Co-reporter:Peng Teng, Ning Ma, Darrell Cole Cerrato, Fengyu She, Timothy Odom, Xiang Wang, Li-June Ming, Arjan van der Vaart, Lukasz Wojtas, Hai Xu, and Jianfeng Cai
Journal of the American Chemical Society May 31, 2017 Volume 139(Issue 21) pp:7363-7363
Publication Date(Web):May 8, 2017
DOI:10.1021/jacs.7b03007
New types of foldamer scaffolds are formidably challenging to design and synthesize, yet highly desirable as structural mimics of peptides/proteins with a wide repertoire of functions. In particular, the development of peptidomimetic helical foldamers holds promise for new biomaterials, catalysts, and drug molecules. Unnatural l-sulfono-γ-AApeptides were recently developed and shown to have potential applications in both biomedical and material sciences. However, d-sulfono-γ-AApeptides, the enantiomers of l-sulfono-γ-AApeptides, have never been studied due to the lack of high-resolution three-dimensional structures to guide structure-based design. Herein, we report the first synthesis and X-ray crystal structures of a series of 2:1 l-amino acid/d-sulfono-γ-AApeptide hybrid foldamers, and elucidate their folded conformation at the atomic level. Single-crystal X-ray crystallography indicates that this class of oligomers folds into well-defined right-handed helices with unique helical parameters. The helical structures were consistent with data obtained from solution 2D NMR, CD studies, and molecular dynamics simulations. Our findings are expected to inspire the structure-based design of this type of unique folding biopolymers for biomaterials and biomedical applications.
Co-reporter:Alekhya Nimmagadda;Olapeju Bolarinwa;Ma Su
Biochemistry January 24, 2017 Volume 56(Issue 3) pp:445-457
Publication Date(Web):December 28, 2016
DOI:10.1021/acs.biochem.6b01132
The intrinsic drawbacks encountered in bioactive peptides in chemical biology and biomedical sciences have diverted research efforts to the development of sequence-specific peptidomimetics that are capable of mimicking the structure and function of peptides and proteins. Modifications in the backbone and/or the side chain of peptides have been explored to develop biomimetic molecular probes or drug leads for biologically important targets. To expand the family of oligomeric peptidomimetics to facilitate their further application, we recently developed a new class of peptidomimetics, AApeptides based on a chiral peptide nucleic acid backbone. AApeptides are resistant to proteolytic degradation and amenable to enormous chemical diversification. Moreover, they could mimic the primary structure of peptides and also fold into discrete secondary structure such as helices and turn-like structures. Furthermore, they have started to show promise in applications in material and biomedical sciences. Herein, we highlight the structural design and some function of AApeptides and present our perspective on their future development.
Co-reporter:Chao Zhang, Xiao Cheng, Mengkun Chen, Jie Sheng, Jing Ren, Zhongying Jiang, Jianfeng Cai, Yong Hu
Colloids and Surfaces B: Biointerfaces 2017 Volume 160(Volume 160) pp:
Publication Date(Web):1 December 2017
DOI:10.1016/j.colsurfb.2017.09.045
•We developed a kind of pH-responsive chlorin e6-conjugated gold nanorods (Ce6-PEG-AuNR).•Ce6-PEG-AuNR showed pH activated fluorescent imaging ability and PTT/PDT properties.•They exhibited high anti-tumour properties in vitro and in vivo.•Ce6-PEG-AuNR could serve as fluorescence guided photothermal/photodynamic therapy agents.Photothermal/photodynamic therapies (PTT/PDT) have been widely accepted as non-invasive therapeutic modalities to erase tumours. However, both therapies face the problem of precisely locating tumours and reducing their side effects. Herein, chlorin e6 conjugated gold nanorod, (Ce6-PEG-AuNR), a type of gold nanorod-photosensitizer conjugate, is designed as a kind of nano-therapeutic agent to simultaneously realize combined PTT/PDT. Compared to free Ce6, the fluorescence of Ce6 adhered to the conjugate is effectively quenched by the longitudinal surface plasmon resonance (LSPR) of in the Ce6-PEG-AuNR. However, the specific fluorescence of Ce6 can be recovered in tumour tissue when Ce6 is separated from the conjugate owing to the cleavage of hydrazone bond between Ce6 and PEG caused by intracellular acidic conditions in tumour tissue. Based on this effect, we can precisely locate tumours and further kill cancer cells by combined PTT/PDT. In addition, the combined therapy (PTT/PDT) function is more efficient in cancer treatment than that of PTT or PDT alone. Therefore, Ce6-PEG-AuNR can serve as a promising dual-modal phototherapeutic agent as well as a tumour-sensitive fluorescent probe to diagnose and treat cancer.Download high-res image (211KB)Download full-size image
Co-reporter:Peng Teng;Alekhya Nimmagadda;Ma Su;Yuzhu Hong;Ning Shen;Chunpu Li;Ling-Yu Tsai;Jessica Cao;Qi Li
Chemical Communications 2017 vol. 53(Issue 87) pp:11948-11951
Publication Date(Web):2017/10/31
DOI:10.1039/C7CC07285F
We designed a class of small dimeric cyclic guanidine derivatives which display potent antibacterial activity against both multidrug-resistant Gram-negative and Gram-positive bacteria. They could compromise bacterial membranes without developing resistance, inhibit biofilms formed by E. coli, and exhibit excellent in vivo activity in the MRSA-infected thigh burden mouse model.
Co-reporter:Alekhya Nimmagadda, Xuan Liu, Peng Teng, Ma Su, Yaqiong Li, Qiao Qiao, Nawal K. Khadka, Xiaoting Sun, Jianjun Pan, Hai XuQi Li, Jianfeng Cai
Biomacromolecules 2017 Volume 18(Issue 1) pp:
Publication Date(Web):November 22, 2016
DOI:10.1021/acs.biomac.6b01385
The resistance developed by life-threatening bacteria toward conventional antibiotics has become a major concern in public health. To combat antibiotic resistance, there has been a significant interest in the development of antimicrobial cationic polymers due to the ease of synthesis and low manufacturing cost compared to host-defense peptides (HDPs). Herein, we report the design and synthesis of amphiphilic polycarbonates containing primary amino groups. These polymers exhibit potent antimicrobial activity and excellent selectivity to Gram-positive bacteria, including multidrug resistant pathogens. Fluorescence and TEM studies suggest that these polymers are likely to kill bacteria by disrupting bacterial membranes. These polymers also show low tendency to elicit resistance in bacteria. Their further development may lead to new antimicrobial agents combating drug-resistance.
