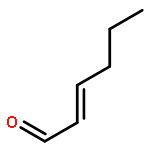
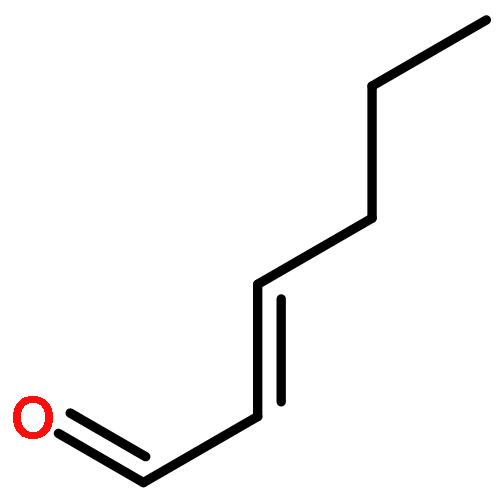
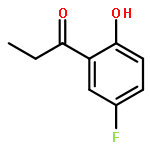
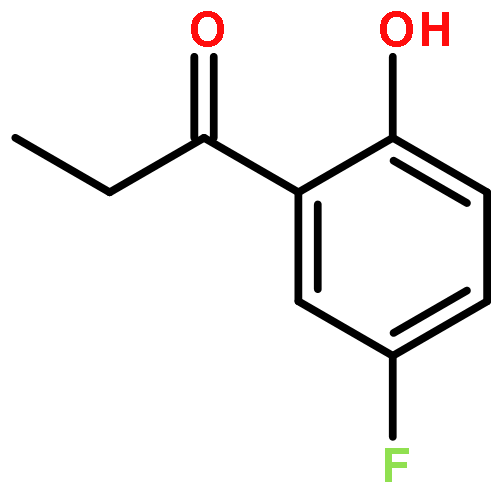
Co-reporter:Junghwa Kim, Nuwan Pannilawithana, and Chae S. Yi
ACS Catalysis December 2, 2016 Volume 6(Issue 12) pp:8395-8395
Publication Date(Web):November 14, 2016
DOI:10.1021/acscatal.6b02186
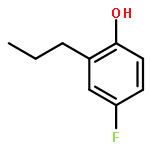
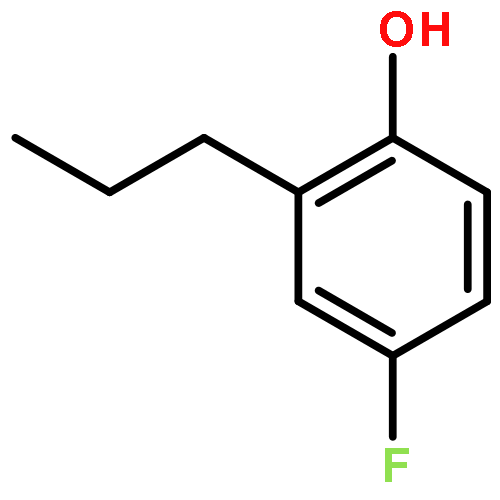
The ruthenium-hydride catalyst has been successfully used for the tandem sp3 C–H dehydrogenation–alkylation reaction of saturated hydrocarbon substrates with alcohols to form the alkyl-substituted alkene and arene products. The analogous one-pot dehydrogenation–insertion of saturated ketones with alkenes and dienes directly yielded synthetically useful 2-alkylphenol and benzopyran products in a highly regio- and stereoselective manner without forming any wasteful byproducts.Keywords: alkylation; dehydrogenation; ruthenium catalyst; saturated hydrocarbon; tandem catalysis;
Co-reporter:Junghwa Kim and Chae S. Yi
ACS Catalysis 2016 Volume 6(Issue 5) pp:3336
Publication Date(Web):April 19, 2016
DOI:10.1021/acscatal.6b00856
The cationic Ru–H complex was found to be an effective catalyst for the intermolecular hydroacylation of aryl-substituted olefins with aldehydes to form branched ketone products. The preliminary kinetic and spectroscopic studies elucidated a ruthenium-acyl complex as the key intermediate species. The catalytic method directly afforded branched ketone products in a highly regioselective manner while tolerating a number of heteroatom functional groups.Keywords: aldehyde; alkene; branched ketone; hydroacylation; ruthenium catalyst
Co-reporter:Hanbin Lee and Chae S. Yi
Organometallics 2016 Volume 35(Issue 11) pp:1973-1977
Publication Date(Web):May 27, 2016
DOI:10.1021/acs.organomet.6b00273
The cationic ruthenium-hydride complex catalyzes the dehydrative C–H coupling reaction of arylamines with 1,2-diols to form the indole products. The analogous coupling of arylamines with 1,3-diols afforded the substituted quinolines. The catalytic method directly forms these coupling products in a highly regioselective manner without generating any toxic byproducts.
