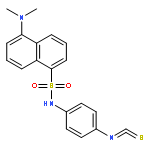
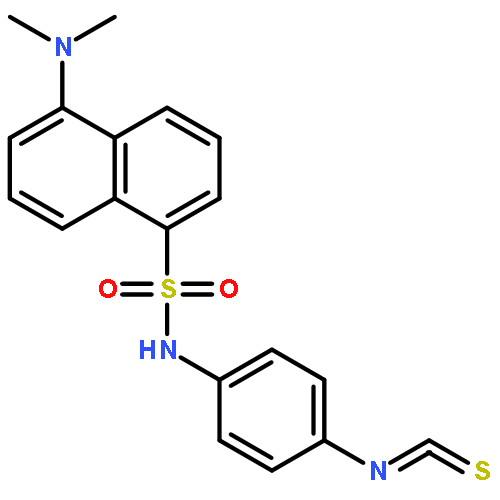
Co-reporter:Shajila Siricilla, Katsuhiko Mitachi, Junshu Yang, Shakiba Eslamimehr, Maddie R. Lemieux, Bernd Meibohm, Yinduo Ji, and Michio Kurosu
Journal of Medicinal Chemistry April 13, 2017 Volume 60(Issue 7) pp:2869-2869
Publication Date(Web):March 14, 2017
DOI:10.1021/acs.jmedchem.6b01805
Multidrug-resistant (MDR) Acinetobacter baumannii is one of the most difficult Gram-negative bacteria to treat and eradicate. In a cell-based screening of pleuromutilin derivatives against a drug sensitive A. baumannii strain, new molecules (2–4) exhibit bacteriostatic activity with 3.13 μg/mL concentration and 1 shows bactericidal activity with an MBC of 6.25 μg/mL. The pleuromutilin derivative 1 displays strong synergistic effects with doxycycline in a wide range of concentrations. A 35/1 ratio of 1 and doxycycline (1-Dox 35/1) kills drug susceptible A. baumannii with the MBC of 2.0 μg/mL and an MDR A. baumannii with the MBC of 3.13 μg/mL. In vitro anti-Acinetobacter activity of 1-Dox 35/1 is superior to that of clinical drugs such as tobramycin, tigecycline, and colistin. The efficacy of 1-Dox 35/1 is evaluated in a mouse septicemia model; treatment of the infected C57BL/6 mice with 1-Dox 35/1 protects from lethal infection of A. baumannii with an ED50 value of <2.0 mg/kg.
Co-reporter:Katsuhiko Mitachi, Bilal A. Aleiwi, Christopher M. Schneider, Shajila Siricilla, and Michio Kurosu
Journal of the American Chemical Society 2016 Volume 138(Issue 39) pp:12975-12980
Publication Date(Web):September 12, 2016
DOI:10.1021/jacs.6b07395
Co-reporter:Katsuhiko Mitachi, Lekh Nath Sharma Gautam, Jeffrey H. Rice, Keiko Eda, Ashutosh Wadhwa, Eiichi Momotani, Joseph P. Hlopak, Shigetoshi Eda, Michio Kurosu
Analytical Biochemistry 2016 Volume 505() pp:29-35
Publication Date(Web):15 July 2016
DOI:10.1016/j.ab.2016.04.001
Abstract
Mycobacterium avium subspecies paratuberculosis (MAP) causes chronic illnesses mostly in ruminants. MAP infection of intestinal tissue triggers a fatal inflammatory disorder, Johne's disease (paratuberculosis). Development of fast and reliable diagnostic methods for Johne's disease in clinically suspected ruminants requires the discovery of MAP-specific antigens that induce immune responses. Despite a longtime interest in finding such antigens that can detect serum antibody responses with high sensitivity, the antigens currently used for a diagnosis of the MAP infections are the crude extracts from the whole cell. We performed the serum antibody response assay-guided purification of the ethanol extract from MAP isolated from an infected cow. With the results of extensive fractionations and in vitro assays, we identified that arachidyl-d-Phe-N-Me-l-Val-l-Ile-l-Phe-l-Ala-OH (named lipopeptide IIß, 3) exhibited the highest antibody binding activity in serum of a MAP-infected cattle compared with the other lipopeptides isolated from MAP. The absolute chemistry of 3 was determined unequivocally via our high-performance liquid chromatography (HPLC)–amino acid databases. α-Amino lipopeptide IIß and its fluorescent probes were synthesized and evaluated in serum antibody binding activity assays. Lipopeptide IIß-(2S)-NH2 (9) and its dansyl and fluorescein isothiocyanate (FITC) probes (10 and 11) exhibited antibody-mediated binding activity; thus, such MAP-specific lipopeptide probes can be potential biomarkers for the development of rapid and accurate diagnosis of Johne's disease.
