Co-reporter:Boning Wu, Min Liang, Mark Maroncelli, and Edward W. Castner Jr.
The Journal of Physical Chemistry B 2015 Volume 119(Issue 46) pp:14790-14799
Publication Date(Web):October 26, 2015
DOI:10.1021/acs.jpcb.5b09216
Ionic liquids with electron-donating anions are used to investigate rates and mechanisms of photoinduced bimolecular electron transfer to the photoexcited acceptor 9,10-dicyanoanthracene (9,10-DCNA). The set of five cyano anion ILs studied comprises the 1-ethyl-3-methylimidazolium cation paired with each of these five anions: selenocyanate, thiocyanate, dicyanamide, tricyanomethanide, and tetracyanoborate. Measurements with these anions dilute in acetonitrile and 1-ethyl-3-methylimidazolium bis(trifluoromethylsulfonyl)amide show that the selenocyanate and tricyanomethanide anions are strong quenchers of the 9,10-DCNA fluorescence, thiocyanate is a moderately strong quencher, dicyanamide is a weak quencher, and no quenching is observed for tetracyanoborate. Quenching rates are obtained from both time-resolved fluorescence transients and time-integrated spectra. Application of a Smoluchowski diffusion-and-reaction model showed that the complex kinetics observed can be fit using only two adjustable parameters, D and V0, where D is the relative diffusion coefficient between donor and acceptor and V0 is the value of the electronic coupling at donor–acceptor contact.
Co-reporter:Sufia Khatun and Edward W. Castner Jr.
The Journal of Physical Chemistry B 2015 Volume 119(Issue 29) pp:9225-9235
Publication Date(Web):November 17, 2014
DOI:10.1021/jp509861g
Intermolecular interactions between a Ru2+(bpy)3 solute and the anions and cations of four different ionic liquids (ILs) are investigated by 2D NMR nuclear Overhauser effect (NOE) techniques, including {1H–19F} HOESY and {1H–1H} ROESY. Four ILs are studied, each having the same bis(trifluoromethylsulfonyl)amide anion in common. Two of the ILs have aliphatic 1-alkyl-1-methylpyrrolidinium cations, while the other two ILs have aromatic 1-alkyl-3-methylimidazolium cations. ILs with both shorter (butyl) and longer (octyl or decyl) cationic alkyl substituents are studied. NOE NMR results suggest that the local environment of IL anions and cations near the Ru2+(bpy)3 solute is rather different from the bulk IL structure. The solute–anion and solute–cation interactions are significantly different both for ILs with short vs long alkyl tails and for ILs with aliphatic vs aromatic cation polar head groups. In particular, the solute–anion interactions are observed to be about 3 times stronger for the cations with shorter alkyl tails relative to the ILs with longer alkyl tails. The Ru2+(bpy)3 solute interacts with both the polar head and the nonpolar tail groups of the 1-butyl-1-methylpyrrolidinium cation but only with the nonpolar tail groups of the 1-decyl-1-methylpyrrolidinium cation.
Co-reporter:Jessalyn A. DeVine, Marena Labib, Megan E. Harries, Rouba Abdel Malak Rached, Joseph Issa, James F. Wishart, and Edward W. Castner Jr.
The Journal of Physical Chemistry B 2015 Volume 119(Issue 34) pp:11336-11345
Publication Date(Web):June 15, 2015
DOI:10.1021/acs.jpcb.5b03320
Intramolecular photoinduced electron transfer from an N,N-dimethyl-p-phenylenediamine donor bridged by a diproline spacer to a coumarin 343 acceptor was studied using time-resolved fluorescence measurements in three ionic liquids and in acetonitrile. The three ionic liquids have the bis[(trifluoromethyl)sulfonyl]amide anion paired with the tributylmethylammonium, 1-butyl-1-methylpyrrolidinium, and 1-decyl-1-methylpyrrolidinium cations. The dynamics in the two-proline donor–bridge–acceptor complex are compared to those observed for the same donor and acceptor connected by a single proline bridge, studied previously by Lee et al. ( J. Phys. Chem. C 2012, 116, 5197). The increased conformational freedom afforded by the second bridging proline resulted in multiple energetically accessible conformations. The multiple conformations have significant variations in donor–acceptor electronic coupling, leading to dynamics that include both adiabatic and nonadiabatic contributions. In common with the single-proline bridged complex, the intramolecular electron transfer in the two-proline system was found to be in the Marcus inverted regime.
