Co-reporter:Takuya Yoshioka, Kazuma Murakami, Kyohei Ido, Mizuho Hanaki, Kanoko Yamaguchi, Satohiro Midorikawa, Shinji Taniwaki, Hiroki Gunji, and Kazuhiro Irie
Journal of Natural Products 2016 Volume 79(Issue 10) pp:2521-2529
Publication Date(Web):October 4, 2016
DOI:10.1021/acs.jnatprod.6b00392
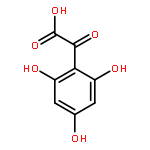
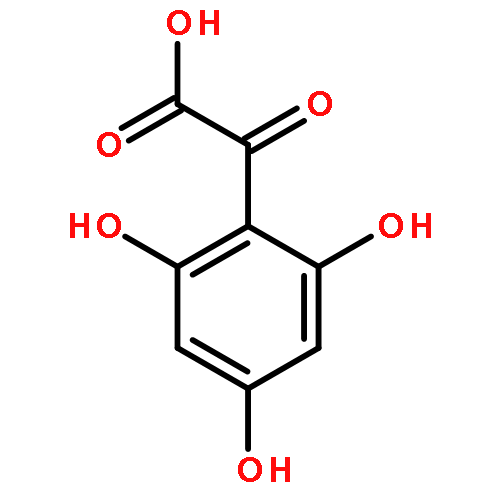
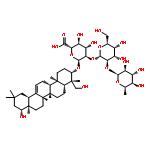
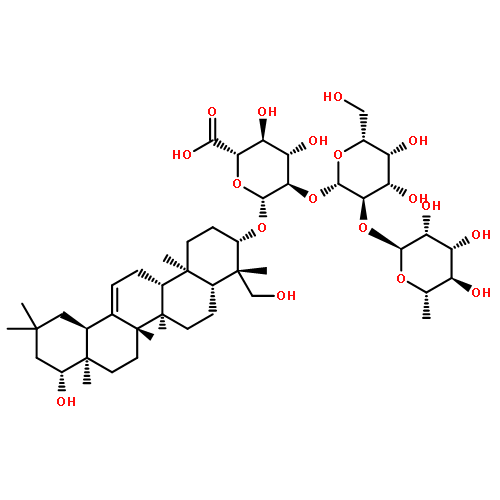
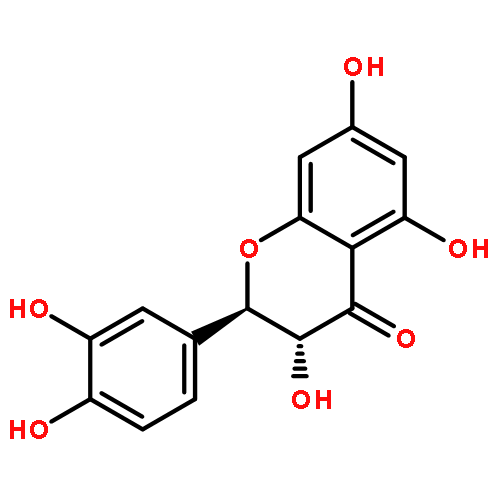
Oligomers of the 42-mer amyloid-β protein (Aβ42), rather than fibrils, cause synaptic dysfunction in the pathology of Alzheimer’s disease (AD). The nucleation phase in a nucleation-dependent aggregation model of Aβ42 is related to the formation of oligomers. Uncaria rhynchophylla is one component of “Yokukansan”, a Kampo medicine, which is widely used for treating AD symptoms. Previously, an extract of U. rhynchophylla was found to reduce the aggregation of Aβ42, but its active principles have yet to be identified. In the present work, uncarinic acid C (3) was identified as an inhibitor of Aβ42 aggregation that is present in U. rhynchophylla. Moreover, compound 3 acted as a specific inhibitor of the nucleation phase of Aβ42 aggregation. Compound 3 was synthesized from saponin A (10), an abundant byproduct of rutin purified from Uncaria elliptica. Comprehensive structure–activity studies on 3 suggest that both a C-27 ferulate and a C-28 carboxylic acid group are required for its inhibitory activity. These findings may aid the development of oligomer-specific inhibitors for AD therapy.
