Co-reporter:Yetsedaw A. Tsegaw, Pritam E. Kadam, Niklas Tötsch, Elsa Sanchez-Garcia, and Wolfram Sander
Journal of the American Chemical Society September 6, 2017 Volume 139(Issue 35) pp:12310-12310
Publication Date(Web):August 8, 2017
DOI:10.1021/jacs.7b06868
p-Tolyl(trifluoromethyl)carbene and the related fluorenyl(trifluoromethyl)carbene were synthesized in solid argon and characterized by IR, UV–vis, and electron paramagnetic resonance spectroscopy as well as by quantum mechanical calculations. The carbenes can be generated in both their triplet and singlet states, and both states coexist under the conditions of matrix isolation. According to our calculations, the singlet and triplet states of these carbenes are energetically nearly degenerate in the gas phase. Warming of matrices containing pure triplet p-tolyl(trifluoromethyl)carbene from 3 to 25 K leads to an interconversion of up to 20% of the triplet into the singlet state. This interconversion is thermally irreversible, and cooling back to 3 K does not change the singlet to triplet ratio. Irradiation at 365 nm results in a complete singlet to triplet interconversion, whereas 450 nm irradiation produces again up to 20% of the singlet state. An alternative way to generate the singlet carbene is the reaction of the triplet with water molecules by annealing water-doped matrices at 25 K. This results in the irreversible formation of a hydrogen-bonded complex between the singlet carbene and water. For fluorenyl(trifluoromethyl)carbene, very similar results are obtained, but the yield of the singlet state is even higher. Magnetic bistability of carbenes seems to be a general phenomenon that only depends on the singlet–triplet gap rather than on the nature of the carbene.
Co-reporter:Paolo Costa, Iris Trosien, Joel Mieres-Perez, and Wolfram Sander
Journal of the American Chemical Society September 20, 2017 Volume 139(Issue 37) pp:13024-13024
Publication Date(Web):August 22, 2017
DOI:10.1021/jacs.7b05807
The reaction of triplet tetrachlorocyclopentadienylidene with BF3 in rare gas matrices yields a zwitterion consisting of a cyclopentadienyl cation bearing a positive charge and a negatively charged BF3 unit. IR and UV–vis spectra as well as the absence of EPR signals demonstrate a singlet ground state of the zwitterion, and its calculated geometry and magnetic properties clearly reveal a strong antiaromatic character. The zwitterion is highly labile and by visible or IR irradiation rearranges via a 1,2-fluorine migration from boron to carbon. Interaction with a second molecule of BF3 stabilizes the zwitterion and suppresses the fluorine migration, thus providing a convenient and efficient synthesis of an antiaromatic molecule under very mild conditions.
Co-reporter:Yetsedaw A. Tsegaw, Sándor Góbi, Marko Förstel, Pavlo Maksyutenko, Wolfram Sander, and Ralf I. Kaiser
The Journal of Physical Chemistry A October 12, 2017 Volume 121(Issue 40) pp:7477-7477
Publication Date(Web):September 11, 2017
DOI:10.1021/acs.jpca.7b07500
We irradiated binary ice mixtures of ammonia (NH3) and oxygen (O2) ices at astrophysically relevant temperatures of 5.5 K with energetic electrons to mimic the energy transfer process that occurs in the track of galactic cosmic rays. By monitoring the newly formed molecules online and in situ utilizing Fourier transform infrared spectroscopy complemented by temperature-programmed desorption studies with single-photon photoionization reflectron time-of-flight mass spectrometry, the synthesis of hydroxylamine (NH2OH), water (H2O), hydrogen peroxide (H2O2), nitrosyl hydride (HNO), and a series of nitrogen oxides (NO, N2O, NO2, N2O2, N2O3) was evident. The synthetic pathway of the newly formed species, along with their rate constants, is discussed exploiting the kinetic fitting of the coupled differential equations representing the decomposition steps in the irradiated ice mixtures. Our studies suggest the hydroxylamine is likely formed through an insertion mechanism of suprathermal oxygen into the nitrogen–hydrogen bond of ammonia at such low temperatures. An isotope-labeled experiment examining the electron-irradiated D3-ammonia–oxygen (ND3–O2) ices was also conducted, which confirmed our findings. This study provides clear, concise evidence of the formation of hydroxylamine by irradiation of interstellar analogue ices and can help explain the question how potential precursors to complex biorelevant molecules may form in the interstellar medium.
Co-reporter:Soumya Radhakrishnan, Joel Mieres-Perez, Murthy S. Gudipati, and Wolfram Sander
The Journal of Physical Chemistry A August 31, 2017 Volume 121(Issue 34) pp:6405-6405
Publication Date(Web):August 3, 2017
DOI:10.1021/acs.jpca.7b05466
The benzhydryl radical is generated in high yields by flash-vacuum thermolysis of 1,1,2,2-tetraphenylethane with subsequent trapping of the product in argon or amorphous water at 3–4 K. Photoionization of the radical with various UV lights and electron sources produces the benzhydryl cation, which was identified by IR and UV–vis spectroscopy. In solid argon, the formation of the benzhydryl cation is irreversible, whereas in amorphous water–ice the electron transfer is reversible, and irradiation into the major absorption band at 443 nm of the cation leads back to the radical by electron attachment. Applications of ionization of organic matter trapped in water–ice to icy environments in astrophysics and planetary sciences, including Earth, are discussed.
Co-reporter:Paolo Costa; Thomas Lohmiller; Iris Trosien; Anton Savitsky; Wolfgang Lubitz; Miguel Fernandez-Oliva; Elsa Sanchez-Garcia
Journal of the American Chemical Society 2016 Volume 138(Issue 5) pp:1622-1629
Publication Date(Web):January 14, 2016
DOI:10.1021/jacs.5b11696
Bis(p-methoxyphenyl)carbene is the first carbene that at cryogenic temperatures can be isolated in both its lowest energy singlet and triplet states. At 3 K, both states coexist indefinitely under these conditions. The carbene is investigated in argon matrices by IR, UV–vis, and X-band EPR spectroscopy and in MTHF glasses by W-band EPR and Q-band ENDOR spectroscopy. UV (365 nm) irradiation of the system results in formation of predominantly the triplet carbene, whereas visible (450 nm) light shifts the photostationary equilibrium toward the singlet state. Upon annealing at higher temperatures (>10 K), the triplet is converted to the singlet; however, cooling back to 3 K does not restore the triplet. Therefore, depending on matrix temperature and irradiation conditions, matrices containing predominantly the triplet or singlet carbene can be generated. Controlling the magnetic and chemical properties of carbenes by using light of different wavelengths might be of general interest for applications such as information storage and radical-initiated polymerization processes.
Co-reporter:Stefan Henkel; Paolo Costa; Linda Klute; Pandian Sokkar; Miguel Fernandez-Oliva; Walter Thiel; Elsa Sanchez-Garcia
Journal of the American Chemical Society 2016 Volume 138(Issue 5) pp:1689-1697
Publication Date(Web):January 13, 2016
DOI:10.1021/jacs.5b12726
The interactions between diphenylcarbene DPC and the halogen bond donors CF3I and CF3Br were investigated using matrix isolation spectroscopy (IR, UV–vis, and EPR) in combination with QM and QM/MM calculations. Both halogen bond donors CF3X form very strong complexes with the singlet state of DPC, but only weakly interact with triplet DPC. This results in a switching of the spin state of DPC, the singlet complexes becoming more stable than the triplet complexes. CF3I forms a second complex (type II) with DPC that is thermodynamically slightly more stable. Calculations predict that in this second complex the DPC···I distance is shorter than the F3C···I distance, whereas in the first (type I) complex the DPC···I distance is, as expected, longer. CF3Br only forms the type I complex. Upon irradiation I or Br, respectively, are transferred to the DPC carbene center and radical pairs are formed. Finally, on annealing, the formal C–X insertion product of DPC is observed. Thus, halogen bonding is a powerful new principle to control the spin state of reactive carbenes.
Co-reporter:Wolfram Ser
Israel Journal of Chemistry 2016 Volume 56( Issue 1) pp:62-65
Publication Date(Web):
DOI:10.1002/ijch.201500047
Abstract
Physical organic chemistry plays a unifying role in chemistry by combining a large variety of experimental and theoretical techniques to create simplified, intuitively understandable concepts of chemical processes. The concept of “antiaromaticity” is used to demonstrate how complex experiments may result in general models that allow chemists to predict the chemical and physical properties of difficult to access molecules and even of transition states. Cyclooctatetraene and the fluorenyl cation are used as examples to illustrate some working methods of physical organic chemistry.
Co-reporter:Geneviève Richter, Enrique Mendez-Vega, and Wolfram Sander
The Journal of Physical Chemistry A 2016 Volume 120(Issue 20) pp:3524-3532
Publication Date(Web):April 27, 2016
DOI:10.1021/acs.jpca.6b02550
Chlorophenylcarbene and fluorophenylcarbene were generated in water-doped argon matrices at cryogenic temperatures by photolysis of the corresponding matrix-isolated diazirines. When diffusion of H2O in solid argon was induced by annealing of the matrices at temperatures above 20 K, hydrogen-bonded complexes between the carbenes and water were formed. UV photolysis of these complexes resulted in the formation of benzaldehyde and hydrogen halides HX. The same products were obtained after photolysis of the diazirines in amorphous water ice. Obviously, the primary insertion product of the carbenes into H–OH is unstable under these conditions, and benzaldehyde is formed via secondary photolysis. The stable primary photochemical insertion product of chlorophenylcarbene into an O–H bond was observed in the reaction of the carbene with methanol.