Co-reporter:Yan Shi, Peng Teng, Peng Sang, Fengyu She, Lulu Wei, and Jianfeng Cai
Accounts of Chemical Research 2016 Volume 49(Issue 3) pp:428
Publication Date(Web):February 22, 2016
DOI:10.1021/acs.accounts.5b00492
The development of sequence-specific peptidomimetics has led to a variety of fascinating discoveries in chemical biology. Many peptidomimetics can mimic primary, secondary, and even tertiary structure of peptides and proteins, and because of their unnatural backbones, they also possess significantly enhanced resistance to enzymatic hydrolysis, improved bioavailability, and chemodiversity. It is known that peptide nucleic acids (PNAs) are peptidic sequences developed for the mimicry of nucleic acids; however, their unique backbone as the molecular scaffold of peptidomimetics to mimic structure and function of bioactive peptides has not been investigated systematically. As such, we recently developed a new class of peptidomimetics, “γ-AApeptides”, based on the chiral γ-PNA backbone. They are termed γ-AApeptides because they are the oligomers of γ-substituted-N-acylated-N-aminoethyl amino acids. Similar to other classes of peptidomimetics, γ-AApeptides are also resistant to proteolytic degradation and possess the potential to enhance chemodiversity. Moreover, in our scientific journey on the exploration of this class of peptidomimetics, we have discovered some intriguing structures and functions of γ-AApeptides. In this Account, we summarize the current development and application of γ-AApeptides with biological potential. Briefly, both linear and cyclic (either through head-to-tail or head-to-side-chain cyclization) γ-AApeptides with diverse functional groups can be synthesized easily on the solid phase using the synthetic protocol we developed. γ-AApeptides could mimic the primary structure of peptides, as they project the same number of side chains as peptides of the same lengths. For instance, they could mimic the Tat peptide to permeate cell membranes and bind to HIV RNA with high specificity and affinity. Certain γ-AApeptides show similar activity to the RGD peptide and target integrin specifically on the cell surface. γ-AApeptides with function akin to fMLF peptides are also identified. More importantly, we found that γ-AApeptides can fold into discrete secondary structures, such as helical and β-turn-like structures. Therefore, they could be rationally designed for a range of biological applications. For instance, γ-AApeptides can mimic host-defense peptides and display potent and broad-spectrum activity toward a panel of drug-resistant bacterial pathogens. Meanwhile, because of their stability against proteolysis and their chemodiversity, γ-AApeptides are also amenable for combinatorial screening. We demonstrate that, through combinatorial selection, certain γ-AApeptides are identified to inhibit Aβ40 peptide aggregation, suggesting their potential use as a molecular probe to intervene in Alzheimer’s disease. In addition, a few γ-AApeptides identified from the γ-AApeptide library have been shown to bind to the DNA-binding domain of STAT3 and antagonize STAT3/DNA interactions. Our studies suggest that, with further studies and exploration on both structures and functions, γ-AApeptides may emerge to be a new class of peptidomimetics that play an important role in chemical biology and biomedical sciences.
Co-reporter:Peng Teng, Da Huo, Alekhya Nimmagadda, Jianfeng Wu, Fengyu She, Ma Su, Xiaoyang Lin, Jiyu Yan, Annie Cao, Chuanwu Xi, Yong Hu, and Jianfeng Cai
Journal of Medicinal Chemistry 2016 Volume 59(Issue 17) pp:7877-7887
Publication Date(Web):August 15, 2016
DOI:10.1021/acs.jmedchem.6b00640
Prevalence of drug-resistant bacteria has emerged to be one of the greatest threats in the 21st century. Herein, we report the development of a series of small molecular antibacterial agents that are based on the acylated reduced amide scaffold. These molecules display good potency against a panel of multidrug-resistant Gram-positive and Gram-negative bacterial strains. Meanwhile, they also effectively inhibit the biofilm formation. Mechanistic studies suggest that these compounds kill bacteria by compromising bacterial membranes, a mechanism analogous to that of host-defense peptides (HDPs). The mechanism is further supported by the fact that the lead compounds do not induce resistance in MRSA bacteria even after 14 passages. Lastly, we also demonstrate that these molecules have therapeutic potential by preventing inflammation caused by MRSA induced pneumonia in a rat model. This class of compounds could lead to an appealing class of antibiotic agents combating drug-resistant bacterial strains.
Co-reporter:H. Xu, S. Q. Zhao, Y. Ren, W. Xu, D. B. Zhu, J. Z. Jiang and J. F. Cai
RSC Advances 2016 vol. 6(Issue 25) pp:21067-21071
Publication Date(Web):16 Feb 2016
DOI:10.1039/C5RA26417K
Cyclo[8]pyrrole (CP) was prepared and its UV/vis spectral and optical limiting properties for ns light pulse were investigated with Z-scan technical. Our experiments show that CP has good solubility in organic solvents, and its Q band absorption shows an obvious red shift to the NIR region at about 1100 nm compared with normal porphyrin. Studies on transient absorption spectra, photophysical parameters and optical limiting properties indicate that the compound exhibits good optical limiting performance under variable conditions. Briefly, it shows a reverse saturable absorber with the limiting threshold of 1.8 J cm−2 at 1064 nm in dichloromethane; interestingly, it displays limiting properties at both 532 nm and 1064 nm after treatment with NaOH.
Co-reporter:Peng Teng;Yan Shi;Peng Sang
Chemistry - A European Journal 2016 Volume 22( Issue 16) pp:5458-5466
Publication Date(Web):
DOI:10.1002/chem.201504936
Abstract
Sequence-specific peptidomimetics are molecules that mimic the structure and function of peptides and proteins. With new backbones and molecular frameworks, peptidomimetics are of considerable interest in addressing challenges encountered in chemical biology and biomedical sciences. Based on the γ-PNA backbone, a new class of peptidomimetics - “γ-AApeptides” were recently developed. Both linear and cyclic γ-AApeptides can be synthesized with high efficiency. Compared with α-peptides, γ-AApeptides are resistant to enzymatic degradation, and amendable to diversification with a variety of chemical groups. Moreover, they could mimic primary and secondary structure, as well as the function of peptides, and show promise in biological applications, such as the development of new agents combating bacteria, cancer, and Alzheimer's disease. A few research outcomes of γ-AApeptides are highlighted in this Concept article, and a future perspective is also proposed.
Co-reporter:Fengyu She, Alekhya Nimmagadda, Peng Teng, Ma Su, Xiaobing Zuo, and Jianfeng Cai
Biomacromolecules 2016 Volume 17(Issue 5) pp:
Publication Date(Web):March 31, 2016
DOI:10.1021/acs.biomac.6b00286
As one of the greatest threats facing the 21st century, antibiotic resistance is now a major public health concern. Host-defense peptides (HDPs) offer an alternative approach to combat emerging multi-drug-resistant bacteria. It is known that helical HDPs such as magainin 2 and its analogs adopt cationic amphipathic conformations upon interaction with bacterial membranes, leading to membrane disruption and subsequent bacterial cell death. We have previously shown that amphipathic sulfono-γ-AApeptides could mimic magainin 2 and exhibit bactericidal activity. In this article, we demonstrate for the first time that amphipathic helical 1:1 α/sulfono-γ-AA heterogeneous peptides, in which regular amino acids and sulfono-γ-AApeptide building blocks are alternatively present in a 1:1 pattern, display potent antibacterial activity against both Gram-positive and Gram-negative bacterial pathogens. Small angle X-ray scattering (SAXS) suggests that the lead sequences adopt defined helical structures. The subsequent studies including fluorescence microscopy and time-kill experiments indicate that these hybrid peptides exert antimicrobial activity by mimicking the mechanism of HDPs. Our findings may lead to the development of HDP-mimicking antimicrobial peptidomimetics that combat drug-resistant bacterial pathogens. In addition, our results also demonstrate the effective design of a new class of helical foldamer, which could be employed to interrogate other important biological targets such as protein–protein interactions in the future.
