Co-reporter:Yugen Zhu, Lakota Cleaver, Wei Wang, Jessica D. Podoll, Shane Walls, Austin Jolly, Xiang Wang
European Journal of Medicinal Chemistry 2017 Volume 125() pp:130-142
Publication Date(Web):5 January 2017
DOI:10.1016/j.ejmech.2016.09.034
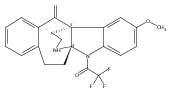
•A novel class of tetracyclic indolines was discovered in a β-lactam potentiator screen in MRSA.•The compound selectively potentiates β-lactams but not any other classes of antibiotics.•This novel class of compounds does not have antibacterial activity on its own, nor directly inhibit the β-lactamase activity.•Structure-activity relationship (SAR) study identified a more potent analogue with reduced mammalian cell toxicity.Antibiotic-resistant bacterial infections have seen a marked increase in recent years, while antibiotic discovery has waned. Resistance-modifying agents (RMA) offer an intriguing alternative strategy to fight against resistant bacteria. Here we report the discovery, antibiotic profiling, and structure-activity relationships of a novel class of RMAs, tetracyclic indolines. These selectively potentiate β-lactam antibiotics in methicillin-resistant Staphylococcus aureus (MRSA) without antibacterial or β-lactamase inhibitory activity on their own. The most potent analogue, 6a, showed strong potentiation of amoxicillin/clavulanic acid in a variety of hospital-acquired and community-acquired MRSA strains with low mammalian toxicity. These compounds may be further developed to extend the clinic life span of β-lactam antibiotics.
Co-reporter:W. He;B. M. Griffiths;W. Wang;X. Wang
Organic & Biomolecular Chemistry 2017 vol. 15(Issue 19) pp:4241-4245
Publication Date(Web):2017/05/16
DOI:10.1039/C7OB00897J
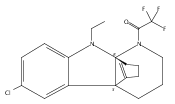
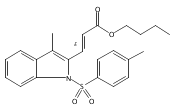
Tricyclic indolines are common in both natural products and synthetic chemical probes. In this study we demonstrated that enantiomerically pure tricyclic indolines can be prepared from an inexpensive commercially available chiral starting material, pyroglutamic acid. The synthesis features a highly diastereoselective gold-catalyzed cyclization of alkyne-tethered indoles and subsequent diastereoselective reductive ring-opening reaction. Using this approach, we synthesized analogs of our previously discovered tricyclic indoline probes that possess antibacterial and resistance-modifying activity. The biological activity against methicillin-resistant Staphylococcus aureus (MRSA) of these analogues was evaluated and reported. The synthetic approach reported may be leveraged in the future to prepare diastereopure chemical probes for the determination of biological targets for drug discovery.
Co-reporter:Dr. Yugen Zhu;Dr. Wei He;Dr. Wei Wang;Chloe E. Pitsch; Dr. Xiaotai Wang; Dr. Xiang Wang
Angewandte Chemie 2017 Volume 129(Issue 40) pp:12374-12377
Publication Date(Web):2017/09/25
DOI:10.1002/ange.201706694
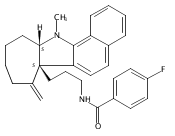
AbstractReported is the enantioselective synthesis of tetracyclic indolines using silver(I)/chiral phosphoric acid catalysis. A variety of alkyne-tethered indoles are suitable for this process. Mechanistic studies suggest that the in situ generated silver(I) chiral phosphate activates both the alkyne and the indole nucleophile in the initial cyclization step through an intermolecular hydrogen bond and the phosphate anion promotes proton transfer. In addition, further modifications of the cyclization products enabled stereochemistry–function studies of a series of bioactive indolines.
Co-reporter:Dr. Yugen Zhu;Dr. Wei He;Dr. Wei Wang;Chloe E. Pitsch; Dr. Xiaotai Wang; Dr. Xiang Wang
Angewandte Chemie International Edition 2017 Volume 56(Issue 40) pp:12206-12209
Publication Date(Web):2017/09/25
DOI:10.1002/anie.201706694
AbstractReported is the enantioselective synthesis of tetracyclic indolines using silver(I)/chiral phosphoric acid catalysis. A variety of alkyne-tethered indoles are suitable for this process. Mechanistic studies suggest that the in situ generated silver(I) chiral phosphate activates both the alkyne and the indole nucleophile in the initial cyclization step through an intermolecular hydrogen bond and the phosphate anion promotes proton transfer. In addition, further modifications of the cyclization products enabled stereochemistry–function studies of a series of bioactive indolines.
Co-reporter:Patrick M. Barbour;Wei Wang;Le Chang;Kasey L. Pickard;Rana Rais;Barbara S. Slusher
Advanced Synthesis & Catalysis 2016 Volume 358( Issue 9) pp:1482-1490
Publication Date(Web):
DOI:10.1002/adsc.201501101
Co-reporter:Laura J. Marholz, Wei Wang, Yu Zheng, and Xiang Wang
ACS Medicinal Chemistry Letters 2016 Volume 7(Issue 2) pp:167
Publication Date(Web):December 7, 2015
DOI:10.1021/acsmedchemlett.5b00366