Co-reporter:Hemant K. Kashyap, Cherry S. Santos, Ryan P. Daly, Jeevapani J. Hettige, N. Sanjeeva Murthy, Hideaki Shirota, Edward W. Castner Jr., and Claudio J. Margulis
The Journal of Physical Chemistry B 2013 Volume 117(Issue 4) pp:1130-1135
Publication Date(Web):December 21, 2012
DOI:10.1021/jp311032p
X-ray scattering experiments and molecular dynamics simulations have been performed to investigate the structure of four room temperature ionic liquids (ILs) comprising the bis(trifluoromethylsulfonyl)amide (NTf2–) anion paired with the triethyloctylammonium (N2228+) and triethyloctylphosphonium (P2228+) cations and their isoelectronic diether analogs, the (2-ethoxyethoxy)ethyltriethylammonium (N222(2O2O2)+) and (2-ethoxyethoxy)ethyltriethylphosphonium (P222(2O2O2)+) cations. Agreement between simulations and experiments is good and permits a clear interpretation of the important topological differences between these systems. The first sharp diffraction peak (or prepeak) in the structure function S(q) that is present in the case of the liquids containing the alkyl-substituted cations is absent in the case of the diether substituted analogs. Using different theoretical partitioning schemes for the X-ray structure function, we show that the prepeak present in the alkyl-substituted ILs arises from polarity alternations between charged groups and nonpolar alkyl tails. In the case of the diether substituted ILs, we find considerable curling of tails. Anions can be found with high probability in two different environments: close to the cationic nitrogen (phosphorus) and also close to the two ether groups. For the two diether systems, anions are found in locations from which they are excluded in the alkyl-substituted systems. This removes the longer range (polar/nonpolar) pattern of alternation that gives rise to the prepeak in alkyl-substituted systems.
Co-reporter:Hemant K. Kashyap, Cherry S. Santos, N. Sanjeeva Murthy, Jeevapani J. Hettige, Kijana Kerr, Sharon Ramati, JinHee Gwon, Masao Gohdo, Sharon I. Lall-Ramnarine, James F. Wishart, Claudio J. Margulis, and Edward W. Castner Jr.
The Journal of Physical Chemistry B 2013 Volume 117(Issue 49) pp:15328-15337
Publication Date(Web):June 10, 2013
DOI:10.1021/jp403518j
X-ray scattering and molecular dynamics simulations have been carried out to investigate structural differences and similarities in the condensed phase between pyrrolidinium-based ionic liquids paired with the bis(trifluoromethylsulfonyl)amide (NTf2–) anion where the cationic tail is linear, branched, or cyclic. This is important in light of the charge and polarity type alternations that have recently been shown to be present in the case of liquids with cations of moderately long linear tails. For this study, we have chosen to use the 1-alkyl-1-methylpyrrolidinium, Pyrr1,n+ with n = 5 or 7, as systems with linear tails, 1-(2-ethylhexyl)-1-methylpyrrolidinium, Pyrr1,EtHx+, as a system with a branched tail, and 1-(cyclohexylmethyl)-1-methylpyrrolidinium, Pyrr1,ChxMe+, as a system with a cyclic tail. We put these results into context by comparing these data with recently published results for the Pyrr1,n+/NTf2– ionic liquids with n = 4, 6, 8, and 10.1,2 General methods for interpreting the structure function S(q) in terms of q-dependent natural partitionings are described. This allows for an in-depth analysis of the scattering data based on molecular dynamics (MD) trajectories that highlight the effect of modifying the cationic tail.
Co-reporter:Heather Y. Lee, Hideaki Shirota, and Edward W. Castner Jr.
The Journal of Physical Chemistry Letters 2013 Volume 4(Issue 9) pp:1477-1483
Publication Date(Web):April 12, 2013
DOI:10.1021/jz400465x
Specific cation–anion interactions for two pairs of isoelectronic ionic liquids (ILs) have been investigated using nuclear Overhauser effect (NOE) 2D NMR methods to explore proximities between ions. The four ILs comprise the bis(trifluoromethylsulfonyl)amide anion paired with the following cations: triethyloctylammonium, (2-ethoxyethoxy)ethyltriethylammonium, triethyloctylphosphonium, and (2-ethoxyethoxy)ethyltriethylphosphonium. Substantial interactions are observed between the anion 19F nuclei and all of the protons on the triethyl chains for each of the four cationic head groups. Significantly different interactions are observed for each of the four cationic tail groups and the anions.Keywords: 2D NMR; HOESY; ion interactions; ionic liquid structure; NOE;
Co-reporter:Alessandro Triolo, Olga Russina, Ruggero Caminiti, Hideaki Shirota, Heather Y. Lee, Cherry S. Santos, N. Sanjeeva Murthy and Edward W. Castner, Jr
Chemical Communications 2012 vol. 48(Issue 41) pp:4959-4961
Publication Date(Web):21 Mar 2012
DOI:10.1039/C2CC31550E
X-ray scattering data from four pairs of ionic liquids (ILs) are compared. The alkyl-substituted cations show a first sharp diffraction peak between 3 and 4 nm−1 that is not observed for ILs having cations with ether- or hydroxy-substitutions. These observations indicate a significant difference in the intermediate range order for these liquids.