Co-reporter:Haifan Wu, Qiao Qiao, Peng Teng, Yaogang Hu, Dimitrios Antoniadis, Xiaobing Zuo, and Jianfeng Cai
Organic Letters 2015 Volume 17(Issue 14) pp:3524-3527
Publication Date(Web):July 8, 2015
DOI:10.1021/acs.orglett.5b01608
A new class of unnatural heterogeneous foldamers is reported to contain alternative α-amino acid and sulfono-γ-AA amino acid residues in a 1:1 repeat pattern. Two-dimensional NMR data show that two 1:1 α/sulfono-γ-AA peptides with diverse side chains form analogous right-handed helical structures in solution. The effects of sequence length, side chain, N-capping, and temperature on folding propensity were further investigated using circular dichroism and small-angle X-ray scattering.
Co-reporter:Yaqiong Li; Haifan Wu; Peng Teng; Ge Bai; Xiaoyang Lin; Xiaobing Zuo; Chuanhai Cao
Journal of Medicinal Chemistry 2015 Volume 58(Issue 11) pp:4802-4811
Publication Date(Web):May 28, 2015
DOI:10.1021/acs.jmedchem.5b00537
Host-defense peptides (HDPs) such as magainin 2 have emerged as potential therapeutic agents combating antibiotic resistance. Inspired by their structures and mechanism of action, herein we report the first example of antimicrobial helical sulfono-γ-AApeptide foldamers. The lead molecule displays broad-spectrum and potent antimicrobial activity against multi-drug-resistant Gram-positive and Gram-negative bacterial pathogens. Time-kill studies and fluorescence microscopy suggest that sulfono-γ-AApeptides eradicate bacteria by taking a mode of action analogous to that of HDPs. Clear structure–function relationships exist in the studied sequences. Longer sequences, presumably adopting more-defined helical structures, are more potent than shorter ones. Interestingly, the sequence with less helical propensity in solution could be more selective than the stronger helix-forming sequences. Moreover, this class of antimicrobial agents are resistant to proteolytic degradation. These results may lead to the development of a new class of antimicrobial foldamers combating emerging antibiotic-resistant pathogens.
Co-reporter:Haifan Wu, Fengyu She, Wenyang Gao, Austin Prince, Yaqiong Li, Lulu Wei, Allison Mercer, Lukasz Wojtas, Shengqian Ma and Jianfeng Cai
Organic & Biomolecular Chemistry 2015 vol. 13(Issue 3) pp:672-676
Publication Date(Web):18 Nov 2014
DOI:10.1039/C4OB02232G
We report an efficient method for the preparation of unprecedented head-to-tail cyclic sulfono-γ-AApeptides. Following this method, a number of cyclic sequences bearing two to five subunits were efficiently synthesized. In addition, the X-ray crystal structure study of a three-membered cyclic sulfono-γ-AApeptide revealed a type II β-turn-like character.
Co-reporter:Haifan Wu;Qiao Qiao;Yaogang Hu;Peng Teng;Wenyang Gao;Xiaobing Zuo;Lukasz Wojtas;Ry W. Larsen;Shengqian Ma
Chemistry - A European Journal 2015 Volume 21( Issue 6) pp:2501-2507
Publication Date(Web):
DOI:10.1002/chem.201406112
Abstract
Foldamers offer an attractive opportunity for the design of novel molecules that mimic the structures and functions of proteins and enzymes including biocatalysis and biomolecular recognition. Herein we report a new class of nonnatural helical sulfono-γ-AApeptide foldamers of varying lengths. The crystal structure of the sulfono-γ-AApeptide monomer S6 illustrates the intrinsic folding propensity of sulfono-γ-AApeptides, which likely originates from the bulkiness of tertiary sulfonamide moiety. The two-dimensional solution NMR spectroscopy data for the longest sequence S1 demonstrates a 10/16 right-handed helical structure. Optical analysis using circular dichroism further supports well- defined helical conformation of sulfono-γ-AApeptides in solution containing as few as five building blocks. Future development of sulfono-γ-AApeptides may lead to new foldamers with discrete functions, enabling expanded application in chemical biology and biomedical sciences.
Co-reporter:Yan Wang, Frankie Costanza, Haifan Wu, Daqian Song, Jianfeng Cai and Qi Li
Journal of Materials Chemistry A 2014 vol. 2(Issue 20) pp:3115-3122
Publication Date(Web):07 Mar 2014
DOI:10.1039/C4TB00041B
Tanshinone IIA is a potent anti-tumor agent; however, its therapeutic applications have been hindered by its intrinsic poor solubility and short half-life. Herein we report the design and synthesis of amphiphilic PEG–poly(amino acid)s (PPAAs), which were used as a nanocarrier to encapsulate tanshinone IIA. PPAA-encapsulated tanshinone IIA formed defined nanoparticles and effectively arrested the growth of the hepatoma tumor in MHCC97-H hepatoma-bearing mice, which has therefore significantly increased their survival time. Moreover, these tanshinone-loaded nanoparticles are also capable of suppressing hepatic tumor cell migration and metastasis. In all cases, they significantly improve efficacy compared to tanshinone alone. Our results show that tanshinone IIA encapsulated by PPAAs is a potential novel nanomedicine for the treatment of hepatoma. Furthermore, this use is only a proof of concept, and PPAAs can be used as a potential drug delivery system for a much broader array of hydrophobic drugs.
Co-reporter:Haifan Wu, Yaqiong Li, Ge Bai, Youhong Niu, Qiao Qiao, Jeremiah D. Tipton, Chuanhai Cao and Jianfeng Cai
Chemical Communications 2014 vol. 50(Issue 40) pp:5206-5208
Publication Date(Web):10 Oct 2013
DOI:10.1039/C3CC46685J
We report the design, synthesis, characterization and evaluation of a novel class of γ-AApeptide one-bead-one-compound (OBOC) library, from which a small γ-AApeptide was identified to effectively prevent and disassemble Aβ aggregation.
Co-reporter:Peng Teng, Xiaolei Zhang, Haifan Wu, Qiao Qiao, Said M. Sebti and Jianfeng Cai
Chemical Communications 2014 vol. 50(Issue 63) pp:8739-8742
Publication Date(Web):09 Jun 2014
DOI:10.1039/C4CC03909B
From a γ-AApeptide-based one-bead-one-compound (OBOC) combinatorial library, we identified γ-AApeptides that can selectively inhibit STAT3–DNA interaction and suppress the expression levels of STAT3 target genes in intact cells. Our results demonstrate that in addition to the SH2 domain, the DNA binding domain of STAT3 is targetable for the development of a new generation of anti-cancer therapeutics.
Co-reporter:Yaqiong Li, Christina Smith, Haifan Wu, Shruti Padhee, Namitha Manoj, Joseph Cardiello, Qiao Qiao, Chuanhai Cao, Hang Yin, and Jianfeng Cai
ACS Chemical Biology 2014 Volume 9(Issue 1) pp:211
Publication Date(Web):October 21, 2013
DOI:10.1021/cb4006613
Antimicrobial peptides (AMPs) are host-defense agents capable of both bacterial membrane disruption and immunomodulation. However, the development of natural AMPs as potential therapeutics is hampered by their moderate activity and susceptibility to protease degradation. Herein we report lipidated cyclic γ-AApeptides that have potent antibacterial activity against clinically relevant Gram-positive and Gram-negative bacteria, many of which are resistant to conventional antibiotics. We show that lipidated cyclic γ-AApeptides mimic the bactericidal mechanism of AMPs by disrupting bacterial membranes. Interestingly, they also harness the immune response and inhibit lipopolysaccharide (LPS) activated Toll-like receptor 4 (TLR4) signaling, suggesting that lipidated cyclic γ-AApeptides have dual roles as novel antimicrobial and anti-inflammatory agents.