Co-reporter:Nishantha Kalutharage;Chae S. Yi
Journal of the American Chemical Society 2015 Volume 137(Issue 34) pp:11105-11114
Publication Date(Web):August 2, 2015
DOI:10.1021/jacs.5b06097
A cationic ruthenium hydride complex, [(C6H6)(PCy3)(CO)RuH]+BF4– (1), with a phenol ligand was found to exhibit high catalytic activity for the hydrogenolysis of carbonyl compounds to yield the corresponding aliphatic products. The catalytic method showed exceptionally high chemoselectivity toward the carbonyl reduction over alkene hydrogenation. Kinetic and spectroscopic studies revealed a strong electronic influence of the phenol ligand on the catalyst activity. The Hammett plot of the hydrogenolysis of 4-methoxyacetophenone displayed two opposite linear slopes for the catalytic system 1/p-X-C6H4OH (ρ = −3.3 for X = OMe, t-Bu, Et, and Me; ρ = +1.5 for X = F, Cl, and CF3). A normal deuterium isotope effect was observed for the hydrogenolysis reaction catalyzed by 1/p-X-C6H4OH with an electron-releasing group (kH/kD = 1.7–2.5; X = OMe, Et), whereas an inverse isotope effect was measured for 1/p-X-C6H4OH with an electron-withdrawing group (kH/kD = 0.6–0.7; X = Cl, CF3). The empirical rate law was determined from the hydrogenolysis of 4-methoxyacetophenone: rate = kobsd[Ru][ketone][H2]−1 for the reaction catalyzed by 1/p-OMe-C6H4OH, and rate = kobsd[Ru][ketone][H2]0 for the reaction catalyzed by 1/p-CF3-C6H4OH. Catalytically relevant dinuclear ruthenium hydride and hydroxo complexes were synthesized, and their structures were established by X-ray crystallography. Two distinct mechanistic pathways are presented for the hydrogenolysis reaction on the basis of these kinetic and spectroscopic data.
Co-reporter:Nishantha Kalutharage and Chae S. Yi
Organic Letters 2015 Volume 17(Issue 7) pp:1778-1781
Publication Date(Web):March 24, 2015
DOI:10.1021/acs.orglett.5b00553
A well-defined cationic Ru–H complex catalyzes reductive etherification of aldehydes and ketones with alcohols. The catalytic method employs environmentally benign water as the solvent and cheaply available molecular hydrogen as the reducing agent to afford unsymmetrical ethers in a highly chemoselective manner.
Co-reporter:Hanbin Lee ;Chae S. Yi
European Journal of Organic Chemistry 2015 Volume 2015( Issue 9) pp:1899-1904
Publication Date(Web):
DOI:10.1002/ejoc.201403518
Abstract
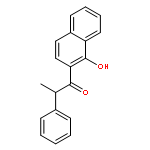
The cationic ruthenium hydride complex [(C6H6)(PCy3)(CO)RuH]+BF4– has been found to be an effective catalyst for the oxidative C–H coupling reaction of phenols with aldehydes to give 2-acylphenol compounds. The coupling of phenols with α,β-unsaturated aldehydes selectively gives the flavene derivatives. The catalytic method mediates direct oxidative C–H coupling of phenol and aldehyde substrates without using any metal oxidants or forming wasteful byproducts.