The discovery of the 5-methylcytosine (5mC) oxidation by the ten–eleven translocation (Tet) protein family was an important advancement in our understanding of DNA-modified epigenetics. Potent inhibitors of these proteins are greatly desired for both the understanding of the functions of these enzymes and to serve as eventual therapeutic leads. So far, the discovery of such small molecules with high affinity has been quite limited. Original tools to screen for activity are greatly needed in order to accelerate this process. Here we present a novel fluorescent probe, and the results of a fluorescence polarization-based binding assay for Naegleria Tet1, a homologue to mammalian Tet. A fluorescence polarization-based competition assay was also established and applied to the rapid and quantitative measurement of the binding affinity of the cofactor αKG and several known Tet1 inhibitors.Keywords: 5-hydroxymethylcytosine; epigenetics; fluorescence polarization (FP); Ten−eleven translocation (Tet) proteins
Co-reporter:Laura J. Marholz, Le Chang, William M. Old, and Xiang Wang
ACS Chemical Biology 2015 Volume 10(Issue 1) pp:129
Publication Date(Web):October 21, 2014
DOI:10.1021/cb5006867
JmjC-domain containing histone demethylases (JHDMs) play critical roles in many key cellular processes and have been implicated in multiple disease conditions. Each enzyme within this family is known to have a strict substrate scope, specifically the position of the lysine within the histone and its degree of methylation. While much progress has been made in determining the substrates of each enzyme, new methods with which to systematically profile each histone mark are greatly needed. Novel chemical tools have the potential to fill this role and, furthermore, can be used as probes to answer fundamental questions about these enzymes and serve as potential therapeutic leads. In this work, we first investigated three small-molecule probes differing in the degree of “methylation state” and their differential bindings to JHDM1A (an H3K36me1/2 demethylase) using a fluorescence polarization-based competition assay. We then applied this specificity toward the “methylation state” and combined it with specificity toward lysine position in the design and synthesis of a peptidic probe targeting H3K36me2 JHDMs. The probe is further functionalized with a benzophenone cross-linking moiety and a biotin for affinity purification. Results showed binding of the peptidic probe to JHDM1A and specific enrichment of this protein in the presence of its native histone substrates. Affinity purification pulldown experiments from nuclear lysate coupled with mass spectrometry revealed the capability of the probe to pull out and enrich JHDMs along with other epigenetic proteins and transcriptional regulators.
Co-reporter:Dr. Wenqing Xu;Dr. Wei Wang ;Dr. Xiang Wang
Angewandte Chemie International Edition 2015 Volume 54( Issue 33) pp:9546-9549
Publication Date(Web):
DOI:10.1002/anie.201503736
Abstract
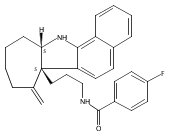
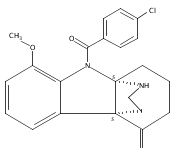
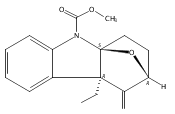
A gold-catalyzed desilylative cyclization was developed for facile synthesis of bridged tetracyclic indolenines, a common motif in many natural indole alkaloids. An antimicrobial screen of the cyclization products identified one compound which selectively potentiates β-lactam antibiotics in methicillin-resistant S. aureus (MRSA), and re-sensitizes a variety of MRSA strains to β-lactams.
Co-reporter:Dr. Wenqing Xu;Dr. Wei Wang ;Dr. Xiang Wang
Angewandte Chemie 2015 Volume 127( Issue 33) pp:9682-9685
Publication Date(Web):
DOI:10.1002/ange.201503736
Abstract
A gold-catalyzed desilylative cyclization was developed for facile synthesis of bridged tetracyclic indolenines, a common motif in many natural indole alkaloids. An antimicrobial screen of the cyclization products identified one compound which selectively potentiates β-lactam antibiotics in methicillin-resistant S. aureus (MRSA), and re-sensitizes a variety of MRSA strains to β-lactams.
Co-reporter:Le Chang ; Jessica D. Podoll ; Wei Wang ; Shane Walls ; Courtney P. O’Rourke
Journal of Medicinal Chemistry 2014 Volume 57(Issue 9) pp:3803-3817
Publication Date(Web):April 2, 2014
DOI:10.1021/jm500146g
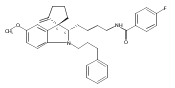
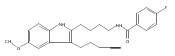
Previously we discovered a tricyclic indoline, N-[2-(6-bromo-4-methylidene-2,3,4,4a,9,9a-hexahydro-1H-carbazol-4a-yl)ethyl]-4-chlorobenzene-1-sulfonamide (1, Of1), from bioinspired synthesis of a highly diverse polycyclic indoline alkaloid library, that selectively resensitizes methicillin-resistant Staphylococcus aureus strains to β-lactam antibiotics. Herein, we report a thorough structure–activity relationship investigation of 1, which identified regions of 1 that tolerate modifications without compromising activity and afforded the discovery of a more potent analogue with reduced mammalian toxicity.
Co-reporter:P. Michael Barbour, Jessica D. Podoll, Laura J. Marholz, Xiang Wang
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 24) pp:5602-5605
Publication Date(Web):15 December 2014
DOI:10.1016/j.bmcl.2014.10.094
Co-reporter:Wenqing Xu ; Jessica D. Podoll ; Xuan Dong ; Anthony Tumber ; Udo Oppermann
Journal of Medicinal Chemistry 2013 Volume 56(Issue 12) pp:5198-5202
Publication Date(Web):May 30, 2013