Co-reporter:Paolo Costa ;Wolfram Ser
Journal of Physical Organic Chemistry 2015 Volume 28( Issue 2) pp:71-74
Publication Date(Web):
DOI:10.1002/poc.3355
The photochemistry of diphenylcarbene 5, matrix-isolated in argon at 3–5 K was investigated by UV–Vis, IR, and EPR spectroscopy. Although carbene 5 proved to be stable toward broadband UV irradiation, it slowly rearranges to 1-phenyl-1,2,4,6-cycloheptatetraene 8 during irradiation with the intense light of a 445-nm diode laser. Other intermediates were not observed. Allene 8 is the key intermediate that was missing in the proposed mechanism of the rearrangement of 5 to fluorene 7. Copyright © 2014 John Wiley & Sons, Ltd.
Co-reporter:Dr. Stefan Henkel ;Dr. Wolfram Ser
Angewandte Chemie 2015 Volume 127( Issue 15) pp:4686-4690
Publication Date(Web):
DOI:10.1002/ange.201410501
Abstract
Carbenes are among the few metal-free molecules that are able to activate molecular hydrogen. Whereas triplet carbenes have been shown to insert into H2 through a two-step mechanism that at low temperature is assisted by quantum mechanical tunneling (QMT), singlet carbenes insert in concerted reactions with considerable activation barriers, and are thus unreactive towards H2 at cryogenic temperatures. Here we show that 1-azulenylcarbene with a singlet ground state readily inserts into H2, and that QMT governs the insertion into both H2 and D2. This is the first example that shows that QMT can also be important for singlet carbenes inserting into dihydrogen.
Co-reporter:Dr. Bishnu Prasad Kar ;Dr. Wolfram Ser
ChemPhysChem 2015 Volume 16( Issue 17) pp:3603-3606
Publication Date(Web):
DOI:10.1002/cphc.201500729
Abstract
The role of N-heterocyclic carbenes in the chemistry of ionic liquids based on imidazolium salts has long been discussed. Here, we present experimental evidence that 1-ethyl-3-methylimidazolium-2-ylidene (EMIm) can coexist with its protonated imidazolium cation (EMImH+) at low temperatures. If the vapor of the ionic liquid [EMImH+][AcO−] is trapped in solid argon or nitrogen at 9 K, only acetic acid (AcOH) and the carbene, but no ionic species, are found by IR spectroscopy. This indicates that during the evaporation of [EMImH+][AcO−] proton transfer occurs to form the neutral species. If the vapor of [EMImH+][AcO−] is trapped at 9 K as film in the absence of a host matrix, a solid consisting of EMImH+, EMIm, AcO−, and AcOH is formed. During warming to room temperature the proton transfer in the solid to form back the IL [EMImH+][AcO−] can be monitored by IR spectroscopy. This clearly demonstrates that evaporation and condensation of the IL [EMImH+][AcO−] results in a double proton transfer, and the carbene EMIm is only metastable even at low temperatures.
Co-reporter:Dr. Stefan Henkel ;Dr. Wolfram Ser
Angewandte Chemie International Edition 2015 Volume 54( Issue 15) pp:4603-4607
Publication Date(Web):
DOI:10.1002/anie.201410501
Abstract
Carbenes are among the few metal-free molecules that are able to activate molecular hydrogen. Whereas triplet carbenes have been shown to insert into H2 through a two-step mechanism that at low temperature is assisted by quantum mechanical tunneling (QMT), singlet carbenes insert in concerted reactions with considerable activation barriers, and are thus unreactive towards H2 at cryogenic temperatures. Here we show that 1-azulenylcarbene with a singlet ground state readily inserts into H2, and that QMT governs the insertion into both H2 and D2. This is the first example that shows that QMT can also be important for singlet carbenes inserting into dihydrogen.
Co-reporter:Joel Mieres-Pérez, Enrique Mendez-Vega, Kavitha Velappan, and Wolfram Sander
The Journal of Organic Chemistry 2015 Volume 80(Issue 24) pp:11926-11931
Publication Date(Web):November 2, 2015
DOI:10.1021/acs.joc.5b01263
Triplet carbenes react with molecular oxygen with rates that approach diffusion control to carbonyl O-oxides, whereas triplet nitrenes react much slower. For investigating the reaction of phenylnitrene with O2, the nitrene was generated by flash vacuum thermolysis (FVT) of phenylazide and subsequently isolated in O2-doped matrices. FVT of the azide produces the nitrene in high yield and with only minor contaminations of the rearranged products that are frequently observed if the nitrene is produced by photolysis. The phenylnitrene was isolated in solid Ar, Xe, mixtures of these rare gases with O2, and even in pure solid O2. At temperatures between 30 and 35 K an extremely slow thermal reaction between the nitrene and O2 was observed, whereas at higher temperatures, solid Ar and O2 rapidly evaporate. Only O2-doped Xe matrices allowed us to anneal at temperatures above 40 K, and at these temperatures, the nitrene reacts with O2 to produce nitroso O-oxide mainly in its syn conformation. Upon visible light irradiation (450 nm), the nitroso oxide rapidly rearranges to nitrobenzene.
Co-reporter:Paolo Costa ; Miguel Fernandez-Oliva ; Elsa Sanchez-Garcia
Journal of the American Chemical Society 2014 Volume 136(Issue 44) pp:15625-15630
Publication Date(Web):September 7, 2014
DOI:10.1021/ja507894x
Diphenylcarbene (DPC) shows a triplet ground-state lying approximately 3 kcal/mol below the lowest singlet state. Under the conditions of matrix isolation at 25 K, DPC reacts with single water molecules embedded in solid argon and switches its ground state from triplet to singlet by forming a strong hydrogen bond. The complex between DPC and water is only metastable, and even at 3 K the carbene center slowly inserts into the OH bond of water to form benzhydryl alcohol via quantum chemical tunneling. Surprisingly, if DPC is generated in amorphous water ice at 3 K, it is protonated instantaneously to give the benzhydryl cation. Under these conditions, the benzhydryl cation is stable, and warming to temperatures above 50 K is required to produce benzhydryl alcohol. Thus, for the first time, a highly electrophilic and extremely reactive secondary carbenium ion can be isolated in a neutral, nucleophilic environment avoiding superacidic conditions.
Co-reporter:Rachel Crespo-Otero, Artur Mardykov, Elsa Sanchez-Garcia, Wolfram Sander and Mario Barbatti
Physical Chemistry Chemical Physics 2014 vol. 16(Issue 35) pp:18877-18887
Publication Date(Web):28 Jul 2014
DOI:10.1039/C4CP02518K
The formation of weakly-bound dimers of N-methylformamide (NMF) and the photochemistry of these dimers after irradiation at 248 nm were explored using matrix-isolation spectroscopy. Calculations were used to characterize the diverse isomers and assign their IR spectra; non-adiabatic dynamics was simulated to understand their photo-deactivation mechanism. The most stable dimers, tt-1 and tt-2, were obtained by trans–trans aggregation (N–H⋯OC interactions) and could be identified in the matrix. The main products formed after irradiation are the trans–cis dimers (tc-3 and tc-4), also stabilized by N–H⋯OC interactions. In contrast to the photochemistry of the monomers, no dissociative products were observed after 248 nm irradiation of the dimers. The absence of dissociative products can be explained by a proton-transfer mechanism in the excited state that is faster than the photo-dissociative mechanism. The fact that hydrogen bonding has such a significant effect on the photochemical stability of NMF has important implications to understand the stability of peptide-bonded systems to UV irradiation.
Co-reporter:Dr. Wolfram Ser
Angewandte Chemie International Edition 2014 Volume 53( Issue 2) pp:362-364
Publication Date(Web):
DOI:10.1002/anie.201305736
Co-reporter:Dr. Wolfram Ser
Angewandte Chemie 2014 Volume 126( Issue 2) pp:370-372
Publication Date(Web):
DOI:10.1002/ange.201305736
Co-reporter:Paolo Costa ;Dr. Wolfram Ser
Angewandte Chemie 2014 Volume 126( Issue 20) pp:5222-5225
Publication Date(Web):
DOI:10.1002/ange.201400176
Abstract
Spin specificity is one of the most important properties of carbenes in their reactions. Alcohols are typically used to probe the reactive spin states of carbenes: OH insertions are assumed to be characteristic of singlet states, whereas CH insertions are typical for the triplets. Surprisingly, the experiments presented here suggest that the spin ground state of diphenylcarbene 1 switches from triplet to singlet if the carbene is allowed to interact with methanol. Carbene 1 and methanol form a strongly hydrogen-bonded singlet ground state complex that was synthesized in low-temperature matrices and characterized by IR spectroscopy. This methanol complex is only metastable, and even at 3 K slowly rearranges to form the product of OH insertion through quantum chemical tunneling. Thus, the ground state triplet (in the gas phase) carbene 1 forms exclusively the products expected from a singlet carbene. Whereas the assumption of spin specific reactions of carbenes is correct, the spin state itself can be changed by solvent interactions, and therefore widely accepted conclusions drawn from earlier experiments have to be revisited.
Co-reporter:Sergei V. Chapyshev, Evgeny N. Ushakov, Patrik Neuhaus, and Wolfram Sander
The Journal of Organic Chemistry 2014 Volume 79(Issue 13) pp:6047-6053
Publication Date(Web):June 6, 2014
DOI:10.1021/jo500677g