Co-reporter:Mizuho Hanaki, Kazuma Murakami, Ken-ichi Akagi, Kazuhiro Irie
Bioorganic & Medicinal Chemistry 2016 Volume 24(Issue 2) pp:304-313
Publication Date(Web):15 January 2016
DOI:10.1016/j.bmc.2015.12.021
The prevention of 42-mer amyloid β-protein (Aβ42) aggregation is promising for the treatment of Alzheimer’s disease. We previously described the site-specific inhibitory mechanism for Aβ42 aggregation by a catechol-type flavonoid, (+)-taxifolin, targeting Lys16,28 after its autoxidation. In contrast, non-catechol-type flavonoids (morin, datiscetin, and kaempferol) inhibited Aβ42 aggregation without targeting Lys16,28 with almost similar potencies to that of (+)-taxifolin. We herein provided structural insights into their mechanisms for inhibiting Aβ42 aggregation. Physicochemical analyses revealed that their inhibition did not require autoxidation. The 1H–15N SOFAST-HMQC NMR of Aβ42 in the presence of morin and datiscetin revealed the significant perturbation of chemical shifts of His13,14 and Gln15, which were close to the intermolecular β-sheet region, Gln15-Ala21. His13,14 also played a role in radical formation at Tyr10, thereby inducing the oxidation of Met35, which has been implicated in Aβ42 aggregation. These results suggest the direct interaction of morin and datiscetin with the Aβ42 monomer. Although only kaempferol was oxidatively-degraded during incubation, its degradation products as well as kaempferol itself suppressed Aβ42 aggregation. However, neither kaempferol nor its decomposed products perturbed the chemical shifts of the Aβ42 monomer. Aggregation experiments using 1,1,1,3,3,3-hexafluoro-2-propanol-treated Aβ42 demonstrated that kaempferol and its degradation products inhibited the elongation rather than nucleation phase, implying that they interacted with small aggregates of Aβ42, but not with the monomer. In contrast, morin and datiscetin inhibited both phases. The position and number of hydroxyl groups on the B-ring of non-catechol-type flavonoids could be important for their inhibitory potencies and mechanisms against Aβ42 aggregation.
Co-reporter:Kazuhiro Irie;Ryo C. Yanagita
The Chemical Record 2014 Volume 14( Issue 2) pp:251-267
Publication Date(Web):
DOI:10.1002/tcr.201300036
Abstract
Protein kinase C (PKC) isozymes play central roles in signal transduction on the cell surface and could serve as promising therapeutic targets of intractable diseases like cancer, Alzheimer's disease, and acquired immunodeficiency syndrome (AIDS). Although natural PKC ligands like phorbol esters, ingenol esters, and teleocidins have the potential to become therapeutic leads, most of them are potent tumor promoters in mouse skin. By contrast, bryostatin-1 (bryo-1) isolated from marine bryozoan is a potent PKC activator with little tumor-promoting activity. Numerous investigations have suggested bryo-1 to be a promising therapeutic candidate for the above intractable diseases. However, there is a supply problem of bryo-1 both from natural sources and by organic synthesis. Recent approaches on the synthesis of bryo-1 have focused on its simplification, without decreasing the ability to activate PKC isozymes, to develop new medicinal leads. Another approach is to use the skeleton of natural PKC ligands to develop bryo-1 surrogates. We have recently identified 10-methyl-aplog-1 (26), a simplified analog of tumor-promoting aplysiatoxin (ATX), as a possible therapeutic lead for cancer. This review summarizes recent investigations on the simplification of natural PKC ligands, bryo-1 and ATX, to develop potential medicinal leads.
Co-reporter:Masayuki Kikumori, Ryo C. Yanagita, Kazuhiro Irie
Tetrahedron 2014 70(52) pp: 9776-9782
Publication Date(Web):
DOI:10.1016/j.tet.2014.11.026
Co-reporter:Hiroaki Kamachi, Keisuke Tanaka, Ryo C. Yanagita, Akira Murakami, Kazuma Murakami, Harukuni Tokuda, Nobutaka Suzuki, Yu Nakagawa, Kazuhiro Irie
Bioorganic & Medicinal Chemistry 2013 Volume 21(Issue 10) pp:2695-2702
Publication Date(Web):15 May 2013
DOI:10.1016/j.bmc.2013.03.013
We have recently developed a simplified analog of aplysiatoxin (aplog-1) as an activator of protein kinase C (PKC) with anti-proliferative activity like bryostain 1. To identify sites in aplog-1 that could be readily modified to optimize therapeutic performance and to develop a molecular probe for examining the analog’s mode of action, substituent effects on the phenol ring were systematically examined. Whereas hydrophilic acetamido derivatives were less active than aplog-1 in inhibiting cancer cell growth and binding to PKCδ, introduction of hydrophobic bromine and iodine atoms enhanced both biological activities. The anti-proliferative activity was found to correlate closely with molecular hydrophobicity, and maximal activity was observed at a log P value of 4.0–4.5. On the other hand, an induction test with Epstein–Barr virus early antigen demonstrated that these derivatives have less tumor-promoting activity in vitro than aplog-1 regardless of the hydrophobicity of their substituents. These results would facilitate rapid preparation of molecular probes to examine the mechanism of the unique biological activities of aplog-1.