DOI:10.1021/jm3018628
We previously reported methylstat as a selective inhibitor of jumonji C domain-containing histone demethylases (JHDMs). Herein, we describe the synthesis of a fluorescent analogue of methylstat and its application as a tracer in fluorescence polarization assays. Using this format, we have evaluated the binding affinities of several known JHDM probes, as well as the native cofactor and substrate of JHDM1A. This fluorophore allowed a highly robust and miniaturized competition assay sufficient for high-throughput screening.
Co-reporter:Jessica D. Podoll;Yongxiang Liu;Le Chang;Shane Walls;Wei Wang
PNAS 2013 Volume 110 (Issue 39 ) pp:15503-15504
Publication Date(Web):2013-09-24
DOI:10.1073/pnas.1310459110
The continuous emergence of resistant bacteria has become a major worldwide health threat. The current development of new
antibacterials has lagged far behind. To discover reagents to fight against resistant bacteria, we initiated a chemical approach
by synthesizing and screening a small molecule library, reminiscent of the polycyclic indole alkaloids. Indole alkaloids are
a class of structurally diverse natural products, many of which were isolated from plants that have been used as traditional
medicine for millennia. Specifically, we adapted an evolutionarily conserved biosynthetic strategy and developed a concise
and unified diversity synthesis pathway. Using this pathway, we synthesized 120 polycyclic indolines that contain 26 distinct
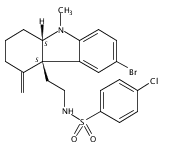
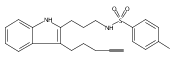
skeletons and a wide variety of functional groups. A tricyclic indoline, Of1, was discovered to selectively potentiate the
activity of β-lactam antibiotics in multidrug-resistant methicillin-resistant Staphylococcus aureus (MRSA), but not in methicillin-sensitive S. aureus. In addition, we found that Of1 itself does not have antiproliferative activity but can resensitize several MRSA strains
to the β-lactam antibiotics that are widely used in the clinic, such as an extended-spectrum β-lactam antibiotic amoxicillin/clavulanic
acid and a first-generation cephalosporin cefazolin. These data suggest that Of1 is a unique selective resistance-modifying
agent for β-lactam antibiotics, and it may be further developed to fight against resistant bacteria in the clinic.
Co-reporter:Se Jeong Yeo, Yongxiang Liu, Xiang Wang
Tetrahedron 2012 68(3) pp: 813-818
Publication Date(Web):
DOI:10.1016/j.tet.2011.11.032
Co-reporter:Xuelai Luo ; Yongxiang Liu ; Stefan Kubicek ; Johanna Myllyharju ; Anthony Tumber ; Stanley Ng ; Ka Hing Che ; Jessica Podoll ; Tom D. Heightman ; Udo Oppermann ; Stuart L. Schreiber
Journal of the American Chemical Society 2011 Volume 133(Issue 24) pp:9451-9456
Publication Date(Web):May 17, 2011
DOI:10.1021/ja201597b
Histone methylations are important chromatin marks that regulate gene expression, genomic stability, DNA repair, and genomic imprinting. Histone demethylases are the most recent family of histone-modifying enzymes discovered. Here, we report the characterization of a small-molecule inhibitor of Jumonji C domain-containing histone demethylases. The inhibitor derives from a structure-based design and preferentially inhibits the subfamily of trimethyl lysine demethylases. Its methyl ester prodrug, methylstat, selectively inhibits Jumonji C domain-containing his-tone demethylases in cells and may be a useful small-molecule probe of chromatin and its role in epigenetics.
Co-reporter:Yongxiang Liu, Wenqing Xu and Xiang Wang
Organic Letters 2010 Volume 12(Issue 7) pp:1448-1451
Publication Date(Web):March 2, 2010
DOI:10.1021/ol100153h
Two highly stereoselective cationic gold(I)-catalyzed tandem cyclization reactions of alkynylindoles are described. These reactions demonstrated a novel and general strategy to rapidly construct highly functionalized polycyclic indolines. This approach was successfully employed for a formal synthesis of the akuammiline alkaloid minfiensine.
Co-reporter:W. He, B. M. Griffiths, W. Wang and X. Wang
Organic & Biomolecular Chemistry 2017 - vol. 15(Issue 19) pp:NaN4245-4245
Publication Date(Web):2017/04/26
DOI:10.1039/C7OB00897J
Tricyclic indolines are common in both natural products and synthetic chemical probes. In this study we demonstrated that enantiomerically pure tricyclic indolines can be prepared from an inexpensive commercially available chiral starting material, pyroglutamic acid. The synthesis features a highly diastereoselective gold-catalyzed cyclization of alkyne-tethered indoles and subsequent diastereoselective reductive ring-opening reaction. Using this approach, we synthesized analogs of our previously discovered tricyclic indoline probes that possess antibacterial and resistance-modifying activity. The biological activity against methicillin-resistant Staphylococcus aureus (MRSA) of these analogues was evaluated and reported. The synthetic approach reported may be leveraged in the future to prepare diastereopure chemical probes for the determination of biological targets for drug discovery.
.jpg)