The key intermediates of decomposition of high-energy 2,4,6-triazidopyrimidine and its 5-chloro-substituted derivative, the detonation of which is used for preparation of carbon nitrides, were investigated using electron paramagnetic resonance (EPR) spectroscopy in combination with quantum chemical calculations. The decomposition of the triazides was carried out photochemically, using the matrix isolation technique. The photodecomposition of both triazides with 254 nm light in argon matrices at 5 K occurred selectively to subsequently give the corresponding triplet 4,6-diazido-2-nitrenopyrimidines, quintet 4-azido-2,6-dinitrenopyrimidines, and septet 2,4,6-trinitrenopyrimidines. The latter were photochemically unstable and decomposed to form triplet nitrenes NCN and NNC as well as triplet carbenes NCCCN, HCCN, and HCCCCN. The results obtained provide important information about exchange interactions in high-spin nitrenes with the pyrimidine ring and the mechanism of the formation of carbon nitrides during thermolysis of 2,4,6-triazidopyrimidine.
Co-reporter:Melanie Ertelt;Dr. David A. Hrovat;Dr. Weston Thatcher Borden;Dr. Wolfram Ser
Chemistry - A European Journal 2014 Volume 20( Issue 16) pp:4713-4720
Publication Date(Web):
DOI:10.1002/chem.201303792
Abstract
The highly strained 1H-bicyclo[3.1.0]-hexa-3,5-dien-2-one 1 is metastable, and rearranges to 4-oxacyclohexa-2,5-dienylidene 2 in inert gas matrices (neon, argon, krypton, xenon, and nitrogen) at temperatures as low as 3 K. The kinetics for this rearrangement show pronounced matrix effects, but in a given matrix, the reaction rate is independent of temperature between 3 and 20 K. This temperature independence means that the activation energy is zero in this temperature range, indicating that the reaction proceeds through quantum mechanical tunneling from the lowest vibrational level of the reactant. At temperatures above 20 K, the rate increases, resulting in curved Arrhenius plots that are also indicative of thermally activated tunneling. These experimental findings are supported by calculations performed at the CASSCF and CASPT2 levels by using the small-curvature tunneling (SCT) approximation.
Co-reporter:Stefan Henkel;Melanie Ertelt ;Dr. Wolfram Ser
Chemistry - A European Journal 2014 Volume 20( Issue 25) pp:7585-7588
Publication Date(Web):
DOI:10.1002/chem.201402064
Abstract
4-Oxocyclohexa-2,5-dienylidene is a highly reactive triplet ground state carbene that is hydrogenated in solid H2, HD, and D2 at temperatures as low as 3 K. The mechanism of the insertion of the carbene into dihydrogen was investigated by IR and EPR spectroscopy and by kinetic studies. H or D atoms were observed as products of the reaction with H2 and D2, respectively, whereas HD produces exclusively D atoms. The hydrogenation shows a very large kinetic isotope effect and remarkable isotope selectivity, as was expected for a tunneling reaction. The experiments, therefore, provide clear evidence for both hydrogen tunneling and the rare deuterium tunneling in an intermolecular reaction.
Co-reporter:Dr. Wolfram Ser;Dr. Saonli Roy;Kenny Bravo-Rodriguez;Dr. Dirk Grote;Dr. Elsa Sanchez-Garcia
Chemistry - A European Journal 2014 Volume 20( Issue 40) pp:12917-12923
Publication Date(Web):
DOI:10.1002/chem.201402459
Abstract
The benzyl radical (1) is a key intermediate in the combustion and tropospheric oxidation of toluene. Because of its relevance, the reaction of 1 with molecular oxygen was investigated by matrix-isolation IR and EPR spectroscopy as well as computational methods. The primary reaction product of 1 and O2 is the benzylperoxyl radical (2), which exists in several conformers that can easily interconvert even at cryogenic temperatures. Photolysis of radical 2 at 365 nm results in a formal [1,3]-H migration and subsequent cleavage of the OO bond to produce a hydrogen-bonded complex between the hydroxyl radical and benzaldehyde (4). Prolonged photolysis produces the benzoyl radical (5) and water, which finally yield the phenyl radical (7), CO, and H2O. Thus, via a sequence of exothermic reactions 1 is transformed into radicals of even higher reactivity, such as OH and 7. Our results have implications for the development of models for the highly complicated process of combustion of aromatic compounds.
Co-reporter:Paolo Costa ;Dr. Wolfram Ser
Angewandte Chemie International Edition 2014 Volume 53( Issue 20) pp:5122-5125
Publication Date(Web):
DOI:10.1002/anie.201400176
Abstract
Spin specificity is one of the most important properties of carbenes in their reactions. Alcohols are typically used to probe the reactive spin states of carbenes: OH insertions are assumed to be characteristic of singlet states, whereas CH insertions are typical for the triplets. Surprisingly, the experiments presented here suggest that the spin ground state of diphenylcarbene 1 switches from triplet to singlet if the carbene is allowed to interact with methanol. Carbene 1 and methanol form a strongly hydrogen-bonded singlet ground state complex that was synthesized in low-temperature matrices and characterized by IR spectroscopy. This methanol complex is only metastable, and even at 3 K slowly rearranges to form the product of OH insertion through quantum chemical tunneling. Thus, the ground state triplet (in the gas phase) carbene 1 forms exclusively the products expected from a singlet carbene. Whereas the assumption of spin specific reactions of carbenes is correct, the spin state itself can be changed by solvent interactions, and therefore widely accepted conclusions drawn from earlier experiments have to be revisited.
Co-reporter:Muhammad Sajid ; Arunlibertsen Lawzer ; Weishi Dong ; Christoph Rosorius ; Wolfram Sander ; Birgitta Schirmer ; Stefan Grimme ; Constantin G. Daniliuc ; Gerald Kehr ;Gerhard Erker
Journal of the American Chemical Society 2013 Volume 135(Issue 49) pp:18567-18574
Publication Date(Web):October 22, 2013
DOI:10.1021/ja408815k
The intramolecular frustrated Lewis pair (FLP) Mes2PCH2CH2B(C6F5)2 4 adds cooperatively to carbon monoxide to form the five-membered heterocyclic carbonyl compound 5. The intramolecular FLP 7 contains an exo-3-B(C6F5)2 Lewis acid and an endo-2-PMes2 Lewis base functionality coordinated at the norbornane framework. This noninteracting FLP adds carbon monoxide in solution at −35 °C cooperatively to yield a five-membered heterocyclic FLP-carbonyl compound 8. In contrast, FLP 7 is carbonylated in a CO-doped argon matrix at 25 K to selectively form a borane carbonyl 9 without involvement of the adjacent phosphanyl moiety. The free FLP 7 was generated in the gas phase from its FLPH2 product 10. A DFT study has shown that the phosphonium hydrido borate zwitterion 10 is formed exergonically in solution but tends to lose H2 when brought into the gas phase.
Co-reporter:Fee Li, Kenny Bravo-Rodriguez, Charlotte Phillips, Rüdiger W. Seidel, Florian Wieberneit, Raphael Stoll, Nikos L. Doltsinis, Elsa Sanchez-Garcia, and Wolfram Sander
The Journal of Physical Chemistry B 2013 Volume 117(Issue 13) pp:3560-3570
Publication Date(Web):March 21, 2013
DOI:10.1021/jp4007334
The cyclic disulfide-bridged tetrapeptide cyclo(Boc-Cys-Pro-Gly-Cys-OMe) (1) was designed as a model for the study of solvent-driven conformational changes in peptides. The three-dimensional structure and dynamics of 1 were studied using a variety of experimental and computational techniques. The crystal structure of 1 reveals a β-turn stabilized by a hydrogen bond between the two cysteine residues. In solution, the UV–CD and NMR analysis of 1 suggest a β-turn II conformation, stable up to 60 °C. The characteristic NMR 13C shifts of the Cβ and Cγ atoms of proline show that the peptide adopts exclusively the energetically favored trans conformation of the peptidyl-prolyl bond. The combination of IR spectroscopy with Car–Parrinello MD simulations and DFT calculations allowed us to assign the absorptions in the amide I region to the individual amino acids. The NH group of Gly, which as hydrogen bond donor competes with the NH group of Cys4 for the carbonyl oxygen atom of Cys1 as hydrogen bond acceptor, plays a relevant role for the structure and spectroscopic properties of the peptide. Since Gly is more exposed to the solvent, its hydrogen-bonding capability can be partially blocked by external solvent molecules in solution or by a second peptide molecule in the crystal. Furthermore, the presence of only one molecule of acetonitrile is sufficient to change the preferred conformation of 1, and even in acetonitrile solution the simulations suggest that on average only one solvent molecule strongly interacts with the cyclic core of the peptide.
Co-reporter:Dr. Rachel Crespo-Otero;Dr. Artur Mardyukov;Dr. Elsa Sanchez-Garcia;Dr. Mario Barbatti;Dr. Wolfram Ser
ChemPhysChem 2013 Volume 14( Issue 4) pp:827-836
Publication Date(Web):
DOI:10.1002/cphc.201200573
Abstract
The photochemistry of N-methylformamide (MF) is elucidated by investigating its photodissociation products generated by UV irradiation (248 nm) in an argon matrix (10 K). We find that, starting from trans-MF, prolonged irradiation produces cis-MF, CH3NH2 and CO fragments as major products. Another photoproduct is identified as methylformimidic acid (FIA). Nonadiabatic dynamics simulations starting from both MF conformers revealed that the internal conversion occurs within 1 ps through a CN dissociation channel. The major product is a weakly bound complex between CH3NH and HCO radicals. This complex owes its existence to the cage effect of the matrix which allows for H-transfer reactions and recombination. By identifying the primary photoisomerization and photodissociation pathways of MF, we gain new insights into the photochemistry of peptide bonds in general, which is a prerequisite for a better understanding of the effect of UV irradiation on living systems.
Co-reporter: Dr. Gautam R. Desiraju; Dr. Mark A. Johnson; Dr. Wolfram Ser