Co-reporter:Junghwa Kim, Dong-Hwan Lee, Nishantha Kalutharage, and Chae S. Yi
ACS Catalysis 2014 Volume 4(Issue 11) pp:3881
Publication Date(Web):September 26, 2014
DOI:10.1021/cs5012537
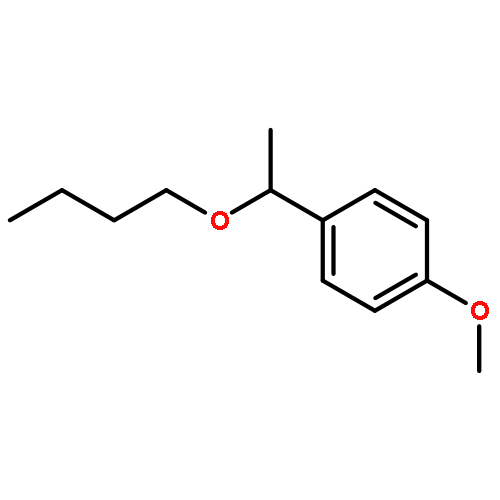
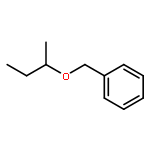
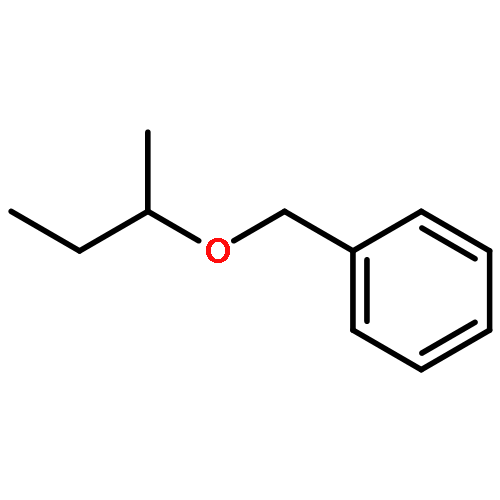
The cationic ruthenium–hydride complex [(C6H6)(PCy3)(CO)RuH]+BF4– catalyzes selective etherification of two different alcohols to form unsymmetrically substituted ethers. The catalytic method exhibits a broad substrate scope while tolerating a range of heteroatom functional groups in forming unsymmetrical ethers, and it is successfully used to directly synthesize a number of highly functionalized chiral nonracemic ethers.Keywords: alcohol; dehydration; ether; ruthenium catalyst
Co-reporter:Ruili Gao, Dale R. Pahls, Thomas R. Cundari, and Chae S. Yi
Organometallics 2014 Volume 33(Issue 23) pp:6937-6944
Publication Date(Web):November 19, 2014
DOI:10.1021/om501019j
The ruthenium hydride complex (PCy3)2(CO)RuHCl was found to be a highly effective catalyst for the regio- and stereoselective hydrosilylation of alkynes to form vinylsilane products. (Z)-Vinylsilane products were selectively formed for sterically nondemanding terminal alkynes, while (E)-vinylsilane products resulted from sterically demanding terminal alkynes. Kinetic data were obtained from the hydrosilylation of phenylacetylene. The phosphine inhibition study showed an uncompetitive Michaelis–Menten type of inhibition kinetics. The empirical rate law rate = kobs[1]1[alkyne]0[silane]0 was established from the reaction rate as a function of both [alkyne] and [silane]. DFT calculations were performed and found that Z/E isomerization is facile via a metallacyclopropene transition state and that the isomerization occurs prior to the silane substrate binding. A detailed mechanistic scheme on the hydrosilylation reaction has been delineated on the basis of both experimental and computational data.