Co-reporter:Heather Y. Lee ; Joseph B. Issa ; Stephan S. Isied ; Edward W. Castner ; Jr.; Yunfeng Pan ; Charles L. Hussey ; Kwang Soon Lee ;James F. Wishart
The Journal of Physical Chemistry C 2012 Volume 116(Issue 8) pp:5197-5208
Publication Date(Web):January 25, 2012
DOI:10.1021/jp208852r
The effect of ionic liquids on photoinduced electron-transfer reactions in a donor–bridge–acceptor system is examined for two ionic liquid solvents, 1-butyl-1-methylpyrrolidinium bis(trifluoromethylsulfonyl)amide and tributylmethylammonium bis(trifluoromethylsulfonyl)amide. The results are compared with those for the same system in methanol and acetonitrile solution. Electron-transfer rates were measured using time-resolved fluorescence quenching for the donor–bridge–acceptor system comprising a 1-N,1-N-dimethylbenzene-1,4-diamine donor, a proline bridge, and a coumarin 343 acceptor. The photoinduced electron-transfer processes are in the inverted regime (−ΔG > λ) in all four solvents, with driving forces of −1.6 to −1.9 eV and estimated reorganization energies of about 1.0 eV. The observed electron-transfer kinetics have broadly distributed rates that are generally slower in the ionic liquids compared to the neutral solvents, which also have narrower rate distributions. To describe the broad distributions of electron-transfer kinetics, we use two different models: a distribution of exponential lifetimes and a discrete sum of exponential lifetimes. Analysis of the donor–acceptor electronic coupling shows that for ionic liquids this intramolecular electron-transfer reaction should be treated using a solvent-controlled electron-transfer model.
Co-reporter:Alessandro Triolo, Olga Russina, Ruggero Caminiti, Hideaki Shirota, Heather Y. Lee, Cherry S. Santos, N. Sanjeeva Murthy and Edward W. Castner, Jr
Chemical Communications 2012 - vol. 48(Issue 41) pp:NaN4961-4961
Publication Date(Web):2012/03/21
DOI:10.1039/C2CC31550E
X-ray scattering data from four pairs of ionic liquids (ILs) are compared. The alkyl-substituted cations show a first sharp diffraction peak between 3 and 4 nm−1 that is not observed for ILs having cations with ether- or hydroxy-substitutions. These observations indicate a significant difference in the intermediate range order for these liquids.
.jpg)









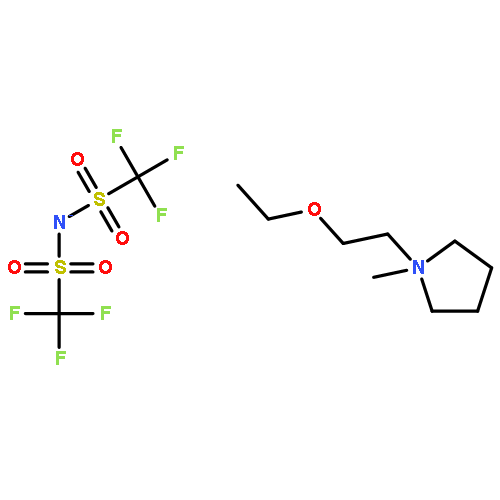








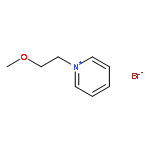
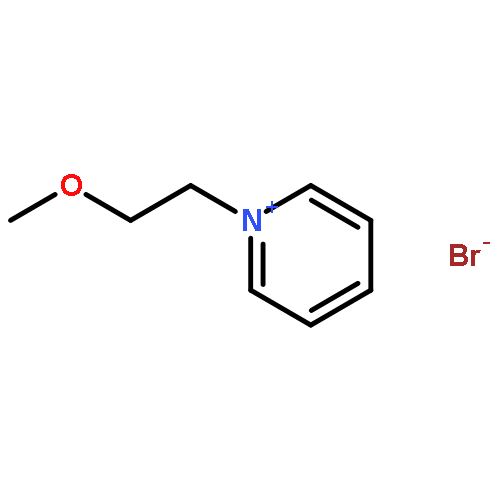
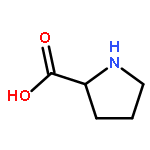





![11-oxo-2,3,5,6,7,11-Hexahydro-1H-pyrano[2,3-f]pyrido[3,2,1-ij]quinoline-10-carboxylic acid](/data/chemimg/86700/55804-65-4.png)
![11-oxo-2,3,5,6,7,11-Hexahydro-1H-pyrano[2,3-f]pyrido[3,2,1-ij]quinoline-10-carboxylic acid](/data/chemimg/86700/55804-65-4_b.png)


![1H,5H,11H-[1]Benzopyrano[6,7,8-ij]quinolizin-11-one, 2,3,6,7-tetrahydro-9-(trifluoromethyl)-](/data/chemimg/905400/53518-18-6.png)
![1H,5H,11H-[1]Benzopyrano[6,7,8-ij]quinolizin-11-one, 2,3,6,7-tetrahydro-9-(trifluoromethyl)-](/data/chemimg/905400/53518-18-6_b.png)