Co-reporter:Frankie Costanza, Shruti Padhee, Haifan Wu, Yan Wang, Jesse Revenis, Chuanhai Cao, Qi Li and Jianfeng Cai
RSC Advances 2014 vol. 4(Issue 4) pp:2089-2095
Publication Date(Web):06 Nov 2013
DOI:10.1039/C3RA44324H
There has been significant interest in the development of antimicrobial cationic polymers due to their low manufacture cost and ease of synthesis compared to small antimicrobial peptides (AMPs). These polymers are designed to mimic amphiphilic structures of AMPs which can disrupt negatively charged bacterial membranes, and can therefore lead to potential antibiotic agents to fight emerging drug resistance. However, the reports of biodegradable antimicrobial polymer nanoparticles are rare. Herein we report the development of antimicrobial PEG-poly(amino acid)s. Some of these multi-block PEG-poly(amino acid)s form defined nanoparticles in solution, and display potent and broad-spectrum antimicrobial activity. Fluorescence and SEM studies show that these polymers are likely to kill bacteria by disrupting bacterial membranes. As these polymers are biodegradable and easy to scale up, they may provide an attractive approach for the development of antibiotic agents.
Co-reporter:Haifan Wu;Peng Teng
European Journal of Organic Chemistry 2014 Volume 2014( Issue 8) pp:1760-1765
Publication Date(Web):
DOI:10.1002/ejoc.201301841
Abstract
Herein we report a highly efficient new method for preparing γ-AApeptides capable of theoretically containing any functionality; the method uses a few common N-alloc γ-AApeptide building blocks. More importantly, using the same approach, new classes of peptidomimetics bearing novel backbone scaffolds can also be readily generated. Our results not only demonstrate the versatility of this new synthetic method, but also highlight the possibility that the resulting novel peptidomimetics may find discrete biomedical/biomaterial applications in the future.
Co-reporter:Yaogang Hu ;Ni Cheng ;Haifan Wu;Samuel Kang;Richard D. Ye ;Dr. Jianfeng Cai
ChemBioChem 2014 Volume 15( Issue 16) pp:2420-2426
Publication Date(Web):
DOI:10.1002/cbic.201402396
Abstract
The tripeptide N-formyl-Met-Leu-Phe (fMLF) is a potent neutrophil chemoattractant and the reference agonist for the G protein-coupled N-formylpeptide receptor (FPR). As it plays a very important role in host defense and inflammation, there has been considerable interest in the development of fMLF analogues in the hope of identifying potential therapeutic agents. Herein we report the design, synthesis, and evaluation of AApeptides that mimic the structure and function of fMLF. The lead AApeptides induced calcium mobilization and mitogen-activated protein kinase (MAPK) signal transduction pathways in FPR-transfected rat basophilic leukemic (RBL) cells. More intriguingly, at high concentrations, certain AApeptides were more effective than fMLF in the induction of calcium mobilization. Their agonistic activity is further supported by their ability to stimulate chemotaxis and the production of superoxide in HL-60 cells. Similarly to fMLF, these AApeptides are much more selective towards FPR1 than FPR2. These results suggest that the fMLF-mimicking AApeptides might emerge as a new class of therapeutic agents that target FPRs.
Co-reporter:Shruti Padhee;Christina Smith;Haifan Wu;Yaqiong Li;Namitha Manoj;Qiao Qiao;Zoya Khan; Chuanhai Cao; Hang Yin; Jianfeng Cai
ChemBioChem 2014 Volume 15( Issue 5) pp:688-694
Publication Date(Web):
DOI:10.1002/cbic.201300709
Abstract
Herein we describe the development of a new class of antimicrobial and anti-inflammatory peptidomimetics: cyclic lipo-α-AApeptides. They have potent and broad-spectrum antibacterial activity against a range of clinically relevant pathogens, including both multidrug-resistant Gram-positive and Gram-negative bacteria. Fluorescence microscopy suggests that cyclic lipo-α-AApeptides kill bacteria by disrupting bacterial membranes, possibly through a mechanism similar to that of cationic host-defense peptides (HDPs). Furthermore, the cyclic lipo-α-AApeptide can mimic cationic host-defense peptides by antagonizing Toll-like receptor 4 (TLR4) signaling responses and suppressing proinflammatory cytokines such as tumor necrosis factor-α (TNF-α). Our results suggest that by mimicking HDPs, cyclic lipo-α-AApeptides could emerge as a new class of antibiotic agents that directly kill bacteria, as well as novel antiinflammatory agents that act through immunomodulation.
Co-reporter:Yaqiong Li;Christina Smith;Haifan Wu;Peng Teng;Yan Shi;Shruti Padhee;Torey Jones;Anh-My Nguyen;Chuanhai Cao;Hang Yin
ChemBioChem 2014 Volume 15( Issue 15) pp:2275-2280
Publication Date(Web):
DOI:10.1002/cbic.201402264
Abstract
The last two decades have seen the rise of antimicrobial peptides (AMPs) to combat emerging antibiotic resistance. Herein we report the solid-phase synthesis of short lipidated α/γ-AA hybrid peptides. This family of lipo-chimeric peptidomimetics displays potent and broad-spectrum antimicrobial activity against a range of multi-drug resistant Gram-positive and Gram-negative bacteria. These lipo-α/γ-AA hybrid peptides also demonstrate high biological specificity, with no hemolytic activity towards red blood cells. Fluorescence microscopy suggests that these lipo-α/γ-AA chimeric peptides can mimic the mode of action of AMPs and kill bacterial pathogens via membrane disintegration. As the composition of these chimeric peptides is simple, therapeutic development could be economically feasible and amenable for a variety of antimicrobial applications.
Co-reporter:Youhong Niu, Haifan Wu, Yaqiong Li, Yaogang Hu, Shruti Padhee, Qi Li, Chuanhai Cao and Jianfeng Cai
Organic & Biomolecular Chemistry 2013 vol. 11(Issue 26) pp:4283-4290
Publication Date(Web):02 May 2013
DOI:10.1039/C3OB40444G
Antibiotic resistance is an increasing public health concern around the world, and is recognized as one of the greatest threats facing humankind in the 21st century. Natural antimicrobial peptides (AMPs) are small cationic amphiphilic peptides found in virtually all living organisms, and play a key role in the defense against bacterial infections. Compared with conventional antibiotics, which target specific metabolic processes, AMPs are able to adopt globally amphipathic conformations, and kill bacteria through disruption of their membranes. As such, AMPs do not readily induce drug-resistance. However, AMPs are associated with intrinsic drawbacks such as low-to-moderate activity, susceptibility to enzymatic degradation, and inconvenience for optimization. Recently, we have developed a new class of peptidomimetics termed “AApeptides”. Such peptide mimics are highly resistant to protease degradation and are straightforward for chemical diversification and development. Our current studies show that AApeptides with globally amphipathic structures can mimic the bactericidal mechanism of AMPs, and display potent and broad-spectrum activity against both Gram-positive and -negative multi-drug-resistant bacteria. In this review, we summarize our current findings of antimicrobial AApeptides, and discuss potential future directions on the development of more potent and specific analogues.