ChemPhysChem 2013 Volume 14( Issue 4) pp:631-633
Publication Date(Web):
DOI:10.1002/cphc.201200980
No abstract is available for this article.
Co-reporter:Stefan Henkel ; Y-am Huynh ; Patrik Neuhaus ; Michael Winkler
Journal of the American Chemical Society 2012 Volume 134(Issue 32) pp:13204-13207
Publication Date(Web):July 16, 2012
DOI:10.1021/ja3050376
1-Azulenylcarbene was synthesized by photolysis of 1-azulenyldiazomethane in argon or neon matrices at 3–10 K. The highly polar singlet carbene is only metastable and undergoes a tunneling rearrangement to 8-methylene-bicyclo[5.3.0]deca-1,3,5,6,9-pentaene. After substitution of the 4 and 8 positions with deuterium, the rearrangement is completely inhibited. This indicates a very large kinetic isotope effect, as expected for a tunneling reaction.
Co-reporter:Wolfram Sander ; Saonli Roy ; Iakov Polyak ; Juan Manuel Ramirez-Anguita ;Elsa Sanchez-Garcia
Journal of the American Chemical Society 2012 Volume 134(Issue 19) pp:8222-8230
Publication Date(Web):April 6, 2012
DOI:10.1021/ja301528w
The phenoxyl radical 1 was generated in high yields by flash vacuum pyrolysis of allyl phenyl ether 2 with subsequent trapping of the products in argon at 3 K. In water-doped argon matrices, an OH···O complex between 1 and water is formed that could be characterized by IR spectroscopy. Several isotopomers of the complex were generated, and the IR spectra compared to results of density functional theory calculations. Other dimers between 1 and water were not found under these conditions. QM/MM calculations in simulated argon matrices reveal that an OH···π complex is unstable even at a time scale of picoseconds. This finding has implications on the related interaction between the tyrosyl radical and the water in biological systems.
Co-reporter:Dirk Grote;Christopher Finke;Patrik Neuhaus ;Wolfram Ser
European Journal of Organic Chemistry 2012 Volume 2012( Issue 17) pp:3229-3236
Publication Date(Web):
DOI:10.1002/ejoc.201101866
Abstract
The photochemistry of 3-iodo-2,5,6-trifluoropyridyl azide (6c), matrix-isolated in argon, was investigated by IR and EPR spectroscopy. The primary photoproduct is 3-iodo-2,5,6-trifluoropyridylnitrene (7c) in its triplet ground state. Further irradiation produced very low yields of nitrene radical 3c, which shows a characteristic EPR spectrum. The yield of 3c, however, was too low for IR detection. Instead, azirinyl radical 11, formed from 3c by ring closure, could clearly be identified in the IR spectra. Other products observed were azirine 8c′ and the ketenimine 9c′, which are formed in a photostationary equilibrium together with nitrine 7c.
Co-reporter:Christopher Finke;Dirk Grote;Rüdiger W. Seidel;Sergei V. Chapyshev;Wolfram Ser
Journal of Physical Organic Chemistry 2012 Volume 25( Issue 6) pp:486-492
Publication Date(Web):
DOI:10.1002/poc.1943
The photochemistry of 2,4,6-triazido-3,5-difluoropyridine 21 was investigated by matrix infrared and electron paramagnetic resonance spectroscopies. Ultraviolet irradiation (>260 nm) of 21 results in the formation of 3,5-difluoropyridyl-2,4,6-trinitrene 26 in yields high enough for characterization by infrared spectroscopy. The experimental infrared spectrum is in good agreement with density functional theory calculations. Under similar conditions, a very strong electron paramagnetic resonance spectrum of septet trinitrene 26 was obtained. Shorter irradiation times resulted in more complex product mixtures containing, in addition, mononitrenes and dinitrenes. Surprisingly, azirines and keteneimines, the typical photoproducts of arylnitrenes, were not observed. Copyright © 2011 John Wiley & Sons, Ltd.
Co-reporter:Dr. Sergei V. Chapyshev;Dr. Denis V. Korchagin;Dr. Mikhail F. Budyka;Dr. Tatiana N. Gavrishova;Dr. Patrik Neuhaus; Dr. Wolfram Ser
ChemPhysChem 2012 Volume 13( Issue 11) pp:2721-2728
Publication Date(Web):
DOI:10.1002/cphc.201200200
Abstract
The UV (λ>305 nm) photolysis of triazide 3 in 2-methyl-tetrahydrofuran glass at 7 K selectively produces triplet mononitrene 4 (g=2.003, DT=0.92 cm−1, ET=0 cm−1), quintet dinitrene 6 (g=2.003, DQ=0.204 cm−1, EQ=0.035 cm−1), and septet trinitrene 8 (g=2.003, DS=−0.0904 cm−1, ES=−0.0102 cm−1). After 45 min of irradiation, the major products are dinitrene 6 and trinitrene 8 in a ratio of ∼1:2, respectively. These nitrenes are formed as mixtures of rotational isomers each of which has slightly different magnetic parameters D and E. The best agreement between the line-shape spectral simulations and the experimental electron paramagnetic resonance (EPR) spectrum is obtained with the line-broadening parameters Γ(EQ)=180 MHz for dinitrene 6 and Γ(ES)=330 MHz for trinitrene 8. According to these line-broadening parameters, the variations of the angles Θ in rotational isomers of 6 and 8 are expected to be about ±1 and ±3°, respectively. Theoretical estimations of the magnetic parameters obtained from PBE/DZ(COSMO)//UB3LYP/6-311+G(d,p) calculations overestimate the E and D values by 1 and 8 %, respectively. Despite the large distances between the nitrene units and the extended π systems, the zero field splitting (zfs) parameters D are found to be close to those in quintet dinitrenes and septet trinitrenes, where the nitrene centers are attached to the same aryl ring. The large D values of branched septet nitrenes are due to strong negative one-center spin–spin interactions in combination with weak positive two-center spin–spin interactions, as predicted by theoretical considerations.
Co-reporter:Patrik Neuhaus;Michael Winkler;Wolfram Ser
Journal of Physical Organic Chemistry 2011 Volume 24( Issue 10) pp:976-992
Publication Date(Web):
DOI:10.1002/poc.1911
The isomeric 2-dehydro- and 4-dehydro-1,3-benzoquinodimethanes, 18 and 19, were generated by irradiation of the corresponding triiodo compounds in cryogenic argon matrices and characterized by electron paramagnetic resonance spectroscopy. In agreement with multireference computations, both systems possess quartet ground states, whereas in the isomeric 5-dehydro-m-xylylene 20 the 2B2 doublet state lies energetically below the 4B2 state, so that no characteristic quartet signals could be observed for this triradical under similar experimental conditions. The best estimates for the adiabatic doublet–quartet energy splittings (CAS(7,7)-AQCC/cc-pVTZ//CAS(9,9)-RS2c/cc-pVTZ) of 18–20 are 10.4, 7.7, and −1.3 kcal/mol, respectively. The measured zero-field splitting parameters of 18 and 19 are discussed in terms of the contributions of carbenoid resonance structures (spin polarization of the π-system) to the resonance hybrid of the title triradicals. Copyright © 2011 John Wiley & Sons, Ltd.
Co-reporter:Sergei V. Chapyshev, Denis V. Korchagin, Mikhail F. Budyka, Tatiana N. Gavrishova, Patrik Neuhaus, and Wolfram Sander
The Journal of Physical Chemistry A 2011 Volume 115(Issue 30) pp:8419-8425
Publication Date(Web):June 17, 2011
DOI:10.1021/jp203038k
Photolysis of 2,6-bis(4′-azidophenyl)-4-phenylpyridine in 2-methyltetrahydrofuran (2MTHF) glass at 7 K leads to quintet 2,6-bis(4′-nitrenophenyl)-4-phenylpyridine as a mixture of rotational isomers. The electron spin resonance (ESR) spectrum of this mixture of rotamers shows a considerable broadening of many transitions in the range of 0–5000 G and cannot be reproduced by computer simulations solely based on the tuning of the spin Hamiltonian parameters g, DQ, and EQ alone or on predictions of DFT calculations. The best modeling of the experimental ESR spectrum is obtained only when the large line-broadening parameter of Γ(EQ) = 1200 MHz along with the spin Hamiltonian g = 2.003, DQ = 0.154 cm–1, and EQ = 0.050 cm–1 is used in the spectral simulations. The most accurate theoretical estimations of the magnetic parameters of the dinitrene in a 2MTHF glass are obtained from the B3LYP/6-311+G(d,p)+PBE/DZ/COSMO calculations of the spin–spin coupling parameters DSS and ESS. Such calculations overestimate the EQ and DQ values of the dinitrene just by 1% and 10%, respectively, demonstrating that contributions of the spin–orbit coupling parameters DSOC and ESOC to the total DQ and EQ values are negligibly small. The research shows that ESR studies of polynuclear high-spin nitrenes, obtained by photolysis of rotational isomers of the starting azides, can only be successful if large EQ strain effects are taken into account in the spectral simulations.
Co-reporter:Artur Mardyukov ;Wolfram Ser
European Journal of Organic Chemistry 2010 Volume 2010( Issue 15) pp:2904-2909
Publication Date(Web):
DOI:10.1002/ejoc.201000153
Abstract
The benzoyl radical 1 was synthesized in argon matrices by the thermal reaction of the phenyl radical 2 with CO. The IR spectrum with the C=O str. vibration at 1824.4 cm–1 is in good agreement with DFT calculations. The formation of 1 is reversible and UV irradiation results in the cleavage back to 2 and CO. The benzoyl radical 1 can react with molecular oxygen in the matrix to produce the benzoylperoxy radical 3. Radical 3 was also characterized by IR spectroscopy in combination with DFT calculations.
Co-reporter:Sergei V. Chapyshev;Patrik Neuhaus;Dirk Grote;Wolfram Ser
Journal of Physical Organic Chemistry 2010 Volume 23( Issue 4) pp:340-346
Publication Date(Web):
DOI:10.1002/poc.1622
Abstract