Co-reporter:Ryo C. Yanagita, Hiroaki Kamachi, Masayuki Kikumori, Harukuni Tokuda, Nobutaka Suzuki, Kiyotake Suenaga, Hiroshi Nagai, Kazuhiro Irie
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 15) pp:4319-4323
Publication Date(Web):1 August 2013
DOI:10.1016/j.bmcl.2013.05.096
Debromoaplysiatoxin (DAT) is a tumor promoter isolated from sea hare and exhibits anti-proliferative activity against several cancer cell lines. To clarify key residues that are responsible for its tumor-promoting activity, we focused on the chiral methoxy group in the side chain, whose role had not yet been discussed or examined before. Demethoxy-DAT (8) was derived from DAT and we evaluated its tumor-promoting activity, anti-proliferative activity, and ability to bind to protein kinase C (PKC) isozymes. Compound 8 showed somewhat weaker tumor-promoting activity than that of DAT both in vitro and in vivo, but showed higher anti-proliferative activity against several cancer cell lines. Although the affinity to novel PKC isozymes of 8 was comparable to that of DAT, the affinity to conventional PKC isozymes decreased slightly. These results suggest that the methoxy group of DAT is one of the key residues critical for tumor-promoting activity but not for anti-proliferative activity. Since the methoxy group has little influence on the molecular hydrophobicity, this is the first report showing that structural factors other than hydrophobicity in the side chain of DAT affected its biological activities.
Co-reporter:Yusuke Hanaki, Masayuki Kikumori, Sayo Ueno, Harukuni Tokuda, Nobutaka Suzuki, Kazuhiro Irie
Tetrahedron 2013 69(36) pp: 7636-7645
Publication Date(Web):
DOI:10.1016/j.tet.2013.02.008
Co-reporter:Masayuki Kikumori ; Ryo C. Yanagita ; Harukuni Tokuda ; Nobutaka Suzuki ; Hiroshi Nagai ; Kiyotake Suenaga
Journal of Medicinal Chemistry 2012 Volume 55(Issue 11) pp:5614-5626
Publication Date(Web):May 24, 2012
DOI:10.1021/jm300566h
Aplog-1, a simplified analogue of tumor-promoting debromoaplysiatoxin, is antiproliferative but not tumor-promoting. Our recent study has suggested that local hydrophobicity around the spiroketal moiety is a crucial determinant for antiproliferative activity. To further clarify the structural features relevant to the activity, we synthesized two methyl derivatives of aplog-1, where a methyl group was installed at position 4 or 10 of the spiroketal moiety. 10-Methyl-aplog-1 (5) bound to the C1B domains of novel PKCs (δ, η, and θ) with subnanomolar Ki values, approximately 10–20 times stronger than aplog-1, and markedly inhibited the growth of many human cancer cell lines, while 4-methyl-aplog-1 (4) had levels of activity similar to those of aplog-1. Interestingly, 5 showed little tumor-promoting activity unlike the tumor promoter debromoaplysiatoxin. These results suggest that 5 is a potent PKC ligand without tumor-promoting activity and could be a therapeutic lead for the treatment of cancer, like bryostatins.
Co-reporter:Naotaka Izuo, Toshiaki Kume, Mizuho Sato, Kazuma Murakami, Kazuhiro Irie, Yasuhiko Izumi, and Akinori Akaike
ACS Chemical Neuroscience 2012 Volume 3(Issue 9) pp:674
Publication Date(Web):June 6, 2012
DOI:10.1021/cn300033k
The 42-mer amyloid β-protein (Aβ42) aggregates to form soluble oligomers that cause memory loss and synaptotoxicity in Alzheimer’s disease (AD). Oxidative stress is closely related to the pathogenesis of AD. We previously identified the toxic conformer of Aβ42 with a turn at positions 22 and 23 (“toxic turn”) by solid-state NMR and demonstrated that a monoclonal antibody (11A1) against the toxic turn in Aβ42 mainly detected the oligomer in the brains of AD patients. Our recent study suggested that oxidative stress is a key factor of the oligomerization and cognitive impairment induced by Aβ overproduction in vivo. However, the involvement of the toxic conformer in Aβ42-induced oxidative damage remains unclear. To investigate this mechanism, we examined the levels of intracellular reactive oxygen species (ROS) and neurotoxicity