Co-reporter:Katsuhiko Mitachi, Shajila Siricilla, Dong Yang, Ying Kong, Karolina Skorupinska-Tudek, Ewa Swiezewska, Scott G. Franzblau, Michio Kurosu
Analytical Biochemistry 2016 Volume 512() pp:78-90
Publication Date(Web):1 November 2016
DOI:10.1016/j.ab.2016.08.008
Abstract
Polyprenyl phosphate-GlcNAc-1-phosphate transferase (WecA) is an essential enzyme for the growth of Mycobacterium tuberculosis (Mtb) and some other bacteria. Mtb WecA catalyzes the transformation from UDP-GlcNAc to decaprenyl-P-P-GlcNAc, the first membrane-anchored glycophospholipid that is responsible for the biosynthesis of mycolylarabinogalactan in Mtb. Inhibition of WecA will block the entire biosynthesis of essential cell wall components of Mtb in both replicating and non-replicating states, making this enzyme a target for development of novel drugs. Here, we report a fluorescence-based method for the assay of WecA using a modified UDP-GlcNAc, UDP-Glucosamine-C6-FITC (1), a membrane fraction prepared from an M. smegmatis strain, and the E. coli B21WecA. Under the optimized conditions, UDP-Glucosamine-C6-FITC (1) can be converted to the corresponding decaprenyl-P-P-Glucosamine-C6-FITC (3) in 61.5% yield. Decaprenyl-P-P-Glucosamine-C6-FITC is readily extracted with n-butanol and can be quantified by ultraviolet–visible (UV–vis) spectrometry. Screening of the compound libraries designed for bacterial phosphotransferases resulted in the discovery of a selective WecA inhibitor, UT-01320 (12) that kills both replicating and non-replicating Mtb at low concentration. UT-01320 (12) also kills the intracellular Mtb in macrophages. We conclude that the WecA assay reported here is amenable to medium- and high-throughput screening, thus facilitating the discovery of novel WecA inhibitors.
Co-reporter:Shajila Siricilla, Katsuhiko Mitachi, Bajoie Wan, Scott G Franzblau and Michio Kurosu
The Journal of Antibiotics 2015 68(4) pp:271-278
Publication Date(Web):October 1, 2014
DOI:10.1038/ja.2014.133
Capuramycin (1) and its analogs are strong translocase I (MurX/MraY) inhibitors. In our structure–activity relationship studies of capuramycin analogs against Mycobacterium tuberculosis (Mtb), we observed for the first time that a capuramycin analog, UT-01320 (3) killed nonreplicating (dormant) Mtb at low concentrations under low oxygen conditions, whereas selective MurX inhibitors killed only replicating Mtb under aerobic conditions. Interestingly, 3 did not exhibit MurX enzyme inhibitory activity even at high concentrations, however, 3 inhibited bacterial RNA polymerases with the IC50 values of 100–150 nM range. A new RNA polymerase inhibitor 3 displayed strong synergistic effects with a MurX inhibitor SQ 641 (2), a promising preclinical tuberculosis drug.
Co-reporter:Shajila Siricilla, Katsuhiko Mitachi, Karolina Skorupinska-Tudek, Ewa Swiezewska, Michio Kurosu
Analytical Biochemistry 2014 Volume 461() pp:36-45
Publication Date(Web):15 September 2014
DOI:10.1016/j.ab.2014.05.018
Abstract
Translocase I (MraY/MurX) is an essential enzyme in growth of the vast majority of bacteria that catalyzes the transformation from UDP-MurNAc-pentapeptide (Park’s nucleotide) to prenyl-MurNAc-pentapeptide (lipid I), the first membrane-anchored peptidoglycan precursor. MurX has received considerable attention in the development of new tuberculosis (TB) drugs due to the fact that the MurX inhibitors kill exponentially growing Mycobacterium tuberculosis (Mtb) much faster than clinically used TB drugs. Lipid I isolated from Mtb contains the C50-prenyl unit that shows very poor water solubility; thus, this chemical characteristic of lipid I renders MurX enzyme assays impractical for screening and lacks reproducibility of the enzyme assays. We have established a scalable chemical synthesis of Park’s nucleotide-Nε-dansylthiourea 2 that can be used as a MurX enzymatic substrate to form lipid I analogues. In our investigation of the minimum structure requirement of the prenyl phosphate in the MraY/MurX-catalyzed lipid I analogue synthesis with 2, we found that neryl phosphate (C10 phosphate) can be recognized by MraY/MurX to generate the water-soluble lipid I analogue in quantitative yield under the optimized conditions. Here, we report a rapid and robust analytical method for quantifying MraY/MurX inhibitory activity of library molecules.