Septet 3,5-dicyanopyridyl-2,4,6-trinitrene was synthesized together with quintet 2-azido-3,5-dicyanopyridyl-4,6-dinitrene, quintet 4-azido-3,5-dicyanopyridyl-2,6-dinitrene, triplet 2,6-diazido-3,5-dicyanopyridyl-4-nitrene, and triplet 2,4-diazido-3,5-dicyanopyridyl-6-nitrene by photolysis of 2,4,6-triazido-3,5-dicyanopyridine in solid argon at 4 K. The electronic and magnetic properties of the matrix-isolated nitrenes were studied using electron paramagnetic resonance (EPR) spectroscopy in combination with density functional theory (DFT) calculations. The fine-structure parameters of the nitrenes were determined with high accuracy from spectral simulations. All signals in the EPR spectra of the nitrenes, randomly oriented in the solid phase, were unambiguously assigned based on eigenfield calculations of the Zeeman energy levels and angular dependences of resonance fields. Copyright © 2009 John Wiley & Sons, Ltd.
Co-reporter:Dirk Grote Dr.;Christopher Finke;Simone Kossmann;Frank Neese Dr.;Wolfram Ser Dr.
Chemistry - A European Journal 2010 Volume 16( Issue 15) pp:4496-4506
Publication Date(Web):
DOI:10.1002/chem.200903285
Abstract
The photochemistry of 2-iodo-3,4,5,6-tetrafluorophenyl azide (7 d) has been investigated in argon and neon matrices at 4 K, and the products characterized by IR and EPR spectroscopy. The primary photochemical step is loss of a nitrogen molecule and formation of phenyl nitrene 1 d. Further irradiation with UV or visible light results in mixtures of 1 d with azirine 5 d′, ketenimine 6 d′, nitreno radical 2 d, and azirinyl radical 9. The relative amounts of these products strongly depend on the matrix and on the irradiation conditions. Nitreno radical 2 d with a quartet ground state was characterized by EPR spectroscopy. Electronic structure calculations in combination with the experimental results allow for a detailed understanding of the properties of this unusual new type of organic high-spin molecules.
Co-reporter:Artur Mardyukov;Rachel Crespo-Otero Dr.;Elsa Sanchez-Garcia Dr.;Wolfram Ser Dr.
Chemistry - A European Journal 2010 Volume 16( Issue 29) pp:8679-8689
Publication Date(Web):
DOI:10.1002/chem.200903362
Abstract
The reaction of the phenyl radical 1 with water has been investigated by using matrix isolation spectroscopy and quantum chemical calculations. The primary thermal product of the reaction between 1 and water is a weakly bound complex stabilized by an OH⋅⋅⋅π interaction. This complex is photolabile, and visible-light irradiation (λ>420 nm) results in hydrogen atom transfer from water to radical 1 and the formation of a highly labile complex between benzene and the OH radical. This complex is stable under the conditions of matrix isolation, however, continuous irradiation with λ>420 nm light results in the complete destruction of the aromatic system and formation of an acylic unsaturated ketene. The mechanisms of all reaction steps are discussed in the light of ab initio and DFT calculations.
Co-reporter:Artur Mardyukov;Rachel Crespo-Otero Dr.;Elsa Sanchez-Garcia Dr.;Wolfram Ser Dr.
Chemistry - A European Journal 2010 Volume 16( Issue 29) pp:
Publication Date(Web):
DOI:10.1002/chem.201090141
Co-reporter:Patrik Neuhaus ;Dr. Wolfram Ser
Angewandte Chemie 2010 Volume 122( Issue 40) pp:7435-7438
Publication Date(Web):
DOI:10.1002/ange.201001393
Co-reporter:Patrik Neuhaus ;Dr. Wolfram Ser
Angewandte Chemie International Edition 2010 Volume 49( Issue 40) pp:7277-7280
Publication Date(Web):
DOI:10.1002/anie.201001393
Co-reporter:Artur Mardyukov;Elsa Sanchez-Garcia Dr.;Rachel Crespo-Otero ;Wolfram Ser Dr.
Angewandte Chemie International Edition 2009 Volume 48( Issue 26) pp:4804-4807
Publication Date(Web):
DOI:10.1002/anie.200806268
Co-reporter:Artur Mardyukov ;Wolfram Ser Dr.
Chemistry - A European Journal 2009 Volume 15( Issue 6) pp:1462-1467
Publication Date(Web):
DOI:10.1002/chem.200801546
Abstract
The phenylperoxy radical 1 has been synthesized by the reaction of the phenyl radical 2 with 3O2. Radical 1 could be either generated in the gas phase and subsequently trapped in solid argon at 10 K, or directly synthesized in argon matrices. By reacting 2 as well as its perdeuterated isotopomer [D5]-2 with 16O2 and with 18O2, respectively, the four isotopomers [H5]-16O2-1, [D5]-16O2-1, [H5]-18O2-1, and [D5]-18O2-1 were matrix-isolated and characterized by IR spectroscopy. The experimental IR spectra are in excellent agreement with results from DFT calculations. Irradiation of 1 with visible light produces the 2-oxepinoxy radical 5 in a clean reaction. Subsequent irradiation results in ring-opening and formation of several conformers of ketoketene 6. The radicals 1, 5, and 6 play an important role in the combustion of aromatic hydrocarbons and could now be isolated and spectroscopically characterized for the first time.
Co-reporter:Artur Mardyukov;Elsa Sanchez-Garcia Dr.;Rachel Crespo-Otero ;Wolfram Ser Dr.
Angewandte Chemie 2009 Volume 121( Issue 26) pp:4898-4901
Publication Date(Web):
DOI:10.1002/ange.200806268
Co-reporter:Rachel Crespo-Otero, Elsa Sánchez-García, Reynier Suardíaz, Luis A. Montero, Wolfram Sander
Chemical Physics 2008 Volume 353(1–3) pp:193-201
Publication Date(Web):3 November 2008
DOI:10.1016/j.chemphys.2008.08.012
Abstract
The interactions of the simple radicals CH3, NH2, OH, and F with water have been studied by DFT (UB3LYP/6-311++G(2d,2p)) and ab initio (RHF-UCCSD(T)/6-311++G(2d,2p)) methods. In this order the number of lone pairs (from zero to three), the electronegativity, and the strength of the X–H bonds increase (X = C, N, and O). The various minima of the radical–water complexes were located using the multiple minima hypersurface (MMH) approach which had previously been proven to be useful for closed-shell molecules. The role of the unpaired electron in hydrogen bonding was investigated using the natural bond orbital (NBO) analysis. A considerable contribution of the unpaired electron to the complex stabilization was only found for the methyl radical and the fluorine atom, whereas in the aminyl and the hydroxyl radical the role of the unpaired electron is negligible.
Co-reporter:Elsa Sánchez-García, Artur Mardyukov, Adem Tekin, Rachel Crespo-Otero, Luis A. Montero, Wolfram Sander, Georg Jansen
Chemical Physics 2008 Volume 343(2–3) pp:168-185
Publication Date(Web):29 January 2008
DOI:10.1016/j.chemphys.2007.09.053
Abstract
Five acetylene–furan dimer structures are identified using ab initio calculations at the second-order Møller–Plesset (MP2) level of theory. The structures are stabilized by two basic types of intermolecular interactions: the CH⋯O and the CH⋯π interaction. The CH⋯π interaction appears in two variants, depending on which molecule provides the hydrogen atom and which molecule the π system. The MP2 results indicate that the CH⋯π interaction between one of the hydrogen atoms of acetylene and the π system of furan as found in structure A is the strongest interaction, followed by the in-plane CH⋯O interaction in the second most stable acetylene–furan dimer structure B. A matrix isolation study shows the acetylene–furan dimer to exist in an argon matrix, but likely rather as structure B than as A. High level coupled cluster calculations with up to triple excitations (CCSD(T)) yield the interaction energy of structure A as about −2.4 kcal/mol in the complete basis set limit and find structure B to be nearly isoenergetic with −2.3 kcal/mol. This is confirmed in calculations employing the density functional theory combined with symmetry adapted intermolecular perturbation theory (DFT-SAPT) approach yielding interaction energies of −2.3 and −2.0 kcal/mol for A and B, respectively. DFT-SAPT also helps to understand the importance of the electrostatic, induction and dispersion interaction energies and their respective exchange counterparts for the stability of the various acetylene–furan dimer structures. The CH⋯O and CH⋯π interactions are furthermore analyzed with the help of the atoms in molecules (AIM) theory.
Co-reporter:Sugumar Venkataramani;Michael Winkler Dr.;Wolfram Ser Dr.
Angewandte Chemie International Edition 2007 Volume 46(Issue 26) pp:
Publication Date(Web):14 JUN 2007
DOI:10.1002/anie.200700536
Three stay, three go: Flash vacuum pyrolysis (FVP) of 1 and subsequent trapping of the products in solid argon at 3 K produces trifluoro-1,3,5-tridehydrobenzene (2). IR spectroscopy, the photochemical behavior, and quantum chemical calculations confirm that for the first time a derivative of the elusive 1,3,5-tridehydrobenzene could be isolated and spectroscopically characterized.
Co-reporter:Sugumar Venkataramani, Michael Winkler,Wolfram Sander
Angewandte Chemie International Edition 2005 44(39) pp:6306-6311
Publication Date(Web):
DOI:10.1002/anie.200501912
Co-reporter:Rachel Crespo-Otero, Artur Mardykov, Elsa Sanchez-Garcia, Wolfram Sander and Mario Barbatti
Physical Chemistry Chemical Physics 2014 - vol. 16(Issue 35) pp:NaN18887-18887
Publication Date(Web):2014/07/28
DOI:10.1039/C4CP02518K
The formation of weakly-bound dimers of N-methylformamide (NMF) and the photochemistry of these dimers after irradiation at 248 nm were explored using matrix-isolation spectroscopy. Calculations were used to characterize the diverse isomers and assign their IR spectra; non-adiabatic dynamics was simulated to understand their photo-deactivation mechanism. The most stable dimers, tt-1 and tt-2, were obtained by trans–trans aggregation (N–H⋯OC interactions) and could be identified in the matrix. The main products formed after irradiation are the trans–cis dimers (tc-3 and tc-4), also stabilized by N–H⋯OC interactions. In contrast to the photochemistry of the monomers, no dissociative products were observed after 248 nm irradiation of the dimers. The absence of dissociative products can be explained by a proton-transfer mechanism in the excited state that is faster than the photo-dissociative mechanism. The fact that hydrogen bonding has such a significant effect on the photochemical stability of NMF has important implications to understand the stability of peptide-bonded systems to UV irradiation.