Co-reporter:Haifan Wu, Youhong Niu, Shruti Padhee, Rongsheng E. Wang, Yaqiong Li, Qiao Qiao, Ge Bai, Chuanhai Cao and Jianfeng Cai
Chemical Science 2012 vol. 3(Issue 8) pp:2570-2575
Publication Date(Web):17 May 2012
DOI:10.1039/C2SC20428B
Antimicrobial drug resistance is one of the greatest threats facing mankind. Antimicrobial peptides (AMPs) can potentially circumvent drug resistance, probably through a bacterial membrane-disruption mechanism. However, they suffer from low in vivo stability, potential immunogenicity, and difficulty in optimization. The development of antimicrobial peptidomimetics is therefore an emerging research area as they avoid the potential disadvantages of AMPs. Cyclic peptidomimetics are of significant interest since constraints induced by cyclization are expected to further improve their antimicrobial activity. Nonetheless, the report of cyclic oligomeric peptidomimetics for antimicrobial development is rare. Herein, for the first time, we report the design and synthesis of cyclic γ-AApeptides via an on-resin cyclization. These cyclic γ-AApeptides are potent and broad-spectrum active against fungus and multi-drug resistant Gram-positive and Gram-negative bacterial pathogens. Our results demonstrate the potential of cyclic γ-AApeptides as a new class of antibiotics to circumvent drug resistance by mimicking the bactericidal mechanism of AMPs. Meanwhile, the facile synthesis of cyclic γ-AApeptides may further expand the applications of γ-AApeptides in biomedical sciences.
Co-reporter:Yunan Yang, Youhong Niu, Hao Hong, Haifan Wu, Yin Zhang, Jonathan W. Engle, Todd E. Barnhart, Jianfeng Cai and Weibo Cai
Chemical Communications 2012 vol. 48(Issue 63) pp:7850-7852
Publication Date(Web):19 Jun 2012
DOI:10.1039/C2CC33620K
A γ-AApeptide-based tracer for positron emission tomography imaging of integrin αvβ3 is reported. Despite its shorter sequence and linear nature, this tracer had comparable integrin αvβ3 binding affinity to the cyclic arginine-glycine-aspartic acid peptide but significantly higher resistance to enzymatic degradation and better stability.
Co-reporter:Youhong Niu ; Shruti Padhee ; Haifan Wu ; Ge Bai ; Qiao Qiao ; Yaogang Hu ; Lacey Harrington ; Whittney N. Burda ; Lindsey N. Shaw ; Chuanhai Cao
Journal of Medicinal Chemistry 2012 Volume 55(Issue 8) pp:4003-4009
Publication Date(Web):April 4, 2012
DOI:10.1021/jm300274p
There is increasing demand to develop antimicrobial peptides (AMPs) as next generation antibiotic agents, as they have the potential to circumvent emerging drug resistance against conventional antibiotic treatments. Non-natural antimicrobial peptidomimetics are an ideal example of this, as they have significant potency and in vivo stability. Here we report for the first time the design of lipidated γ-AApeptides as antimicrobial agents. These lipo-γ-AApeptides show potent broad-spectrum activities against fungi and a series of Gram-positive and Gram-negative bacteria, including clinically relevant pathogens that are resistant to most antibiotics. We have analyzed their structure–function relationship and antimicrobial mechanisms using membrane depolarization and fluorescent microscopy assays. Introduction of unsaturated lipid chain significantly decreases hemolytic activity and thereby increases the selectivity. Furthermore, a representative lipo-γ-AApeptide did not induce drug resistance in S. aureus, even after 17 rounds of passaging. These results suggest that the lipo-γ-AApeptides have bactericidal mechanisms analogous to those of AMPs and have strong potential as a new class of novel antibiotic therapeutics.
Co-reporter:Ge Bai, Shruti Padhee, Youhong Niu, Rongsheng E. Wang, Qiao Qiao, Robert Buzzeo, Chuanhai Cao and Jianfeng Cai
Organic & Biomolecular Chemistry 2012 vol. 10(Issue 6) pp:1149-1153
Publication Date(Web):30 Nov 2011
DOI:10.1039/C2OB06679C
Some short and cationic peptides such as the Tat peptide can cross the cell membrane and function as vectors for intracellular delivery. Here we show that an α-AApeptide is able to penetrate the membranes of living cells from an extracellular environment and enter the endosome and cytoplasm of cells. The efficiency of the cellular uptake is comparable to a Tat peptide (48–57) of the same length and is unexpectedly superior to an α-peptide with identical functional groups. The mechanism of uptake is similar to that of the Tat peptide and is through endocytosis by an energy-dependent pathway. Due to the easy synthesis of the α-AApeptides, their resistance to proteolytic hydrolysis, and their low cytotoxicity, α-AApeptides represent a new class of transporters for the delivery of drugs.
Co-reporter:Youhong Niu, Ge Bai, Haifan Wu, Rongsheng E. Wang, Qiao Qiao, Shruti Padhee, Robert Buzzeo, Chuanhai Cao, and Jianfeng Cai
Molecular Pharmaceutics 2012 Volume 9(Issue 5) pp:1529-1534
Publication Date(Web):March 13, 2012
DOI:10.1021/mp300070w
Cell-penetrating peptides including the trans-activating transcriptional activator (Tat) from HIV-1 have been used as carriers for intracellular delivery of a myriad of cargoes including drugs, molecular probes, DNAs and nanoparticles. Utilizing fluorescence flow cytometry and confocal fluorescence microscopy, we demonstrate that a γ-AApeptide mimetic of Tat (48–57) can cross the cell membranes and enter the cytoplasm and nucleus of cells, with efficiency comparable to or better than that of Tat peptide (48–57). Deletion of the four side chains of the γ-AApeptide attenuates translocation capability. We also establish that the γ-AApeptide is even less toxic than the Tat peptide against mammalian cells. In addition to their low toxicity, γ-AApeptides are resistant to protease degradation, which may prove to be advantageous over α-peptides for further development of molecular transporters for intracellular delivery.Keywords: cell penetrating peptide (CPP); cellular uptake; peptidomimetics; Tat; γ-AApeptides;
Co-reporter:Yaogang Hu, Mohamad Nassir Amin, Shruti Padhee, Rongsheng E. Wang, Qiao Qiao, Ge Bai, Yaqong Li, Archana Mathew, Chuanhai Cao, and Jianfeng Cai
ACS Medicinal Chemistry Letters 2012 Volume 3(Issue 8) pp:683
Publication Date(Web):July 12, 2012
DOI:10.1021/ml3001215
We report a series of lipidated α-AApeptides that mimic the structure and function of natural antimicrobial lipopeptides. Several short lipidated α-AApeptides show broad-spectrum activity against a range of clinically related Gram-positive and Gram-negative bacteria as well as fungus. Their antimicrobial activity and selectivity are comparable or even superior to the clinical candidate pexiganan as well as previously reported linear α-AApeptides. The further development of lipidated α-AApeptides will lead to a new class of antibiotics to combat drug resistance.Keywords: antimicrobial peptides; drug resistance; lipidation; peptidomimetics; α-AApeptides
Co-reporter:Youhong Niu, Haifan Wu, Rongfu Huang, Qiao Qiao, Frankie Costanza, Xi-Sen Wang, Yaogang Hu, Mohamad Nassir Amin, Anh-My Nguyen, James Zhang, Edward Haller, Shengqian Ma, Xiao Li, and Jianfeng Cai
Macromolecules 2012 Volume 45(Issue 18) pp:7350-7355
Publication Date(Web):September 7, 2012
DOI:10.1021/ma3015992
Although peptide amphiphiles have been explored as nanomaterials for different applications, nanostructures formed by hierarchical molecular assembly of sequence-specific peptidomimetics are much less developed. Such protein-like nanomaterials could enhance the current application of peptide-based amphiphiles by enriching the diversity of nanostructures, increasing in vivo stability for biomedical applications, and facilitating the understanding of biomacromolecular self-assembly. Herein we present a biomimetic γ-AApeptide amphiphile which forms nanorods. Our results demonstrate the capability of γ-AApeptide amphiphiles as a potential scaffold for the preparation of biomimetic and bioinspired nanostructures. The programmability and biocompatibility of γ-AApeptides could lead to novel nanomaterials for a wide variety of applications.