in rat primary neurons using E22P-Aβ42, a mutant that induces a turn at positions 22 and 23, and E22V-Aβ42, a turn-preventing mutant. E22P-Aβ42, but not E22V-Aβ42, induced greater ROS production than Wt-Aβ42 in addition to potent neurotoxicity. Interestingly, the formation of the toxic conformer in both E22P-Aβ42 and Wt-Aβ42 probed by the 11A1 antibody preceded Aβ42-induced neurotoxicity. Trolox (a radical scavenger) and Congo red (an aggregation inhibitor) significantly prevented the neurotoxicity and intracellular ROS induced by E22P-Aβ42 and Wt-Aβ42, respectively. These results suggest that Aβ42-mediated toxicity is caused by the turn that favors toxic oligomers, which increase generation of ROS.Keywords: Alzheimer’s disease; amyloid β; neurotoxicity; oxidative stress; toxic conformer; turn structure
Co-reporter:Kazuma Murakami, Yuko Horikoshi-Sakuraba, Nakaba Murata, Yoshihiro Noda, Yuichi Masuda, Noriaki Kinoshita, Hiroyuki Hatsuta, Shigeo Murayama, Takuji Shirasawa, Takahiko Shimizu, and Kazuhiro Irie
ACS Chemical Neuroscience 2010 Volume 1(Issue 11) pp:747
Publication Date(Web):September 28, 2010
DOI:10.1021/cn100072e
Aggregation of the 42-mer amyloid β-protein (Aβ42) plays a critical role in the pathogenesis of Alzheimer’s disease (AD). We have proposed a toxic conformer with a turn at positions 22 and 23, as well as a nontoxic conformer with a turn at positions 25 and 26, in Aβ42 aggregates from systematic proline scanning and solid-state NMR studies. Although recent clinical trials of immunization targeting Aβ42 aggregates have proved useful, some adverse effects were reported. One of the reasons was hypothesized to be excessive immunoreactions derived from the unintended removal of nontoxic Aβ42, which plays an important role in the physiological function. To develop a monoclonal antibody for toxic Aβ42, E22P-Aβ10-35, a minimum moiety for neurotoxicity containing the turn at positions 22 and 23, was used for the generation of antibodies, following the selection of clones using Aβ42 mutants of E22P (turn-inducing) and E22V (turn-preventing). The obtained clone (11A1) showed a high binding affinity (KD = 10.3 nM) for Aβ42 using surface plasmon resonance. 11A1 also inhibited the neurotoxicity of Aβ42 in PC12 cells. Immunohistochemical studies showed that not only extracellular but intracellular amyloid was stained in human AD brains. In Western blotting analyses using human brains, low-molecular weight-oligomers rather than the monomer of Aβ were readily recognized by 11A1. These results imply that 11A1 could detect toxic Aβ42 oligomers with the turn at positions 22 and 23 and that 11A1 could be applicable for the therapeutic targeting of toxic Aβ42 in AD.Keywords (keywords): Alzheimer’s disease; amyloid; human brain; neurotoxicity; transgenic mice; turn
Co-reporter:Ryo C. Yanagita, Hiroaki Kamachi, Keisuke Tanaka, Akira Murakami, Yu Nakagawa, Harukuni Tokuda, Hiroshi Nagai, Kazuhiro Irie
Bioorganic & Medicinal Chemistry Letters 2010 Volume 20(Issue 20) pp:6064-6066
Publication Date(Web):15 October 2010
DOI:10.1016/j.bmcl.2010.08.051
The 18-deoxy derivative (3) of a simplified analogue (1) of aplysiatoxin with antiproliferative activity was synthesized to examine the role of the phenolic hydroxyl group at position 18 in the biological activities of 1. Compound 3 as well as 1 showed significant affinity for protein kinase Cδ (PKCδ), and the antiproliferative activity of 3 was slightly higher than that of 1. However, the anti-tumor-promoting activity of 3 was less than that of 1 in vitro, suggesting that the phenolic hydroxyl group of 1 is necessary for the anti-tumor-promoting activity but not for the binding of PKCδ and antiproliferative activity. Moreover, PKC isozyme selectivity of 3 was similar to that of 1, suggesting non-PKC receptors for these compounds to play some roles in the anti-tumor-promoting activity of 1.
Co-reporter:Yuichi Masuda;Satoko Uemura;Ryutaro Ohashi Dr.;Azusa Nakanishi;K. Takegoshi ;Takahiko Shimizu Assoc. Dr.;Takuji Shirasawa Dr.
