+)-nonactin’ and ‘all-(−)-nonactin’, respectively. Both non-natural diastereoisomers were 500-fold less active against Gram positive organisms than nonactin confirming that the natural stereochemistry is necessary for biological activity. We used isothermal calorimetry to obtain the Ka, ΔG, ΔH, and ΔS of formation for the K+, Na+, and NH4+ complexes of nonactin and all-(−)-nonactin; the natural diastereoisomer bound K+ 880-fold better than all-(−)-nonactin. A picrate partitioning assay confirmed that all-(−)-nonactin, unlike nonactin, could not partition K+ ions into organic solvent. To complement the thermodynamic data we used a simple model system to show that K+ transport was facilitated by nonactin but not by all-(−)-nonactin. Modeling of the K+ complexes of nonactin and all-(−)-nonactin suggested that poor steric interactions in the latter complex precluded tight binding to K+. Overall, the data show that both enantiomers of nonactic acid are needed for the formation of a nonactin diastereoisomer that can act as an ionophore and has antibacterial activity.
Co-reporter:John Hoody, Jeremy B. Alverson, Santosh Keshipeddy, Patrick A. Barney, Larissa Walker, Amy C. Anderson, Dennis L. Wright, Nigel D. Priestley
Journal of Chromatography B 2017 Volume 1051(Volume 1051) pp:
Publication Date(Web):15 April 2017
DOI:10.1016/j.jchromb.2017.02.026
Antimicrobial resistance to current antibiotics is a significant public health problem and the need for new antibiotics is a compelling one. We have been developing a new series of antibiotics, propargyl-linked diaminopyrimidines, based on the structure of trimethoprim. To date we have discovered compounds that are effective inhibitors of dihydrofolate reductase (the target of trimethoprim), that are potent antibiotics in vitro against a range of Gram-positive pathogens including methicillin-resistant S. aureus, and that are non-toxic in mammalian cell culture. In this study we report the development of an LC–MS-based protocol for the quantification of our lead antibiotic 37D1-UCP1099 and the application of this assay to follow the concentration of the compound in mouse plasma after intraperitoneal administration. Extraction of 37D1-UCP1099 from mouse plasma was achieved through a liquid-liquid extraction with ethyl acetate. Separation was performed utilizing a reverse-phase C18 column with a ten minute isocratic elution using 47:53 (v/v) 10 mM NH4HCO3:acetonitrile. The lower limit of quantitation for 37D1-UCP1099 was 50 ng mL−1 and the assay showed a dynamic range of 50–4000 ng mL−1 with good linearity (r2 ≥ 0.996 for all fits). Intra-day and inter-day precision and accuracy were within 11.3% (%RSD) and 6.6% (%RE) respectably. We have demonstrated that the compound is stable under the assay procedures. The compound was shown to have a mean residence time of 26.2 ± 1.0 min and a half-life of 18.2 ± 0.7 min after intraperitoneal delivery at 5 mg kg−1. These studies now form the foundation of our work to develop additional analogs of 37D1-UCP1099 with improved pharmacokinetic properties.
Co-reporter:Jian Rong, Micheal E. Nelson, Brian Kusche, and Nigel D. Priestley
Journal of Natural Products 2010 Volume 73(Issue 12) pp:2009-2012
Publication Date(Web):December 7, 2010
DOI:10.1021/np100421v
The polyketide nonactin, a polyketide possessing antitumor and antibacterial activity, is produced by an unusual biosynthesis pathway in Streptomyces griseus that uses both enantiomers of the nonactin precursor, nonactic acid. Despite many studies with labeled precursors, much of the biosynthesis pathway remains unconfirmed, particularly the identity of the last achiral intermediate in the pathway, which is believed to be 4,6-diketoheptanoyl-CoA. We set out to confirm the latter hypothesis with feeding studies employing [4,5-13C2]-, [5,6-13C2]-, and [6,7-13C2]-4,6-diketoheptanoate thioester derivatives. In each case the isotopic label was incorporated efficiently into nonactin; however, at positions inconsistent with the currently accepted biosynthesis pathway. To resolve the discrepancy, we conducted additional feeding studies with a [3,4-13C2]levulinate thioester derivative and again observed efficient label incorporation. The latter result was intriguing, as levulinate is not an obvious precursor to nonactin. Levulinate, however, is known to be efficiently degraded into propionate even though the pathway for the conversion is not known. On the basis of both our levulinate and diketoheptanoate isotope incorporation data we can now postulate a pathway from levulinate to propionate that can also account for the conversion of 4,6-diketoheptanoate into levulinate in S. griseus.