Co-reporter:Youhong Niu, Shruti Padhee, Haifan Wu, Ge Bai, Lacey Harrington, Whittney N. Burda, Lindsey N. Shaw, Chuanhai Cao and Jianfeng Cai
Chemical Communications 2011 vol. 47(Issue 44) pp:12197-12199
Publication Date(Web):03 Oct 2011
DOI:10.1039/C1CC14476F
We report the identification of a new class of antimicrobial peptidomimetics-γ-AApeptides with potent and broad-spectrum activity, including clinically-relevant strains that are unresponsive to most antibiotics. They are also not prone to select for drug-resistance.
Co-reporter:Shruti Padhee, Yaogang Hu, Youhong Niu, Ge Bai, Haifan Wu, Frankie Costanza, Leigh West, Lacey Harrington, Lindsey N. Shaw, Chuanhai Cao and Jianfeng Cai
Chemical Communications 2011 vol. 47(Issue 34) pp:9729-9731
Publication Date(Web):21 Jul 2011
DOI:10.1039/C1CC13684D
We report a new class of peptide mimetics, α-AApeptides, that display broad-spectrum activity against both Gram-negative and Gram-positive bacteria and fungi. With non-hemolytic activity, resistance to protease hydrolysis, and easy sequence programmability, α-AApeptides may emerge as a novel class of antibiotics.
Co-reporter:Youhong Niu, Alisha “Jonesy” Jones, Haifan Wu, Gabriele Varani and Jianfeng Cai
Organic & Biomolecular Chemistry 2011 vol. 9(Issue 19) pp:6604-6609
Publication Date(Web):01 Jul 2011
DOI:10.1039/C1OB05738C
The interactions between proteins and RNAs are of vital importance for many cellular processes, including transcription and processing of RNA, translation, and viral infections. Here we report an γ-AApeptide that can mimic HIV-1 Tat protein and bind to TAR RNAs of HIV and BIV with nanomolar affinity, comparable to that of the RNA-binding fragment of Tat (amino acids 49–58). The interaction is resistant to the presence of a large excess of tRNA. With resistance to proteolytic hydrolysis and limitless potential for diversification, γ-AApeptides may emerge as a new class of peptidomimetics to modulate RNA-protein interactions.
Co-reporter:Youhong Niu, Yaogang Hu, Xiaolong Li, Jiandong Chen and Jianfeng Cai
New Journal of Chemistry 2011 vol. 35(Issue 3) pp:542-545
Publication Date(Web):31 Jan 2011
DOI:10.1039/C0NJ00943A
A new class of peptide mimics termed “γ-AApeptides” has been described. The design and synthesis of γ-AApeptides, and potential bioactivities towards p53/MDM2 interaction were demonstrated. γ-AApeptides were also found to be highly resistant to proteolysis. The development of sequence-specific γ-AApeptides may lead to a family of peptidomimetics with a new framework for drug discovery or peptide/protein mimicry.
Co-reporter:Yaogang Hu, Xiaolong Li, Said M. Sebti, Jiandong Chen, Jianfeng Cai
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 5) pp:1469-1471
Publication Date(Web):1 March 2011
DOI:10.1016/j.bmcl.2011.01.005
A new family of peptide mimics termed ‘AApeptides’, which are oligomers of N-acylated-N-aminoethyl amino acids, was proposed. The design and efficient synthesis of AApeptides are described. As proof-of-the-concept, we show that AApeptides can inhibit p53/MDM2 protein–protein interaction with significant activity (IC50 = 38 μM) and specificity. Preliminary data also demonstrates that AApeptides are resistant to enzymatic hydrolysis. With the ease of synthesis and diversification, potent bioactivity, and resistance to proteolysis, the development of sequence-specific AApeptides may expand the potential biomedical applications of peptidomimetics.A new class of oligomeric peptidomimics were described and synthesized.
Co-reporter:Pavanjeet Kaur, Yaqiong Li, Jianfeng Cai, Likai Song
Biophysical Journal (26 April 2016) Volume 110(Issue 8) pp:
Publication Date(Web):26 April 2016
DOI:10.1016/j.bpj.2016.02.038
γ-AApeptides are a new class of antibacterial peptidomimetics that are not prone to antibiotic resistance and are highly resistant to protease degradation. It is not clear how γ-AApeptides interact with bacterial membranes and alter lipid assembly, but such information is essential to understanding their antimicrobial activities and guiding future design of more potent and specific antimicrobial agents. Using electron paramagnetic resonance techniques, we characterized the membrane interaction and destabilizing mechanism of a lipo-cyclic-γ-AApeptide (AA1), which has broad-spectrum antibacterial activities. The analyses revealed that AA1 binding increases the membrane permeability of POPC/POPG liposomes, which mimic negatively charged bacterial membranes. AA1 binding also inhibits membrane fluidity and reduces solvent accessibility around the lipid headgroup region. Moreover, AA1 interacts strongly with POPC/POPG liposomes, inducing significant lipid lateral-ordering and membrane thinning. In contrast, minimal membrane property changes were observed upon AA1 binding for liposomes mimicking mammalian cell membranes, which consist of neutral lipids and cholesterol. Our findings suggest that AA1 interacts and disrupts bacterial membranes through a carpet-like mechanism. The results showed that the intrinsic features of γ-AApeptides are important for their ability to disrupt bacterial membranes selectively, the implications of which extend to developing new antibacterial biomaterials.
Co-reporter:Hai Xu, Siqi Zhao, Xiang Xiong, Jinzhi Jiang, Wei Xu, Daoben Zhu, Yi Zhang, Wenjie Liang, Jianfeng Cai
Chemical Physics Letters (16 April 2017) Volume 674() pp:
Publication Date(Web):16 April 2017
DOI:10.1016/j.cplett.2017.02.063
•In summary, CP is found to self-assemble into distinct morphological structures under different environmental conditions: this macro cyclic conjugated molecule self organized into nanospheres and nanofibers accompanying natural evaporation of its butanol and toluene solutions on mica, silica and HOPG, respectively.•Intermolecular π-π bonding and electronic attraction is believed to be involved in the self-association processes, and affinity or likeness of the solvent molecules and the molecule-surface interactions may be responsible for the molecules to vary their assembly structures in response to the changes in their solvent and substrate.•The constructions of nanosized fibers may found applications in the field of supramolecular electronics.Cyclo [8] pyrrole (CP) is a porphyrin analogue containing eight α-conjugated pyrrole units which are arranged in a nearly coplanar conformation. The π-π interactions between CP molecules lead to regular aggregations through a solution casting process. Using tapping mode atomic force microscope (AFM), we investigated the morphology of self-assembled aggregates formed by deposition of different CP solutions on different substrates. We found that in the n-butanol solution, nanofibrous structures could be formed on the silicon or mica surface. Interestingly, on the highly oriented pyrolytic graphite (HOPG) surface, or silicon and mica surface with a toluene solution, only irregular spherical structures were identified. The difference in the nanomorphology may be attributed to distinct interactions between molecule-molecule, molecule-solvent and molecule-substrate.Cyclo [8] pyrrole (CP) is found to self-assemble into distinct morphological structures under different environmental conditions: this macro cyclic conjugated molecule self-organize into nanospheres and nanofibers accompanying natural evaporation of its n-butanol and toluene solutions on mica, silica and HOPG, respectively. Intermolecular π-π bonding and electronic attraction are believed to play key roles in the self-association processes, and affinity or likeness of the solvent molecules and the molecule-surface interactions may be responsible for the molecules to vary their assembly structures in response to the changes in solvent and substrate. The constructions of nanosized fibers may find applications in the field of supramolecular electronics.