Co-reporter:Nishantha Kalutharage ;Dr. Chae S. Yi
Angewandte Chemie International Edition 2013 Volume 52( Issue 51) pp:13651-13655
Publication Date(Web):
DOI:10.1002/anie.201307766
Co-reporter:Nishantha Kalutharage ;Dr. Chae S. Yi
Angewandte Chemie 2013 Volume 125( Issue 51) pp:13896-13900
Publication Date(Web):
DOI:10.1002/ange.201307766
Co-reporter:Dong-Hwan Lee ; Ki-Hyeok Kwon ;Chae S. Yi
Journal of the American Chemical Society 2012 Volume 134(Issue 17) pp:7325-7328
Publication Date(Web):April 11, 2012
DOI:10.1021/ja302710v
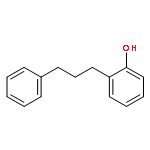
A well-defined cationic Ru–H complex catalyzes the dehydrative C–H alkylation reaction of phenols with alcohols to form ortho-substituted phenol products. Benzofuran derivatives are efficiently synthesized from the dehydrative C–H alkenylation and annulation reaction of phenols with 1,2-diols. The catalytic C–H coupling method employs cheaply available phenols and alcohols, exhibits a broad substrate scope, tolerates carbonyl and amine functional groups, and liberates water as the only byproduct.
Co-reporter:Ki-Hyeok Kwon, Do W. Lee, and Chae S. Yi
Organometallics 2012 Volume 31(Issue 1) pp:495-504
Publication Date(Web):December 14, 2011
DOI:10.1021/om201190v
The cationic ruthenium-hydride complex [(C6H6)(PCy3)(CO)RuH]+BF4– (1) was found to be a highly effective catalyst for the intermolecular conjugate addition of simple alkenes to α,β-unsaturated carbonyl compounds to give (Z)-selective tetrasubstituted olefin products. The analogous coupling reaction of cinnamides with electron-deficient olefins led to the oxidative coupling of two olefinic C–H bonds in forming (E)-selective diene products. The intramolecular version of the coupling reaction efficiently produced indene and bicyclic fulvene derivatives. The empirical rate law for the coupling reaction of ethyl cinnamate with propene was determined as follows: rate = k[1]1[propene]0[cinnamate]−1. A negligible deuterium kinetic isotope effect (kH/kD = 1.1 ± 0.1) was measured from both (E)-C6H5CH═C(CH3)CONHCH3 and (E)-C6H5CD═C(CH3)CONHCH3 with styrene. In contrast, a significant normal isotope effect (kH/kD = 1.7 ± 0.1) was observed from the reaction of (E)-C6H5CH═C(CH3)CONHCH3 with styrene and styrene-d8. A pronounced carbon isotope effect was measured from the coupling reaction of (E)-C6H5CH═CHCO2Et with propene (13C(recovered)/13C(virgin) at Cβ = 1.019(6)), while a negligible carbon isotope effect (13C(recovered)/13C(virgin) at Cβ = 0.999(4)) was obtained from the reaction of (E)-C6H5CH═C(CH3)CONHCH3 with styrene. Hammett plots from the correlation of para-substituted p-X-C6H4CH═CHCO2Et (X = OCH3, CH3, H, F, Cl, CO2Me, CF3) with propene and from the treatment of (E)-C6H5CH═CHCO2Et with a series of para-substituted styrenes p-Y-C6H4CH═CH2 (Y = OCH3, CH3, H, F, Cl, CF3) gave the positive slopes for both cases (ρ = +1.1 ± 0.1 and +1.5 ± 0.1, respectively). Eyring analysis of the coupling reaction led to the thermodynamic parameters, ΔH⧧ = 20 ± 2 kcal mol–1 and ΔS⧧ = −42 ± 5 eu. Two separate mechanistic pathways for the coupling reaction have been proposed on the basis of these kinetic and spectroscopic studies.