![7-Azabicyclo[4.1.0]hepta-2,4,6-triene, 1,2,3,5-tetrafluoro-4-iodo-](http://img.cochemist.com/ccimg/923300/923294-34-2.png)
![7-Azabicyclo[4.1.0]hepta-2,4,6-triene, 1,2,3,5-tetrafluoro-4-iodo-](http://img.cochemist.com/ccimg/923300/923294-34-2_b.png)




![7-Azabicyclo[4.1.0]hepta-2,4,6-triene, 1,3,4,5-tetrafluoro-2-iodo-](http://img.cochemist.com/ccimg/923300/923294-28-4.png)
![7-Azabicyclo[4.1.0]hepta-2,4,6-triene, 1,3,4,5-tetrafluoro-2-iodo-](http://img.cochemist.com/ccimg/923300/923294-28-4_b.png)













![Ethyl, 1-[(fluorocarbonyl)imino]-](http://img.cochemist.com/ccimg/880200/880134-81-6.png)
![Ethyl, 1-[(fluorocarbonyl)imino]-](http://img.cochemist.com/ccimg/880200/880134-81-6_b.png)


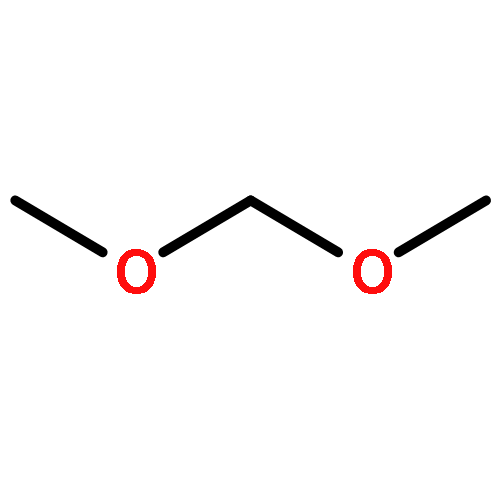
![FORMIC ACID, COMPD. WITH OXYBIS[METHANE] (1:1)](http://img.cochemist.com/ccimg/866400/866344-09-4.png)
![FORMIC ACID, COMPD. WITH OXYBIS[METHANE] (1:1)](http://img.cochemist.com/ccimg/866400/866344-09-4_b.png)
![L-Cysteine, L-phenylalanyl-S-[(acetylamino)methyl]-, methyl ester](http://img.cochemist.com/ccimg/852300/852286-63-6.png)
![L-Cysteine, L-phenylalanyl-S-[(acetylamino)methyl]-, methyl ester](http://img.cochemist.com/ccimg/852300/852286-63-6_b.png)