ChemBioChem 2009 Volume 10( Issue 2) pp:287-295
Publication Date(Web):
DOI:10.1002/cbic.200800411
Abstract
Aggregation of the 42-residue amyloid β-protein (Aβ42) plays a crucial role in the pathogenesis of Alzheimer's disease (AD). Despite numerous structural studies on Aβ aggregates, the relationship between tertiary structure and toxicity remains unclear. Our proline scanning and solid-state NMR studies suggested that aggregates both of wild-type Aβ42 and of E22K-Aβ42 (one of the mutants related to cerebral amyloid angiopathy) contain two conformers: a major one with a turn at positions 25 and 26, and a minor one with a turn at positions 22 and 23. To identify the toxic conformer, the derivative Aβ42-lactam(22K–23E), in which the side chains at positions 22 and 23 were covalently linked, was synthesized as a minor conformer surrogate, along with Aβ42-lactam(25K–26E) as a major conformer surrogate. The Aβ42-lactam(22K–23E) showed stronger aggregation, neurotoxicity, radical generation, and oligomerization than wild-type Aβ42, whereas in Aβ42-lactam(25K–26E) were weak. The transition from the physiological conformation with a turn at positions 25 and 26 to the toxic conformation with a turn at positions 22 and 23 might be a key event in the pathogenesis of AD.
Co-reporter:Ryo C. Yanagita ; Yu Nakagawa ; Nobuhiro Yamanaka ; Kaori Kashiwagi ; Naoaki Saito
Journal of Medicinal Chemistry 2008 Volume 51(Issue 1) pp:46-56
Publication Date(Web):December 12, 2007
DOI:10.1021/jm0706719
Conventional and novel protein kinase C (PKC) isozymes are the main targets of tumor promoters. We developed 1-hexylindolactam-V10 (5) as a selective activator for novel PKC isozymes that play important roles in various cellular processes related to tumor promotion, ischemia−reperfusion injury in the heart, and Alzheimer’s disease. The compound existed as a mixture of three conformers. The trans-amide restricted analogues of 5 (14 and 15) hardly bound to PKC isozymes, suggesting that the active conformation of 5 could be that with a cis-amide. Compound 5 selectively translocated novel PKC isozymes over conventional PKC isozymes in HeLa cells at 0.1–1 μM. These results suggest that 5 could be useful for the functional analysis of novel PKC isozymes.
Co-reporter:Yuichi Masuda, Satoko Uemura, Azusa Nakanishi, Ryutaro Ohashi, K. Takegoshi, Takahiko Shimizu, Takuji Shirasawa, Kazuhiro Irie
Bioorganic & Medicinal Chemistry Letters 2008 Volume 18(Issue 11) pp:3206-3210
Publication Date(Web):1 June 2008
DOI:10.1016/j.bmcl.2008.04.060
Structural analysis of 42-residue amyloid β (Aβ42) aggregates using rotational resonance in solid-state NMR verified that Cβ and/or Cγ of Met-35 and the carboxyl carbon of Ala-42 are proximal enough to form an intramolecular antiparallel β-sheet in the C-terminus. The S-oxidized radical cation at Met-35, an ultimate radical species responsible for neurotoxicity, could be stabilized by the carboxylate anion at the C-terminus, resulting in aggregation to cause long-term oxidative stress.
Co-reporter:Kazuma Murakami Dr.;Hideyuki Hara Dr.;Yuichi Masuda;Hajime Ohigashi
ChemBioChem 2007 Volume 8(Issue 18) pp:
Publication Date(Web):19 NOV 2007
DOI:10.1002/cbic.200700240
The neurotoxicity of the 42-mer and 40-mer amyloid β peptides (Aβ42 and Aβ40) is closely related to the radicalization at both Tyr10 and Met35. Aβ42 is more neurotoxic than Aβ40. Our previous structural analyses of Aβ42 suggested that Tyr10 and Met35 are brought closer together by the turn at positions 22 and 23, and the S-oxidized radical cation at position 35, which is the ultimate toxic radical species, can be produced effectively through oxidation by the phenoxy radical at position 10. To verify this idea, their separation was measured by site-directed spin labeling (MTSSL) by using ESR spectroscopy. Among the three kinds of Aβ42 derivatives, which are doubly or singly spin-labeled at position 10 and 35, only 10,35-MTSSL-Aβ42 showed a clear dipole coupling in continuous-wave ESR; this suggests that the intramolecular spin labels at position 10 and 35 in Aβ42 are located within ∼15 Å. In contrast, 10,35-MTSSL-Aβ40 did not give such signals. The distance between Tyr10 and Met35 in 10,35-MTSSL-Aβ40, which was successfully measured by pulsed ESR spectroscopy was 30 Å long. The difference in the distance between Aβ42 and Aβ40 could explain in part the stronger neurotoxicity of Aβ42 compared to Aβ40.