Co-reporter:Ruili Gao and Chae S. Yi
ACS Catalysis 2011 Volume 1(Issue 5) pp:544
Publication Date(Web):April 4, 2011
DOI:10.1021/cs200087c
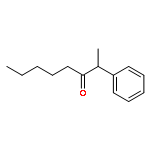
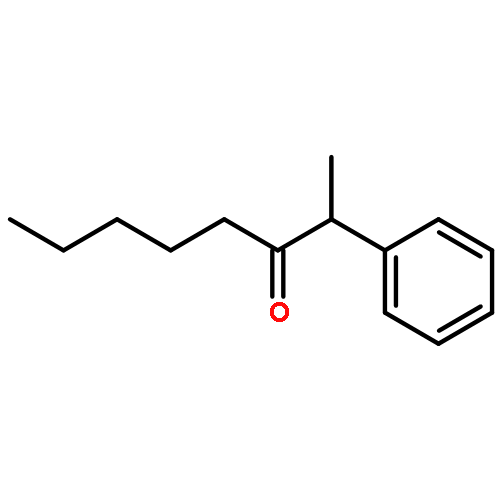
A new catalytic method for the synthesis of silyl enol ethers has been developed from the coupling reaction of ketones with CH2═CHSiMe3 by using a ruthenium hydride catalyst (PCy3)2(CO)RuHCl. The synthetic utility of silyl enol ethers was demonstrated for both Mukaiyama aldol condensation and aminomethylation reactions in forming β-hydroxyketones and β-aminoketones, respectively.Keywords: aldol condensation; ruthenium catalyst; silyl enol ether; vinylsilane
Co-reporter:Chae S. Yi
Journal of Organometallic Chemistry 2011 696(1) pp: 76-80
Publication Date(Web):
DOI:10.1016/j.jorganchem.2010.08.002
Co-reporter:Ki-Hyeok Kwon;Dr. Do W. Lee ;Dr. Chae S. Yi
Angewandte Chemie International Edition 2011 Volume 50( Issue 7) pp:1692-1695
Publication Date(Web):
DOI:10.1002/anie.201006411
Co-reporter:Ki-Hyeok Kwon;Dr. Do W. Lee ;Dr. Chae S. Yi
Angewandte Chemie 2011 Volume 123( Issue 7) pp:1730-1733
Publication Date(Web):
DOI:10.1002/ange.201006411
Co-reporter:Ki-Hyeok Kwon;Chae S. Yi;Dong-Hwan Lee
Science 2011 Volume 333(Issue 6049) pp:1613-1616
Publication Date(Web):16 Sep 2011
DOI:10.1126/science.1208839
A ruthenium catalyst forms carbon-carbon bonds between olefins and alcohols while liberating only water as a by-product.
Co-reporter:Chae S. Yi and Do W. Lee
Organometallics 2010 Volume 29(Issue 8) pp:1883-1885
Publication Date(Web):March 19, 2010
DOI:10.1021/om100051h
The cationic ruthenium−hydride complex [(η6-C6H6)(PCy3)(CO)RuH]+BF4− was found to be a highly effective catalyst for the intermolecular olefination reaction of aryl ketones with cycloalkenes. The preliminary mechanistic analysis revealed that an electrophilic ruthenium−vinyl complex is the key species for mediating both vinyl C−H bond activation and the dehydrative olefination steps of the coupling reaction.
Co-reporter:Ruili Gao and Chae S. Yi
The Journal of Organic Chemistry 2010 Volume 75(Issue 9) pp:3144-3146
Publication Date(Web):April 13, 2010
DOI:10.1021/jo100269y
The cationic ruthenium catalyst Ru3(CO)12/NH4PF6 was found to be highly effective for the intermolecular coupling reaction of pyrroles and terminal alkynes to give gem-selective α-vinylpyrroles. The carbon isotope effect on the α-pyrrole carbon and the Hammett correlation from a series of para-substituted N-arylpyrroles (ρ = −0.90) indicate a rate-limiting C−C bond formation step of the coupling reaction.