Co-reporter:Dr. Katsuhiko Mitachi;Dr. Priya Mohan;Shajila Siricilla ; Michio Kurosu
Chemistry - A European Journal 2014 Volume 20( Issue 16) pp:4554-4558
Publication Date(Web):
DOI:10.1002/chem.201400307
Abstract
(2,6-Dichloro-4-methoxyphenyl)(2,4-dichlorophenyl)methyl trichloroacetimidate (3) and its polymer-supported reagent 4 can be successfully applied to a one-pot protection-glycosylation reaction to form the disaccharide derivative 7 d for the synthesis of lipid II analogues. The temporary protecting group or linker at the C-6 position and N-Troc protecting group of 7 d can be cleaved simultaneously through a reductive condition. Overall yields of syntheses of lipid II (1) and neryl-lipid II Nε-dansylthiourea are significantly improved by using the described methods.
Co-reporter:Bilal A. Aleiwi, Katsuhiko Mitachi, Michio Kurosu
Tetrahedron Letters 2013 Volume 54(Issue 16) pp:2077-2081
Publication Date(Web):17 April 2013
DOI:10.1016/j.tetlet.2013.02.013
We have realized that N-formylations of free amines of some drug leads can improve PK/PD property of parent molecules without decreasing their biological activities. In order to selectively formylate primary amines of polyfunctional molecules, we have sought a mild and convenient formylation reaction. In our screening of N-formylation of an α-amino acid, l-phenylalanine, none of formylation conditions reported to date yielded the desired HCO-l-Phe-OH with satisfactory yield. N-formylations of amino acids with HCO2H require a water-containing media and suppress polymerization reactions due to the competitive reactions among carboxylic acids. We found that N-formylations of α-amino acids could be achieved with a water-soluble peptide coupling additive, an Oxyma derivative, (2,2-dimethyl-1,3-dioxolan-4-yl)methyl-2-cyano-2-(hydroxyimino)acetate (2), EDCI, and NaHCO3 in water or a mixture of water and DMF system, yielding N-formylated α-amino acids with excellent yields. Moreover, these conditions could selectively formylate primary amines over secondary amines at a controlled temperature. A usefulness of these conditions was demonstrated by selective formylation of daptomycin antibiotic which contains three different amino groups.
Co-reporter:Dr. Yong Wang;Shajila Siricilla;Dr. Bilal A. Aleiwi ; Michio Kurosu
Chemistry - A European Journal 2013 Volume 19( Issue 41) pp:13847-13858
Publication Date(Web):
DOI:10.1002/chem.201302389
Abstract
Capuramycin and its congeners are considered to be important lead molecules for the development of a new drug for multidrug-resistant (MDR) Mycobacterium tuberculosis infections. Extensive structure–activity relationship studies of capuramycin to improve the efficacy have been limited because of difficulties in selectively chemically modifying the desired position(s) of the natural product with biologically interesting functional groups. We have developed efficient syntheses of capuramycin and its analogues by using new protecting groups, derived from the chiral (chloro-4-methoxyphenyl)(chlorophenyl)methanols, for the uridine ureido nitrogen and primary alcohol. The chiral nonracemic (2,6-dichloro-4-methoxyphenyl)(2,4-dichlorophenyl)methanol derivative is a useful reagent to resolve rac-3-amino-1,3-dihydro-5-phenyl-2H-1,4-benzodiazepin-2-one, the (S)-configuration isomer of which plays a significant role in improving the mycobactericidal activity of capuramycin.