Co-reporter:Kazuhiro Irie;Yu Nakagawa;Hajime Ohigashi
The Chemical Record 2005 Volume 5(Issue 4) pp:
Publication Date(Web):22 JUL 2005
DOI:10.1002/tcr.20044
Tumor promoters such as phorbol esters bind strongly to protein kinase C (PKC) isozymes to induce their activation. Since each PKC isozyme is involved in diverse biological events in addition to tumor promotion, the isozymes serve as promising therapeutic targets. Tumor promoters bind to the C1A and/or C1B domain of conventional (α, βI, βII, and γ) and novel PKC isozymes (δ, ε, η, and θ). As these C1 domains play differential roles in PKC activation and their translocation in cells, the development of agents with binding selectivity for individual C1 domains is a pressing need. For this purpose, we established a synthetic C1 peptide library of all PKC isozymes. The library enabled us to identify indolactam-V (1) as a promising lead compound. Our diverse structure–activity studies on 1 indicated that the position of the hydrophobic substituent on the indole ring dominates the PKC isozyme- and C1 domain-selective binding rather than conformation of the nine-membered lactam. Moreover, we suggested that the indole ring of 1 could be involved in the CH/π interaction with Pro-11 of the C1B domain of PKCδ. This invaluable information will lead to the structural optimization of the PKCδ ligand as exemplified by the design and synthesis of naphtholactam-V8 (21). © 2005 The Japan Chemical Journal Forum and Wiley Periodicals, Inc. Chem Rec 5: 185–195; 2005: Published online in Wiley InterScience (www.interscience.wiley.com) DOI 10.1002/tcr.20044
Co-reporter:Kazuhiro Irie, Akiko Masuda, Mayumi Shindo, Yu Nakagawa, Hajime Ohigashi
Bioorganic & Medicinal Chemistry 2004 Volume 12(Issue 17) pp:4575-4583
Publication Date(Web):1 September 2004
DOI:10.1016/j.bmc.2004.07.008
Recent investigations discovered nonkinase-type phorbol ester receptors, RasGRPs, chimaerins, and Unc13s. Phorbol ester binding occurs at the cysteine-rich sequences of about 50 residues in the C1 domains of these receptors. Fifty-one-residue RasGRP C1 peptides except for RasGRP2 showed significant phorbol 12,13-dibutyrate (PDBu) binding, but the Kd values of the RasGRP1 and RasGRP3 C1 peptides were about 10-fold larger than those for the corresponding whole enzymes. Addition of the C-terminal basic amino acid cluster decreased their Kd values about 10-fold, suggesting that the positive charges of these C1 peptides play an important role in the PDBu binding in the presence of negatively-charged phosphatidylserine. The 51-mer chimaerin C1 peptides showed potent PDBu binding, while the Unc13 and Munc13-1 C1 peptides without sufficient positive charges hardly bound PDBu. By the rapid screening system using this C1 peptide library, 5-prenyl-indolactam-V was identified as a promising lead for the novel protein kinase C isozyme specific ligands.A C1 peptide library of nonPKC-type phorbol ester receptors has been established.
Co-reporter:Mayumi Shindo, Kazuhiro Irie, Hiroyuki Fukuda, Hajime Ohigashi
Bioorganic & Medicinal Chemistry 2003 Volume 11(Issue 23) pp:5075-5082
Publication Date(Web):17 November 2003
DOI:10.1016/j.bmc.2003.08.024
Effect of zinc and other metal ions on the folding of the protein kinase C (PKC) surrogate peptide (PKCη-C1B) was analyzed intact under neutral conditions by electrospray ionization mass spectrometry (ESI–MS). ESI–MS spectrum of 64ZnCl2-folded PKCη-C1B clearly showed that PKCη-C1B coordinates specifically two atoms of zinc, and that the two thiol protons are lost in each zinc ion coordinate center. 113CdCl2-folded PKCη-C1B also showed stoichiometry of two cadmium atoms that was proved by addition of EDTA. The dissociation constants of zinc- and cadmium-folded PKCη-C1B in the phorbol 12,13-dibutyrate binding (PDBu) were similar (0.66 and 0.81 nM) with different Bmax values (46.4 and 71.4%). The difference would reflect higher coordination potency of cadmium ion that was demonstrated by ESI–MS when PKCη-C1B was folded by 1:1 mixture of zinc and cadmium ions. In contrast, 63CuCl2-treated PKCη-C1B did not show any copper-coordinated peak, instead a molecular mass less than 6 mass units smaller than that of apo-PKCη-C1B was observed. The multiple charge mass envelope of copper-treated PKCη-C1B shifted to that of the lower mass charge state like zinc-treated PKCη-C1B. These data suggest that the copper treatment formed three intramolecular S–S bonds to abolish the PDBu binding of PKCη-C1B.Effect of zinc and other metal ions on the folding of the protein kinase C surrogate peptide (PKCη-C1B) was analyzed by the electrospray ionization mass spectrometry.