Co-reporter:Do W. Lee and Chae S. Yi
Organometallics 2010 Volume 29(Issue 15) pp:3413-3417
Publication Date(Web):July 8, 2010
DOI:10.1021/om100468q
The cationic ruthenium hydride complex [(η6-C6H6)(PCy3)(CO)RuH]+BF4− was found to be a highly regioselective catalyst for the ethylene dimerization reaction to give 2-butene products (TOF = 1910 h−1, >95% selectivity for 2-butenes). The dimerization of styrene exclusively produced the head-to-tail dimer (E)-PhCH(CH3)CH═CHPh at an initial turnover rate of 2300 h−1. A rapid and extensive H/D exchange between the vinyl hydrogens of styrene-d8 and 4-methoxystyrene was observed within 10 min without forming the dimer products at room temperature. The inverse deuterium isotope effect of kH/kD = 0.77 ± 0.10 was measured from the first-order plots on the dimerization reaction of styrene and styrene-d8 in chlorobenzene at 70 °C. The pronounced carbon isotope effect on both vinyl carbons of styrene as measured by using Singleton’s method (13C(recovered)/13C(virgin) at C1 = 1.096 and C2 = 1.042) indicates that the C−C bond formation is the rate-limiting step for the dimerization reaction. The Eyring plot of the dimerization of styrene in the temperature range of 50−90 °C led to ΔH⧧ = 3.3(6) kcal/mol and ΔS⧧ = −35.5(7) eu. An electrophilic addition mechanism has been proposed for the dimerization of styrene.
Co-reporter:Ki-Hyeok Kwon, Do W. Lee, and Chae S. Yi
Organometallics 2010 Volume 29(Issue 22) pp:5748-5750
Publication Date(Web):October 21, 2010
DOI:10.1021/om100764c
The cationic ruthenium hydride complex [(η6-C6H6)(PCy3)(CO)RuH]+BF4− was found to be a highly regioselective catalyst for the oxidative C−H coupling reaction of aryl-substituted amides and unactivated alkenes to give o-alkenylamide products. The kinetic and spectroscopic analyses support a mechanism involving a rapid vinyl C−H activation followed by a rate-limiting C−C bond formation step.
Co-reporter:Chae S. Yi and Do W. Lee
Organometallics 2009 Volume 28(Issue 4) pp:947-949
Publication Date(Web):January 29, 2009
DOI:10.1021/om8010883
The tetranuclear ruthenium-μ-oxo-μ-hydroxo-hydride complex {[(PCy3)(CO)RuH]4(μ4-O)(μ3-OH)(μ2-OH)} (1) was found to be a highly effective catalyst for the transfer dehydrogenation of amines and carbonyl compounds. For example, the initial turnover rate of the dehydrogenation of 2-methylindoline was measured to be 1.9 s−1 with a TON of 7950 after 1 h at 200 °C. The extensive H/D scrambling patterns observed from the dehydrogenation reaction of indoline-N-d1 and indoline-α-d2 suggest a monohydride mechanistic pathway with the C−H bond activation rate-limiting step.
Co-reporter:Chae S. Yi and Do W. Lee
Organometallics 2009 Volume 28(Issue 15) pp:4266-4268
Publication Date(Web):July 6, 2009
DOI:10.1021/om900416k
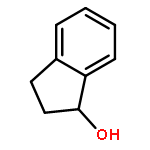
The cationic ruthenium hydride complex, formed in situ from the treatment of the tetranuclear ruthenium hydride complex {[(PCy3)(CO)RuH]4(μ4-O)(μ3-OH)(μ2-OH)} with HBF4·OEt2, was found to be a highly effective catalyst for the intermolecular coupling reaction of arylketones and 1-alkenes to give the substituted indene and ortho-C−H insertion products. The formation of the indene products resulted from the initial alkene isomerization followed by regioselective ortho-C−H insertion of 2-alkene and dehydrative cyclization. The preliminary mechanistic studies revealed a rapid and reversible ortho-C−H bond activation followed by the rate-limiting C−C bond formation step for the coupling reaction.