Co-reporter:Qinghui Wang, Yong Wang, and Michio Kurosu
Organic Letters 2012 Volume 14(Issue 13) pp:3372-3375
Publication Date(Web):June 14, 2012
DOI:10.1021/ol3013556
An Oxyma derivative, (2,2-dimethyl-1,3-dioxolan-4-yl)methyl 2-cyano-2-(hydroxyimino)acetate (2), displayed remarkable physicochemical properties as a peptide-coupling additive for peptide-forming reactions in water. Short peptides to oligopeptides could be synthesized by using 2, EDCI, and NaHCO3 in water without measurable racemization. Significantly, a simple basic and acidic aqueous workup procedure can remove all reagents utilized in the reactions to afford only coupling products in consistently excellent yields.
Co-reporter:Yong Wang, Bilal A. Aleiwi, Qinghui Wang, and Michio Kurosu
Organic Letters 2012 Volume 14(Issue 18) pp:4910-4913
Publication Date(Web):August 31, 2012
DOI:10.1021/ol3022337
Oxyma and an oxyma derivative, (2,2-dimethyl-1,3-dioxolan-4-yl)methyl 2-cyano-2-(hydroxyimino)acetate (5b), displayed a remarkable effect on selective esterifications of primary alcohols. A wide range of carboxylic acids could be esterified with primary alcohols by using EDCI, NaHCO3, and Oxyma or Oxyma derivative 5b in 5% H2O–CH3CN. Oxyma derivative 5b is particularly useful, since it could be removed after the reaction via a simple basic or an acidic aqueous workup procedure.
Co-reporter:Joy Debnath ; Shajila Siricilla ; Bajoie Wan ; Dean C. Crick ; Anne J. Lenaerts ; Scott G. Franzblau
Journal of Medicinal Chemistry 2012 Volume 55(Issue 8) pp:3739-3755
Publication Date(Web):March 26, 2012
DOI:10.1021/jm201608g
Aurachin RE (1) is a strong antibiotic that was recently found to possess 1,4-dihydroxy-2-naphthoate prenyltransferase (MenA) and bacterial electron transport inhibitory activities. Aurachin RE is the only molecule in a series of aurachin natural products that has the chiral center in the alkyl side chain at C9′-position. To identify selective MenA inhibitors against Mycobacterium tuberculosis, a series of chiral molecules were designed based on the structures of previously identified MenA inhibitors and 1. The synthesized molecules were evaluated in in vitro assays, including MenA enzyme and bacterial growth inhibitory assays. We could identify novel MenA inhibitors that showed significant increase in potency of killing nonreplicating M. tuberculosis in the low oxygen recovery assay (LORA) without inhibiting other Gram-positive bacterial growth even at high concentrations. The MenA inhibitors reported here are useful new pharmacophores for the development of selective antimycobacterial agents with strong activity against nonreplicating M. tuberculosis.
Co-reporter:Bilal A. Aleiwi, Michio Kurosu
Tetrahedron Letters 2012 Volume 53(Issue 29) pp:3758-3762
Publication Date(Web):18 July 2012
DOI:10.1016/j.tetlet.2012.05.035
The benzyloxymethyl (BOM) group has been utilized widely in syntheses of a variety of natural and non-natural products. The BOM group is also one of few choices to protect uridine ureido nitrogen. However, hydrogenolytic cleavage of the BOM group of uridine derivatives has been unreliably performed via heterogeneous conditions using Pd catalysts. One of the undesirable by-products formed by Pd-mediated hydrogenation conditions is the over-reduced product in which the C5–C6 double bond of the uracil moiety was saturated. To date, we have generated a wide range of uridine-containing antibacterial agents, where the BOM group has been utilized in their syntheses. In screening of deprotection conditions of the BOM group of uridine ureido nitrogen under Pd-mediated hydrogenation conditions, we realized that the addition of water to the iPrOH-based hydrogenation conditions can suppress the formation of over-reduced uridine derivatives and the addition of HCO2H (0.5%) dramatically improve the reaction rate. An optimized hydrogenation condition described here can be applicable to the BOM-deprotections of a wide range of uridine derivatives.
Co-reporter:Bilal A. Aleiwi, Christopher M. Schneider, and Michio Kurosu
The Journal of Organic Chemistry 2012 Volume 77(Issue 8) pp:3859-3867
Publication Date(Web):March 29, 2012
DOI:10.1021/jo300205b
One of the key constituents of the muraymycins is the 6-membered cyclic guanidine, (2S,3S)-muraymycidine (or epi-capreomycidine). In order to diversify the structure of the oligopeptide moiety of the muraymycins for thorough structure–activity relationship studies, we have developed a highly stereoselective synthesis of ureidomuraymycidine derivatives with the lactone 4a.