![4,7,10,13-Tetraoxa-16-azaheneicosanoicacid, 21-[(3aS,4S,6aR)-hexahydro-2-oxo-1H-thieno[3,4-d]imidazol-4-yl]-17-oxo-](http://img.cochemist.com/ccimg/721500/721431-18-1.png)
![4,7,10,13-Tetraoxa-16-azaheneicosanoicacid, 21-[(3aS,4S,6aR)-hexahydro-2-oxo-1H-thieno[3,4-d]imidazol-4-yl]-17-oxo-](http://img.cochemist.com/ccimg/721500/721431-18-1_b.png)
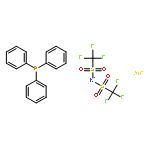
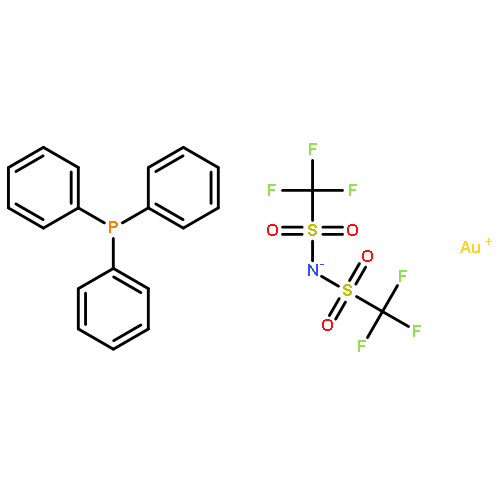