Co-reporter:Yu Nakagawa, Kazuhiro Irie, Nobuhiro Yamanaka, Hajime Ohigashi, Ken-ichiro Tsuda
Bioorganic & Medicinal Chemistry Letters 2003 Volume 13(Issue 18) pp:3015-3019
Publication Date(Web):15 September 2003
DOI:10.1016/S0960-894X(03)00637-1
Benzolactone-V8 (4) is a lactone analogue of the artificial tumor promoter benzolactam-V8 (1). To investigate the effect of hydrophobic substituents at positions 7 and 15 of 4 on binding selectivity for protein kinase C (PKC) isozymes, 7- and 15-decylbenzolactone-V8 (7, 8) were synthesized and their binding affinities for synthetic PKC isozyme C1 peptides were examined. Compound 8 showed moderate selectivity for novel PKC isozymes similar to 9-decylbenzolactone-V8 (5), while 7 was less selective. Compounds 7 and 8 showed no significant selectivity among novel PKC isozymes unlike 8-decylbenzolactone-V8 (6). These results indicate that the introduction of a hydrophobic substituent at position 8 of 4 is most effective in the development of PKCε- and PKCη-selective binders.7- and 15-decylbenzolactone-V8 (7, 8) were synthesized, and their conformation and binding affinities for synthetic C1 peptides of all PKC isozymes were examined.
Co-reporter:Mayumi Shindo, Kazuhiro Irie, Akifumi Nakahara, Hajime Ohigashi, Hiroaki Konishi, Ushio Kikkawa, Hiroyuki Fukuda, Paul A. Wender
Bioorganic & Medicinal Chemistry 2001 Volume 9(Issue 8) pp:2073-2081
Publication Date(Web):August 2001
DOI:10.1016/S0968-0896(01)00100-6
Conventional and novel protein kinase C (PKC) isozymes contain two cysteine-rich C1 domains (C1A and C1B), both of which are candidate phorbol-12,13-dibutyrate (PDBu) binding sites. We previously synthesized C1 peptides (of approximately 50 residues) corresponding to all PKC isozymes and measured their PDBu binding affinity. While many of these peptide receptors exhibited PDBu affinities comparable to the respective complete isozyme, some of the C1A peptides could not be used because they undergo temperature dependent inactivation. This problem was however eliminated by 4 °C incubation or elongation of the 50-mer C1 peptides at both N- and C-termini to increase their folding efficiency and stability. These findings enabled us to determine the Kd's of PDBu for all PKC C1 peptides (except for θ-C1A) and establish the value of these peptides as readily available, stable, and easily handled surrogates of the individual isozymes. The resultant C1 peptide receptor library can be used to screen for new ligands with PKC isozyme and importantly C1 domain selectivity. Most of the C1 peptide receptors showed strong PDBu binding affinities with Kd's in the nanomolar range (0.45–7.4 nM). Two peptides (δ-C1A and θ-C1A) bound PDBu over 100-fold less tightly. To identify the residues that contribute to this affinity difference, several mutants of δ-C1A and θ-C1A were synthesized. Both the G9K mutant of δ-C1A and the P9K mutant of θ-C1A showed Kd's of 2–3 nM. This approach provides a useful procedure to determine the role of each C1 domain of the PKC isozymes by point mutation.Graphic
Co-reporter:Yu Nakagawa, Kazuhiro Irie, Yoshimasa Nakamura, Hajime Ohigashi
Bioorganic & Medicinal Chemistry Letters 2001 Volume 11(Issue 5) pp:723-728
Publication Date(Web):12 March 2001
DOI:10.1016/S0960-894X(01)00047-6
To investigate the role of the amide hydrogen of (−)-indolactam-V (1) and benzolactam-V8’s on protein kinase C (PKC) binding and tumor promotion, 8-decylbenzolactone-V8 (6), a new lactone analogue of 8-decylbenzolactam-V8 (4), was synthesized from 2-nitrophenylpyruvic acid (7) in 11 steps. The PKC binding ability and tumor-promoting activities in vitro of 6 were much lower than those of 1 and 4, suggesting that the amide hydrogen of 1 and benzolactam-V8’s plays a critical role in tumor promotion. However, it is noteworthy that 6 showed significant selectivity in the PKC isozyme surrogate binding.Synthesis, PKC surrogate binding, and tumor-promoting activities in vitro of the new lactone analogue (6) of 8-decylbenzolactam-V8 (4) are described.