Co-reporter:Yong Wang, Michio Kurosu
Tetrahedron 2012 68(24) pp: 4797-4804
Publication Date(Web):
DOI:10.1016/j.tet.2012.03.121

![Uridine 5'-(trihydrogendiphosphate), P'-[2-(acetylamino)-2-deoxy-a-D-glucopyranosyl] ester](http://img.cochemist.com/ccimg/600/528-04-1.png)
![Uridine 5'-(trihydrogendiphosphate), P'-[2-(acetylamino)-2-deoxy-a-D-glucopyranosyl] ester](http://img.cochemist.com/ccimg/600/528-04-1_b.png)
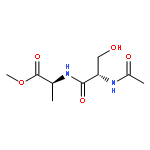
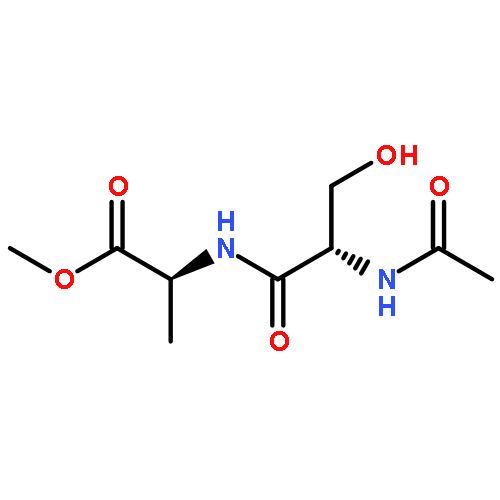




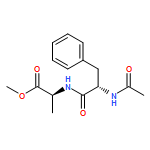
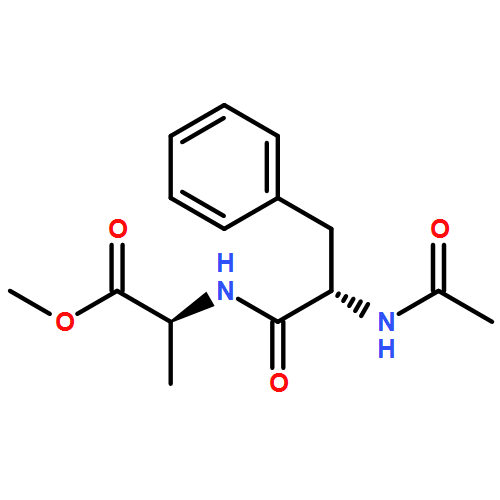


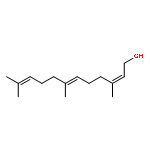
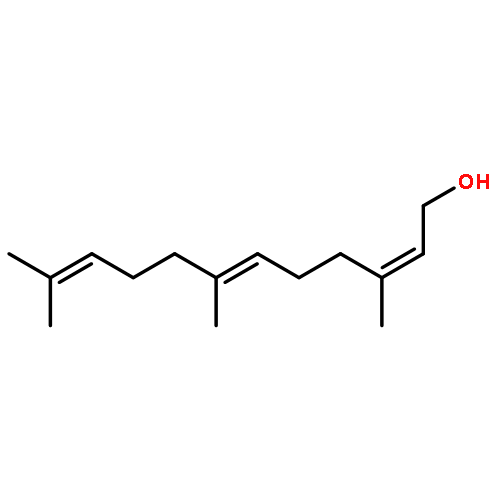


![a-L-Talofuranuronamide,1-deoxy-5-O-[4-deoxy-N-[(3S)-hexahydro-2-oxo-1H-azepin-3-yl]-b-L-erythro-hex-4-enopyranuronamidosyl]-1-(3,4-dihydro-2,4-dioxo-1(2H)-pyrimidinyl)-3-O-methyl-](http://img.cochemist.com/ccimg/102800/102770-00-3.png)
![a-L-Talofuranuronamide,1-deoxy-5-O-[4-deoxy-N-[(3S)-hexahydro-2-oxo-1H-azepin-3-yl]-b-L-erythro-hex-4-enopyranuronamidosyl]-1-(3,4-dihydro-2,4-dioxo-1(2H)-pyrimidinyl)-3-O-methyl-](http://img.cochemist.com/ccimg/102800/102770-00-3_b.png)