![Butanal, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/87200/87184-81-4.png)
![Butanal, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/87200/87184-81-4_b.png)






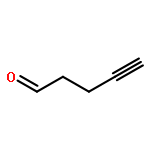
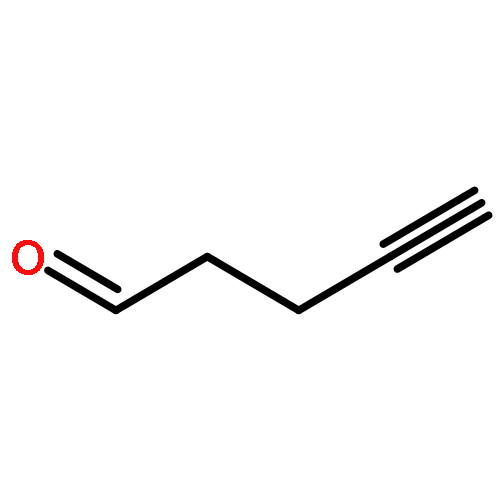




















































































































































































































































































































































































































































































































































![Benzenemethanol, 2-[(trimethylsilyl)ethynyl]-](http://img.cochemist.com/ccimg/229100/229027-92-3.png)
![Benzenemethanol, 2-[(trimethylsilyl)ethynyl]-](http://img.cochemist.com/ccimg/229100/229027-92-3_b.png)





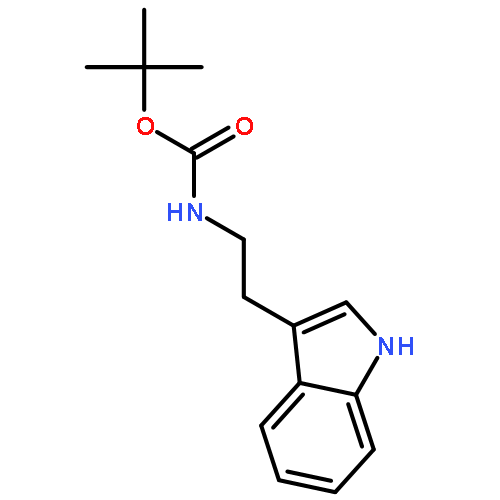

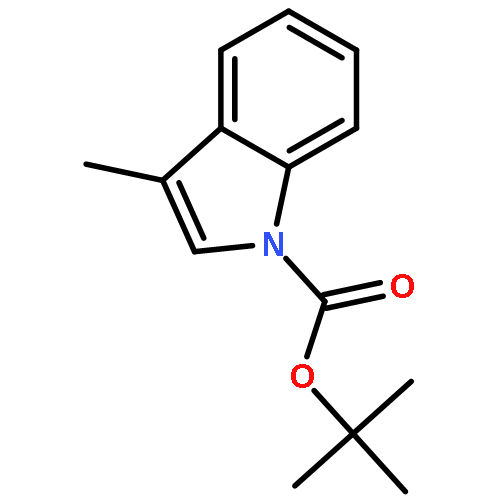


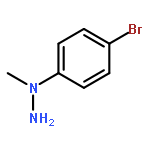



![N-[2-(5-methoxy-2-methyl-1H-indol-3-yl)ethyl]acetamide](http://img.cochemist.com/ccimg/69000/68935-42-2.png)
![N-[2-(5-methoxy-2-methyl-1H-indol-3-yl)ethyl]acetamide](http://img.cochemist.com/ccimg/69000/68935-42-2_b.png)