![BENZENE, [[(PHENYLSULFINYL)CARBONYL]SULFONYL]-](http://img.cochemist.com/ccimg/581100/581050-18-2.png)
![BENZENE, [[(PHENYLSULFINYL)CARBONYL]SULFONYL]-](http://img.cochemist.com/ccimg/581100/581050-18-2_b.png)
![Ethylthio, 2-carboxy-2-[[(1,1-dimethylethoxy)carbonyl]amino]-, (2R)-](http://img.cochemist.com/ccimg/557800/557796-85-7.png)
![Ethylthio, 2-carboxy-2-[[(1,1-dimethylethoxy)carbonyl]amino]-, (2R)-](http://img.cochemist.com/ccimg/557800/557796-85-7_b.png)




![Tricyclo[3.1.0.02,6]hex-3-ene-1,3-diyl](http://img.cochemist.com/ccimg/477400/477336-48-4.png)
![Tricyclo[3.1.0.02,6]hex-3-ene-1,3-diyl](http://img.cochemist.com/ccimg/477400/477336-48-4_b.png)











![Cyclohexane, [(cyclohexylsulfonylmethyl)sulfonyl]-](http://img.cochemist.com/ccimg/392300/392296-54-7.png)
![Cyclohexane, [(cyclohexylsulfonylmethyl)sulfonyl]-](http://img.cochemist.com/ccimg/392300/392296-54-7_b.png)
![Benzene, 1-chloro-4-[[(4-chlorophenyl)sulfonylmethyl]sulfonyl]-](http://img.cochemist.com/ccimg/392300/392296-52-5.png)
![Benzene, 1-chloro-4-[[(4-chlorophenyl)sulfonylmethyl]sulfonyl]-](http://img.cochemist.com/ccimg/392300/392296-52-5_b.png)
![Benzene, 1-methyl-4-[[(4-methylphenyl)sulfonylmethyl]sulfonyl]-](http://img.cochemist.com/ccimg/392300/392296-50-3.png)
![Benzene, 1-methyl-4-[[(4-methylphenyl)sulfonylmethyl]sulfonyl]-](http://img.cochemist.com/ccimg/392300/392296-50-3_b.png)
![Bicyclo[4.1.0]heptane, 7-[bis[(4-methylphenyl)sulfonyl]methyl]-](http://img.cochemist.com/ccimg/392300/392296-44-5.png)
![Bicyclo[4.1.0]heptane, 7-[bis[(4-methylphenyl)sulfonyl]methyl]-](http://img.cochemist.com/ccimg/392300/392296-44-5_b.png)
![Benzene, 1,1'-[(cyclohexylmethylene)bis(sulfonyl)]bis[4-methyl-](http://img.cochemist.com/ccimg/392300/392296-42-3.png)
![Benzene, 1,1'-[(cyclohexylmethylene)bis(sulfonyl)]bis[4-methyl-](http://img.cochemist.com/ccimg/392300/392296-42-3_b.png)



















![Benzo[c]thiophene, 5,7-dimethyl-1-(2,4,6-trimethylphenyl)-](http://img.cochemist.com/ccimg/201200/201156-88-9.png)
![Benzo[c]thiophene, 5,7-dimethyl-1-(2,4,6-trimethylphenyl)-](http://img.cochemist.com/ccimg/201200/201156-88-9_b.png)






![Bicyclo[3.1.0]hexa-2,4,6-trien-2-ol](http://img.cochemist.com/ccimg/197800/197796-02-4.png)
![Bicyclo[3.1.0]hexa-2,4,6-trien-2-ol](http://img.cochemist.com/ccimg/197800/197796-02-4_b.png)






![1,2-Dioxaspiro[2.5]octa-4,7-diene-5-carboxylic acid, 7-methyl-6-oxo-](http://img.cochemist.com/ccimg/197800/197795-92-9.png)
![1,2-Dioxaspiro[2.5]octa-4,7-diene-5-carboxylic acid, 7-methyl-6-oxo-](http://img.cochemist.com/ccimg/197800/197795-92-9_b.png)




















![7-Oxa-8-azabicyclo[4.2.0]octa-1,3,5-triene](http://img.cochemist.com/ccimg/186400/186353-13-9.png)
![7-Oxa-8-azabicyclo[4.2.0]octa-1,3,5-triene](http://img.cochemist.com/ccimg/186400/186353-13-9_b.png)



![7-AZABICYCLO[4.1.0]HEPTA-2,4,6-TRIENE, 1,5-DIFLUORO-](http://img.cochemist.com/ccimg/177900/177859-58-4.png)
![7-AZABICYCLO[4.1.0]HEPTA-2,4,6-TRIENE, 1,5-DIFLUORO-](http://img.cochemist.com/ccimg/177900/177859-58-4_b.png)


![Methyl, [(1Z)-1,2-ethenediyldi-4,1-phenylene]bis-](http://img.cochemist.com/ccimg/177300/177266-71-6.png)
![Methyl, [(1Z)-1,2-ethenediyldi-4,1-phenylene]bis-](http://img.cochemist.com/ccimg/177300/177266-71-6_b.png)





















![Acetic acid, oxo[(2-thioxo-1(2H)-pyridinyl)oxy]-, ethyl ester](http://img.cochemist.com/ccimg/133700/133616-54-3.png)
![Acetic acid, oxo[(2-thioxo-1(2H)-pyridinyl)oxy]-, ethyl ester](http://img.cochemist.com/ccimg/133700/133616-54-3_b.png)

















![1,2-DIOXASPIRO[2.5]OCTA-4,7-DIEN-6-ONE](/data/chemimg/365900/113567-94-5.png)
![1,2-DIOXASPIRO[2.5]OCTA-4,7-DIEN-6-ONE](/data/chemimg/365900/113567-94-5_b.png)
![Naphtho[1,2-c]furan-1,3-dione,9-methyl-](http://img.cochemist.com/ccimg/111100/111013-07-1.png)
![Naphtho[1,2-c]furan-1,3-dione,9-methyl-](http://img.cochemist.com/ccimg/111100/111013-07-1_b.png)