Co-reporter:Yan Wang, Frankie Costanza, Haifan Wu, Daqian Song, Jianfeng Cai and Qi Li
Journal of Materials Chemistry A 2014 - vol. 2(Issue 20) pp:NaN3122-3122
Publication Date(Web):2014/03/07
DOI:10.1039/C4TB00041B
Tanshinone IIA is a potent anti-tumor agent; however, its therapeutic applications have been hindered by its intrinsic poor solubility and short half-life. Herein we report the design and synthesis of amphiphilic PEG–poly(amino acid)s (PPAAs), which were used as a nanocarrier to encapsulate tanshinone IIA. PPAA-encapsulated tanshinone IIA formed defined nanoparticles and effectively arrested the growth of the hepatoma tumor in MHCC97-H hepatoma-bearing mice, which has therefore significantly increased their survival time. Moreover, these tanshinone-loaded nanoparticles are also capable of suppressing hepatic tumor cell migration and metastasis. In all cases, they significantly improve efficacy compared to tanshinone alone. Our results show that tanshinone IIA encapsulated by PPAAs is a potential novel nanomedicine for the treatment of hepatoma. Furthermore, this use is only a proof of concept, and PPAAs can be used as a potential drug delivery system for a much broader array of hydrophobic drugs.
Co-reporter:Youhong Niu, Alisha “Jonesy” Jones, Haifan Wu, Gabriele Varani and Jianfeng Cai
Organic & Biomolecular Chemistry 2011 - vol. 9(Issue 19) pp:NaN6609-6609
Publication Date(Web):2011/07/01
DOI:10.1039/C1OB05738C
The interactions between proteins and RNAs are of vital importance for many cellular processes, including transcription and processing of RNA, translation, and viral infections. Here we report an γ-AApeptide that can mimic HIV-1 Tat protein and bind to TAR RNAs of HIV and BIV with nanomolar affinity, comparable to that of the RNA-binding fragment of Tat (amino acids 49–58). The interaction is resistant to the presence of a large excess of tRNA. With resistance to proteolytic hydrolysis and limitless potential for diversification, γ-AApeptides may emerge as a new class of peptidomimetics to modulate RNA-protein interactions.
Co-reporter:Yunan Yang, Youhong Niu, Hao Hong, Haifan Wu, Yin Zhang, Jonathan W. Engle, Todd E. Barnhart, Jianfeng Cai and Weibo Cai
Chemical Communications 2012 - vol. 48(Issue 63) pp:NaN7852-7852
Publication Date(Web):2012/06/19
DOI:10.1039/C2CC33620K
A γ-AApeptide-based tracer for positron emission tomography imaging of integrin αvβ3 is reported. Despite its shorter sequence and linear nature, this tracer had comparable integrin αvβ3 binding affinity to the cyclic arginine-glycine-aspartic acid peptide but significantly higher resistance to enzymatic degradation and better stability.
Co-reporter:Youhong Niu, Haifan Wu, Yaqiong Li, Yaogang Hu, Shruti Padhee, Qi Li, Chuanhai Cao and Jianfeng Cai
Organic & Biomolecular Chemistry 2013 - vol. 11(Issue 26) pp:NaN4290-4290
Publication Date(Web):2013/05/02
DOI:10.1039/C3OB40444G
Antibiotic resistance is an increasing public health concern around the world, and is recognized as one of the greatest threats facing humankind in the 21st century. Natural antimicrobial peptides (AMPs) are small cationic amphiphilic peptides found in virtually all living organisms, and play a key role in the defense against bacterial infections. Compared with conventional antibiotics, which target specific metabolic processes, AMPs are able to adopt globally amphipathic conformations, and kill bacteria through disruption of their membranes. As such, AMPs do not readily induce drug-resistance. However, AMPs are associated with intrinsic drawbacks such as low-to-moderate activity, susceptibility to enzymatic degradation, and inconvenience for optimization. Recently, we have developed a new class of peptidomimetics termed “AApeptides”. Such peptide mimics are highly resistant to protease degradation and are straightforward for chemical diversification and development. Our current studies show that AApeptides with globally amphipathic structures can mimic the bactericidal mechanism of AMPs, and display potent and broad-spectrum activity against both Gram-positive and -negative multi-drug-resistant bacteria. In this review, we summarize our current findings of antimicrobial AApeptides, and discuss potential future directions on the development of more potent and specific analogues.
Co-reporter:Peng Teng, Xiaolei Zhang, Haifan Wu, Qiao Qiao, Said M. Sebti and Jianfeng Cai
Chemical Communications 2014 - vol. 50(Issue 63) pp:NaN8742-8742
Publication Date(Web):2014/06/09
DOI:10.1039/C4CC03909B
From a γ-AApeptide-based one-bead-one-compound (OBOC) combinatorial library, we identified γ-AApeptides that can selectively inhibit STAT3–DNA interaction and suppress the expression levels of STAT3 target genes in intact cells. Our results demonstrate that in addition to the SH2 domain, the DNA binding domain of STAT3 is targetable for the development of a new generation of anti-cancer therapeutics.
Co-reporter:Shruti Padhee, Yaogang Hu, Youhong Niu, Ge Bai, Haifan Wu, Frankie Costanza, Leigh West, Lacey Harrington, Lindsey N. Shaw, Chuanhai Cao and Jianfeng Cai
Chemical Communications 2011 - vol. 47(Issue 34) pp:NaN9731-9731
Publication Date(Web):2011/07/21
DOI:10.1039/C1CC13684D
We report a new class of peptide mimetics, α-AApeptides, that display broad-spectrum activity against both Gram-negative and Gram-positive bacteria and fungi. With non-hemolytic activity, resistance to protease hydrolysis, and easy sequence programmability, α-AApeptides may emerge as a novel class of antibiotics.
Co-reporter:Youhong Niu, Shruti Padhee, Haifan Wu, Ge Bai, Lacey Harrington, Whittney N. Burda, Lindsey N. Shaw, Chuanhai Cao and Jianfeng Cai
Chemical Communications 2011 - vol. 47(Issue 44) pp:NaN12199-12199
Publication Date(Web):2011/10/03
DOI:10.1039/C1CC14476F
We report the identification of a new class of antimicrobial peptidomimetics-γ-AApeptides with potent and broad-spectrum activity, including clinically-relevant strains that are unresponsive to most antibiotics. They are also not prone to select for drug-resistance.