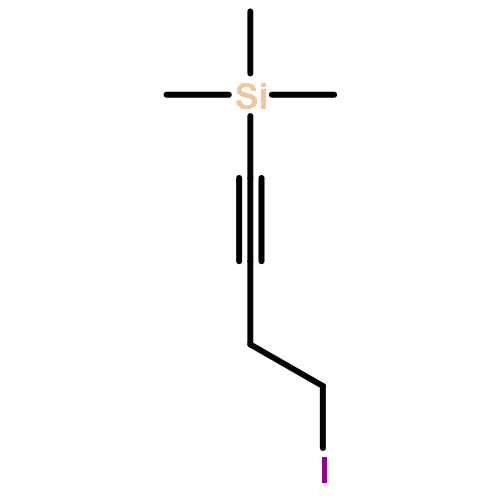
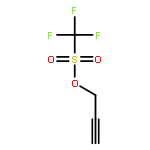
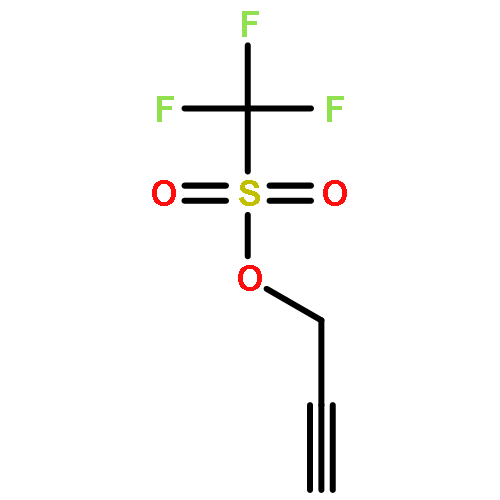
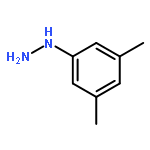















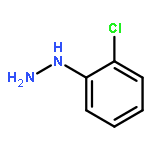



![N-[2-(1H-INDOL-3-YL)ETHYL]ACETAMIDE](http://img.cochemist.com/ccimg/1100/1016-47-3.png)
![N-[2-(1H-INDOL-3-YL)ETHYL]ACETAMIDE](http://img.cochemist.com/ccimg/1100/1016-47-3_b.png)








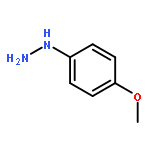
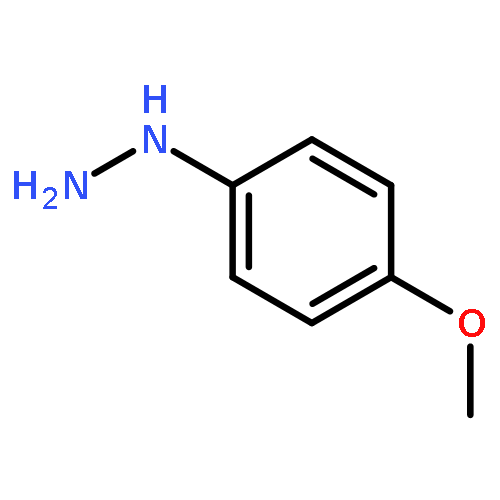


![4-Thia-1-azabicyclo[3.2.0]heptane-2-carboxylicacid, 6-[(2,6-dimethoxybenzoyl)amino]-3,3-dimethyl-7-oxo-, (2S,5R,6R)-](http://img.cochemist.com/ccimg/100/61-32-5.png)
![4-Thia-1-azabicyclo[3.2.0]heptane-2-carboxylicacid, 6-[(2,6-dimethoxybenzoyl)amino]-3,3-dimethyl-7-oxo-, (2S,5R,6R)-](http://img.cochemist.com/ccimg/100/61-32-5_b.png)