![Bicyclo[3.1.0]hex-2-ene, 4,6-bis(methylene)-](http://img.cochemist.com/ccimg/97600/97521-44-3.png)
![Bicyclo[3.1.0]hex-2-ene, 4,6-bis(methylene)-](http://img.cochemist.com/ccimg/97600/97521-44-3_b.png)




















![Alanine, N-[1-[(1,1-dimethylethoxy)carbonyl]-L-prolyl]-2-methyl-](http://img.cochemist.com/ccimg/79200/79118-34-6.png)
![Alanine, N-[1-[(1,1-dimethylethoxy)carbonyl]-L-prolyl]-2-methyl-](http://img.cochemist.com/ccimg/79200/79118-34-6_b.png)






















![2-Azabicyclo[2.2.0]hexa-1,3,5-triene](http://img.cochemist.com/ccimg/65600/65501-40-8.png)
![2-Azabicyclo[2.2.0]hexa-1,3,5-triene](http://img.cochemist.com/ccimg/65600/65501-40-8_b.png)











































![7bH-Cyclopropa[a]naphthalene](http://img.cochemist.com/ccimg/38700/38617-06-0.png)
![7bH-Cyclopropa[a]naphthalene](http://img.cochemist.com/ccimg/38700/38617-06-0_b.png)




























![Benzene, 1,1'-[(diazomethylene)bis(sulfonyl)]bis[4-chloro-](http://img.cochemist.com/ccimg/28400/28343-24-0.png)
![Benzene, 1,1'-[(diazomethylene)bis(sulfonyl)]bis[4-chloro-](http://img.cochemist.com/ccimg/28400/28343-24-0_b.png)






![Methanesulfonic acid, 1,1,1-trifluoro-, diphenyl[[(trifluoromethyl)sulfonyl]oxy]silyl ester](/data/chemimg/1173900/27607-80-3.png)
![Methanesulfonic acid, 1,1,1-trifluoro-, diphenyl[[(trifluoromethyl)sulfonyl]oxy]silyl ester](/data/chemimg/1173900/27607-80-3_b.png)
![Methanesulfonic acid,1,1,1-trifluoro-, dimethyl[[(trifluoromethyl)sulfonyl]oxy]silyl ester](http://img.cochemist.com/ccimg/27700/27607-78-9.png)
![Methanesulfonic acid,1,1,1-trifluoro-, dimethyl[[(trifluoromethyl)sulfonyl]oxy]silyl ester](http://img.cochemist.com/ccimg/27700/27607-78-9_b.png)



![1H-Cyclobuta[de]naphthalene](http://img.cochemist.com/ccimg/25000/24973-91-9.png)
![1H-Cyclobuta[de]naphthalene](http://img.cochemist.com/ccimg/25000/24973-91-9_b.png)




















































![7-Azabicyclo[4.2.0]octa-1,3,5-triene](http://img.cochemist.com/ccimg/7100/7095-75-2.png)
![7-Azabicyclo[4.2.0]octa-1,3,5-triene](http://img.cochemist.com/ccimg/7100/7095-75-2_b.png)
![1aH-Cyclopropa[a]naphthalene](http://img.cochemist.com/ccimg/7100/7078-38-8.png)
![1aH-Cyclopropa[a]naphthalene](http://img.cochemist.com/ccimg/7100/7078-38-8_b.png)








































































![Bicyclo[3.1.0]hexa-1,3,5-triene](http://img.cochemist.com/ccimg/1600/1552-99-4.png)
![Bicyclo[3.1.0]hexa-1,3,5-triene](http://img.cochemist.com/ccimg/1600/1552-99-4_b.png)
![Bicyclo[2.2.0]hexane](http://img.cochemist.com/ccimg/1600/1552-98-3.png)
![Bicyclo[2.2.0]hexane](http://img.cochemist.com/ccimg/1600/1552-98-3_b.png)




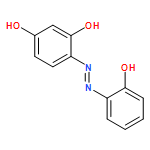
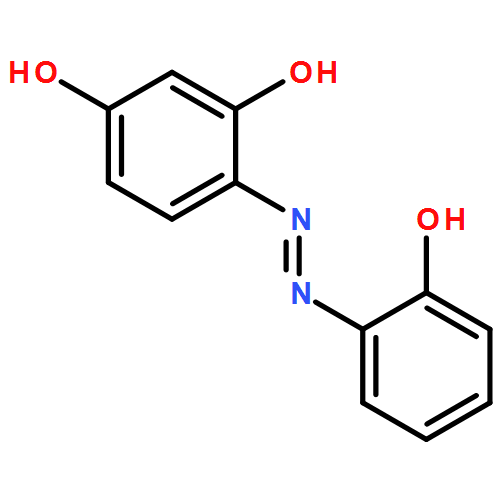
![[bis(4-methoxyphenyl)methylidene]diazenium](http://img.cochemist.com/ccimg/1300/1221-72-3.png)
![[bis(4-methoxyphenyl)methylidene]diazenium](http://img.cochemist.com/ccimg/1300/1221-72-3_b.png)




































![Benzene, [(phenylsulfonyl)sulfonylmethyl]-](http://img.cochemist.com/ccimg/312700/312613-12-0.png)
![Benzene, [(phenylsulfonyl)sulfonylmethyl]-](http://img.cochemist.com/ccimg/312700/312613-12-0_b.png)



























































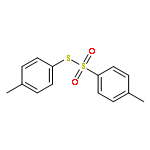
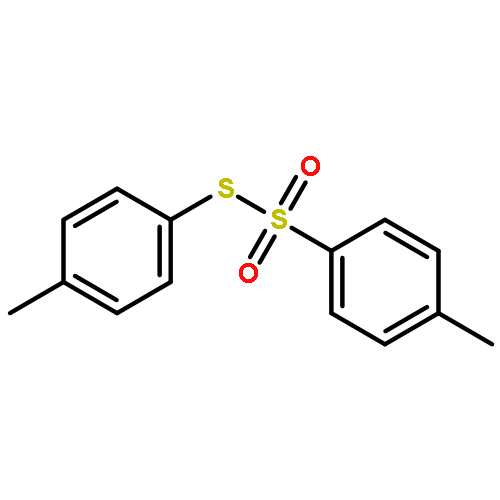
![Benzene,1,1'-[(diazomethylene)bis(sulfonyl)]bis[4-methyl-](http://img.cochemist.com/ccimg/14200/14159-45-6.png)
![Benzene,1,1'-[(diazomethylene)bis(sulfonyl)]bis[4-methyl-](http://img.cochemist.com/ccimg/14200/14159-45-6_b.png)

![N~2~-butyl-N-[2-(3,4-dimethoxyphenyl)ethyl]-N-(thiophen-2-ylmethyl)-N~2~-{[3-(trifluoromethyl)phenyl]carbamoyl}glycinamide](http://img.cochemist.com/ccimg/6000/5919-15-3.png)
![N~2~-butyl-N-[2-(3,4-dimethoxyphenyl)ethyl]-N-(thiophen-2-ylmethyl)-N~2~-{[3-(trifluoromethyl)phenyl]carbamoyl}glycinamide](http://img.cochemist.com/ccimg/6000/5919-15-3_b.png)






![Benzene, 1,1'-[(diazomethylene)bis(sulfonyl)]bis-](http://img.cochemist.com/ccimg/1900/1886-74-4.png)
![Benzene, 1,1'-[(diazomethylene)bis(sulfonyl)]bis-](http://img.cochemist.com/ccimg/1900/1886-74-4_b.png)









![BICYCLO[3.1.0]HEXA-1(6),2-DIEN-4-ONE](http://img.cochemist.com/data/images/133985-05-4.png)
![BICYCLO[3.1.0]HEXA-1(6),2-DIEN-4-ONE](http://img.cochemist.com/data/images/133985-05-4_b.png)


![L-Cysteine,S-[(acetylamino)methyl]-, methyl ester, hydrochloride (1:1)](http://img.cochemist.com/ccimg/33400/33375-68-7.png)
![L-Cysteine,S-[(acetylamino)methyl]-, methyl ester, hydrochloride (1:1)](http://img.cochemist.com/ccimg/33400/33375-68-7_b.png)