Co-reporter:Haifan Wu, Yaqiong Li, Ge Bai, Youhong Niu, Qiao Qiao, Jeremiah D. Tipton, Chuanhai Cao and Jianfeng Cai
Chemical Communications 2014 - vol. 50(Issue 40) pp:NaN5208-5208
Publication Date(Web):2013/10/10
DOI:10.1039/C3CC46685J
We report the design, synthesis, characterization and evaluation of a novel class of γ-AApeptide one-bead-one-compound (OBOC) library, from which a small γ-AApeptide was identified to effectively prevent and disassemble Aβ aggregation.
Co-reporter:Ge Bai, Shruti Padhee, Youhong Niu, Rongsheng E. Wang, Qiao Qiao, Robert Buzzeo, Chuanhai Cao and Jianfeng Cai
Organic & Biomolecular Chemistry 2012 - vol. 10(Issue 6) pp:NaN1153-1153
Publication Date(Web):2011/11/30
DOI:10.1039/C2OB06679C
Some short and cationic peptides such as the Tat peptide can cross the cell membrane and function as vectors for intracellular delivery. Here we show that an α-AApeptide is able to penetrate the membranes of living cells from an extracellular environment and enter the endosome and cytoplasm of cells. The efficiency of the cellular uptake is comparable to a Tat peptide (48–57) of the same length and is unexpectedly superior to an α-peptide with identical functional groups. The mechanism of uptake is similar to that of the Tat peptide and is through endocytosis by an energy-dependent pathway. Due to the easy synthesis of the α-AApeptides, their resistance to proteolytic hydrolysis, and their low cytotoxicity, α-AApeptides represent a new class of transporters for the delivery of drugs.
Co-reporter:Haifan Wu, Youhong Niu, Shruti Padhee, Rongsheng E. Wang, Yaqiong Li, Qiao Qiao, Ge Bai, Chuanhai Cao and Jianfeng Cai
Chemical Science (2010-Present) 2012 - vol. 3(Issue 8) pp:NaN2575-2575
Publication Date(Web):2012/05/17
DOI:10.1039/C2SC20428B
Antimicrobial drug resistance is one of the greatest threats facing mankind. Antimicrobial peptides (AMPs) can potentially circumvent drug resistance, probably through a bacterial membrane-disruption mechanism. However, they suffer from low in vivo stability, potential immunogenicity, and difficulty in optimization. The development of antimicrobial peptidomimetics is therefore an emerging research area as they avoid the potential disadvantages of AMPs. Cyclic peptidomimetics are of significant interest since constraints induced by cyclization are expected to further improve their antimicrobial activity. Nonetheless, the report of cyclic oligomeric peptidomimetics for antimicrobial development is rare. Herein, for the first time, we report the design and synthesis of cyclic γ-AApeptides via an on-resin cyclization. These cyclic γ-AApeptides are potent and broad-spectrum active against fungus and multi-drug resistant Gram-positive and Gram-negative bacterial pathogens. Our results demonstrate the potential of cyclic γ-AApeptides as a new class of antibiotics to circumvent drug resistance by mimicking the bactericidal mechanism of AMPs. Meanwhile, the facile synthesis of cyclic γ-AApeptides may further expand the applications of γ-AApeptides in biomedical sciences.
Co-reporter:Haifan Wu, Fengyu She, Wenyang Gao, Austin Prince, Yaqiong Li, Lulu Wei, Allison Mercer, Lukasz Wojtas, Shengqian Ma and Jianfeng Cai
Organic & Biomolecular Chemistry 2015 - vol. 13(Issue 3) pp:NaN676-676
Publication Date(Web):2014/11/18
DOI:10.1039/C4OB02232G
We report an efficient method for the preparation of unprecedented head-to-tail cyclic sulfono-γ-AApeptides. Following this method, a number of cyclic sequences bearing two to five subunits were efficiently synthesized. In addition, the X-ray crystal structure study of a three-membered cyclic sulfono-γ-AApeptide revealed a type II β-turn-like character.

![Benzoic acid, 4,4',4'',4'''-[1,4,7,10-tetraazacyclododecane-1,4,7,10-tetrayltetrakis(methylene)]tetrakis-](/data/chemimg/3718800/1421944-62-8.png)
![Benzoic acid, 4,4',4'',4'''-[1,4,7,10-tetraazacyclododecane-1,4,7,10-tetrayltetrakis(methylene)]tetrakis-](/data/chemimg/3718800/1421944-62-8_b.png)
![N-(4-Methoxy-3-(4-methylpiperazin-1-yl)phenyl)-2'-methyl-4'-(5-methyl-1,2,4-oxadiazol-3-yl)-[1,1'-biphenyl]-4-carboxamide](http://img.cochemist.com/ccimg/148700/148672-13-3.png)
![N-(4-Methoxy-3-(4-methylpiperazin-1-yl)phenyl)-2'-methyl-4'-(5-methyl-1,2,4-oxadiazol-3-yl)-[1,1'-biphenyl]-4-carboxamide](http://img.cochemist.com/ccimg/148700/148672-13-3_b.png)





![Carbamic acid, [(1S)-1-methyl-2-oxoethyl]-, 2-propenyl ester](http://img.cochemist.com/ccimg/191200/191171-73-0.png)
![Carbamic acid, [(1S)-1-methyl-2-oxoethyl]-, 2-propenyl ester](http://img.cochemist.com/ccimg/191200/191171-73-0_b.png)















![N-[(Allyloxy)carbonyl]glycine](http://img.cochemist.com/ccimg/90800/90711-56-1.png)
![N-[(Allyloxy)carbonyl]glycine](http://img.cochemist.com/ccimg/90800/90711-56-1_b.png)
![L-Phenylalanine, N-[(2-propenyloxy)carbonyl]-](http://img.cochemist.com/ccimg/90600/90508-20-6.png)
![L-Phenylalanine, N-[(2-propenyloxy)carbonyl]-](http://img.cochemist.com/ccimg/90600/90508-20-6_b.png)


![4-Amino-1-[(5S)-5-(hydroxymethyl)tetrahydro-2-furanyl]-2(1H)-pyri midinone](http://img.cochemist.com/ccimg/83900/83869-56-1.png)
![4-Amino-1-[(5S)-5-(hydroxymethyl)tetrahydro-2-furanyl]-2(1H)-pyri midinone](http://img.cochemist.com/ccimg/83900/83869-56-1_b.png)


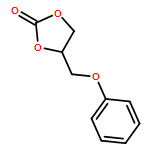



![1,3-Dioxolan-2-one, 4-[(2-propen-1-yloxy)methyl]-](/data/chemimg/2314900/826-29-9.png)
![1,3-Dioxolan-2-one, 4-[(2-propen-1-yloxy)methyl]-](/data/chemimg/2314900/826-29-9_b.png)





![3,5,9-Trioxa-4-phosphaheptacos-18-en-1-aminium,4-hydroxy-N,N,N-trimethyl-10-oxo-7-[[(9Z)-1-oxo-9-octadecen-1-yl]oxy]-, innersalt, 4-oxide, (7R,18Z)-](http://img.cochemist.com/ccimg/4300/4235-95-4.png)
![3,5,9-Trioxa-4-phosphaheptacos-18-en-1-aminium,4-hydroxy-N,N,N-trimethyl-10-oxo-7-[[(9Z)-1-oxo-9-octadecen-1-yl]oxy]-, innersalt, 4-oxide, (7R,18Z)-](http://img.cochemist.com/ccimg/4300/4235-95-4_b.png)