Co-reporter:Minoru Tanaka, Kazuhiro Irie, Yu Nakagawa, Yoshimasa Nakamura, Hajime Ohigashi, Paul A Wender
Bioorganic & Medicinal Chemistry Letters 2001 Volume 11(Issue 5) pp:719-722
Publication Date(Web):12 March 2001
DOI:10.1016/S0960-894X(01)00045-2
To investigate the role of the hydroxyl group at position 4 of the phorbol esters in protein kinase C (PKC) binding and function, 4β-deoxy-phorbol-12,13-dibutyrate (4β-deoxy-PDBu, 5a) and 4β-deoxy-phorbol-13-acetate (6a) were synthesized from phorbol (1). The binding affinities of these 4β-deoxy compounds (5a, 6a) to the 13 PKC isozyme C1 domains were quite similar to those of the corresponding 4β-hydroxy compounds (4a, 4b), suggesting that the C4 hydroxyl group of phorbol esters is not necessary for PKC binding. Moreover, functional assays showed that 4β-deoxy-PDBu (5a) exhibited biological activities (Epstein-Barr virus induction and superoxide generation) equally potent to those of PDBu (4a). These solution phase results differ from expectations based on the previously reported solid-phase structure of the complex of PKCδ-C1B and phorbol-13-acetate (4b).Synthesis of 4β-deoxy-phorbol-12,13-dibutyrate (5a) and its biological activities related to tumor promotion are reported.
Co-reporter:Yu Nakagawa, Kazuhiro Irie, Hajime Ohigashi, Hideo Hayashi, Paul A Wender
Bioorganic & Medicinal Chemistry Letters 2000 Volume 10(Issue 18) pp:2087-2090
Publication Date(Web):September 2000
DOI:10.1016/S0960-894X(00)00411-X
To investigate the role of the amide group of (−)-indolactam-V (1) on PKC binding, we synthesized (−)-indothiolactam-V (2), a new thioamide analogue of 1, by microbial conversion using Streptomyces blastmyceticum. Compounds 2 and 1 showed similar binding affinities to conventional PKCs but 2 had lower affinities to novel PKCs, suggesting that novel PKCs recognize amide modifications more effectively than conventional PKCs.





![Carbamic acid, [3-(2-hydroxyethyl)phenyl]-, 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/180100/180002-10-2.png)
![Carbamic acid, [3-(2-hydroxyethyl)phenyl]-, 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/180100/180002-10-2_b.png)
![L-Leucinamide,N-[(phenylmethoxy)carbonyl]-L-leucyl-N-[(1S)-1-formyl-3-methylbutyl]-](http://img.cochemist.com/ccimg/133500/133407-82-6.png)
![L-Leucinamide,N-[(phenylmethoxy)carbonyl]-L-leucyl-N-[(1S)-1-formyl-3-methylbutyl]-](http://img.cochemist.com/ccimg/133500/133407-82-6_b.png)












![BRYOSTATIN 1;(1S,3S,5Z,7R,8E,11S,12S,13E,15S,17R,21R,23R,25S)-25-(ACETYLOXY)-1,11,21-TRIHYDROXY-17-[(1R)-1-HYDROXYETHYL]-5,13-BIS(2-METHOXY-2-OXOETHYLIDENE)-10,10,26,26-TETRAMETHYL-19-OXO-18,27,28,29-TETRAOXATETRACYCLO[21.3.1.13,7.111,15]NONACOS-8-EN-12-Y](http://img.cochemist.com/ccimg/83400/83314-01-6.png)
![BRYOSTATIN 1;(1S,3S,5Z,7R,8E,11S,12S,13E,15S,17R,21R,23R,25S)-25-(ACETYLOXY)-1,11,21-TRIHYDROXY-17-[(1R)-1-HYDROXYETHYL]-5,13-BIS(2-METHOXY-2-OXOETHYLIDENE)-10,10,26,26-TETRAMETHYL-19-OXO-18,27,28,29-TETRAOXATETRACYCLO[21.3.1.13,7.111,15]NONACOS-8-EN-12-Y](http://img.cochemist.com/ccimg/83400/83314-01-6_b.png)
![2,6,10,17-Tetraoxatricyclo[11.3.1.11,5]octadecane-7,11-dione,3-[(1S,4S)-4-(2-bromo-5-hydroxyphenyl)-4-methoxy-1-methylbutyl]-13-hydroxy-9-[(1R)-1-hydroxyethyl]-4,14,16,16-tetramethyl-,(1S,3R,4S,5S,9R,13S,14R)-](http://img.cochemist.com/ccimg/52700/52659-57-1.png)
![2,6,10,17-Tetraoxatricyclo[11.3.1.11,5]octadecane-7,11-dione,3-[(1S,4S)-4-(2-bromo-5-hydroxyphenyl)-4-methoxy-1-methylbutyl]-13-hydroxy-9-[(1R)-1-hydroxyethyl]-4,14,16,16-tetramethyl-,(1S,3R,4S,5S,9R,13S,14R)-](http://img.cochemist.com/ccimg/52700/52659-57-1_b.png)