Co-reporter:Joshua B. Phillips, Adrienne E. Smith, Brian R. Kusche, Bradley A. Bessette Jr., P. Whitney Swain III, Stephen C. Bergmeier, Mark C. McMills, Dennis L. Wright, Nigel D. Priestley
Bioorganic & Medicinal Chemistry Letters 2010 Volume 20(Issue 19) pp:5936-5938
Publication Date(Web):1 October 2010
DOI:10.1016/j.bmcl.2010.06.146
We have shown that the intentional engineering of a natural product biosynthesis pathway is a useful way to generate stereochemically complex scaffolds for use in the generation of combinatorial libraries that capture the structural features of both natural products and synthetic compounds. Analysis of a prototype library based upon nonactic acid lead to the discovery of triazole-containing nonactic acid analogs, a new structural class of antibiotic that exhibits bactericidal activity against drug resistant, Gram-positive pathogens including Staphylococcus aureus and Enterococcus faecalis.
Co-reporter:Brian R. Kusche, Joshua B. Phillips, Nigel D. Priestley
Bioorganic & Medicinal Chemistry Letters 2009 Volume 19(Issue 4) pp:1233-1235
Publication Date(Web):15 February 2009
DOI:10.1016/j.bmcl.2008.12.096
Nonactin, produced by Streptomyces griseus ETH A7796, is a macrotetrolide assembled from nonactic acid. It is an effective inhibitor of drug efflux in multidrug resistant erythroleukemia K562 cells at sub-toxic concentrations and has been shown to possess both antibacterial and antitumor activity. As total synthesis is impractical for the generation of nonactin analogs we have studied precursor-directed biosynthesis as an alternative as it is known that nonactic acid can serve as a nonactin precursor in vivo. To determine the scope of the approach we prepared and evaluated a furan-based nonactic acid derivative, 11. Although no new nonactin analogs were detected when 11 was administered to S. griseus fermentative cultures, a significant inhibition of nonactin biosynthesis was noted (IC50 ∼ 100 μM). Cell mass, nonactic acid production and the generation of other secondary metabolites in the culture were unaffected by 11 demonstrating that 11 selectively inhibited the assembly of nonactin from nonactic acid. While we were unable to generate new nonactin analogs we have discovered, however, a useful inhibitor that we can use to probe the mechanism of nonactin assembly with the ultimate goal of developing more successful precursor-directed biosynthesis transformations.
Co-reporter:Brian R. Kusche ; Adrienne E. Smith ; Michele A. McGuirl
Journal of the American Chemical Society () pp:
Publication Date(Web):November 10, 2009
DOI:10.1021/ja9050235
Nonactin is a polyketide antibiotic produced by Streptomyces griseus ETH A7796 and is an ionophore that is selective for K+ ions. It is a cyclic tetraester generated from two monomers of (+)-nonactic acid and two of (−)-nonactic acid, arranged (+)-(−)-(+)-(−) so that nonactin has S4 symmetry and is achiral. To understand why achiral nonactin is the naturally generated diastereoisomer, we generated two alternate diastereoisomers of nonactin, one prepared solely from (+)-nonactic acid and one prepared solely from (−)-nonactic acid, referred to here as ‘all-(+)-nonactin’ and ‘all-(−)-nonactin’, respectively. Both non-natural diastereoisomers were 500-fold less active against Gram positive organisms than nonactin confirming that the natural stereochemistry is necessary for biological activity. We used isothermal calorimetry to obtain the Ka, ΔG, ΔH, and ΔS of formation for the K+, Na+, and NH4+ complexes of nonactin and all-(−)-nonactin; the natural diastereoisomer bound K+ 880-fold better than all-(−)-nonactin. A picrate partitioning assay confirmed that all-(−)-nonactin, unlike nonactin, could not partition K+ ions into organic solvent. To complement the thermodynamic data we used a simple model system to show that K+ transport was facilitated by nonactin but not by all-(−)-nonactin. Modeling of the K+ complexes of nonactin and all-(−)-nonactin suggested that poor steric interactions in the latter complex precluded tight binding to K+. Overall, the data show that both enantiomers of nonactic acid are needed for the formation of a nonactin diastereoisomer that can act as an ionophore and has antibacterial activity.
.gif)


![4,13,22,31,37,38,39,40-Octaoxapentacyclo[32.2.1.17,10.116,19.125,28]tetracontane-3,12,21,30-tetrone,2,5,11,14,20,23,29,32-octamethyl-, (1R,2R,5R,7R,10S,11S,14S,16S,19R,20R,23R,25R,28S,29S,32S,34S)-](http://img.cochemist.com/ccimg/6900/6833-84-7.png)
![4,13,22,31,37,38,39,40-Octaoxapentacyclo[32.2.1.17,10.116,19.125,28]tetracontane-3,12,21,30-tetrone,2,5,11,14,20,23,29,32-octamethyl-, (1R,2R,5R,7R,10S,11S,14S,16S,19R,20R,23R,25R,28S,29S,32S,34S)-](http://img.cochemist.com/ccimg/6900/6833-84-7_b.png)
![4H-Furo[3,2-c]pyran-2(6H)-one,4-hydroxy-](http://img.cochemist.com/ccimg/200/149-29-1.png)
![4H-Furo[3,2-c]pyran-2(6H)-one,4-hydroxy-](http://img.cochemist.com/ccimg/200/149-29-1_b.png)