Co-reporter:Chae S. Yi and Ruili Gao
Organometallics 2009 Volume 28(Issue 22) pp:6585-6592
Publication Date(Web):October 30, 2009
DOI:10.1021/om9007357
The ruthenium-hydride complex (PCy3)2(CO)RuHCl was found to be a highly effective catalyst for the alkyne-to-carboxylic acid coupling reaction to give synthetically useful enol ester products. A strong solvent effect was observed for the ruthenium catalyst in modulating the activity and selectivity; the coupling reaction in CH2Cl2 led to the regioselective formation of gem-enol ester products, while the stereoselective formation of (Z)-enol esters was obtained in THF. The coupling reaction was found to be strongly inhibited by PCy3. The coupling reaction of both PhCO2H/PhC≡CD and PhCO2D/PhC≡CH led to extensive deuterium incorporation on the vinyl positions of the enol ester products. An opposite Hammett value was observed when the correlation of a series of para-substituted p-X-C6H4CO2H (X = OMe, CH3, H, CF3, CN) with phenylacetylene was examined in CDCl3 (ρ = +0.30) and THF (ρ = −0.68). Catalytically relevant Ru-carboxylate and -vinylidene-carboxylate complexes, (PCy3)2(CO)(Cl)Ru(κ2-O2CC6H4-p-OMe) and (PCy3)2(CO)(Cl)RuC(═CHPh)O2CC6H4-p-OMe, were isolated, and the structure of both complexes was completely established by X-ray crystallography. A detailed mechanism of the coupling reaction involving a rate-limiting C−O bond formation step was proposed on the basis of these kinetic and structural studies. The regioselective formation of the gem-enol ester products in CH2Cl2 was rationalized by a direct migratory insertion of the terminal alkyne via a Ru-carboxylate species, whereas the stereoselective formation of (Z)-enol ester products in THF was explained by invoking a Ru-vinylidene species.
Co-reporter:Chae S. Yi and Jie Zhang
Chemical Communications 2008 (Issue 20) pp:2349-2351
Publication Date(Web):21 Apr 2008
DOI:10.1039/B804263B
Substituted bicyclic pyrroles are produced directly from the coupling reaction of 2,5-disubstituted pyrroles with terminal alkynes, involving the activation of multiple C–H bonds and regioselective cyclisation.
Co-reporter:Chae S. Yi ; Tonya N. Zeczycki ;Sergey V. Lindeman
Organometallics 2008 Volume 27(Issue 9) pp:2030-2035
Publication Date(Web):April 9, 2008
DOI:10.1021/om800053q
The tetranuclear ruthenium-oxo-hydroxo-hydride complex {[(PCy3)(CO)RuH]4(μ4-O)(μ3-OH)(μ2-OH)} (1) was found to be a highly cooperative catalyst for the nitrile hydration reaction. The cooperative mechanism of the hydration of benzonitrile was established by Hill inhibition kinetics. The treatment of a nitrile substrate with complex 1 led to the catalytically relevant nitrile-coordinated tetraruthenium complex 3. The X-ray structure of the nitrile-coordinated complex 3 showed a considerably “relaxed” tetrameric core structure compared to that of 1. The hydration of para-substituted benzonitriles p-X-C6H4CN with an electron-withdrawing group (X = Cl, Br, CO2H, CF3) exhibited cooperative kinetics, as indicated by the sigmoidal saturation kinetics, while the hydration of nitriles with an electron-donating group (X = OH, OMe, t-Bu, CH3) obeyed Michaelis–Menten saturation kinetics. The formation of a ruthenium hydride species was observed during the hydration of methacrylonitrile, and its monomeric nature was established by using DOSY NMR techniques.
Co-reporter:Chae S. Yi and Jie Zhang
Chemical Communications 2008(Issue 20) pp:NaN2351-2351
Publication Date(Web):2008/04/21
DOI:10.1039/B804263B
Substituted bicyclic pyrroles are produced directly from the coupling reaction of 2,5-disubstituted pyrroles with terminal alkynes, involving the activation of multiple C–H bonds and regioselective cyclisation.


![Silane, triethyl[(1E)-2-(6-methoxy-2-naphthalenyl)ethenyl]-](http://img.cochemist.com/ccimg/866400/866364-00-3.png)
![Silane, triethyl[(1E)-2-(6-methoxy-2-naphthalenyl)ethenyl]-](http://img.cochemist.com/ccimg/866400/866364-00-3_b.png)
![BENZENE, 1-METHOXY-4-[1-(PHENYLMETHOXY)ETHYL]-](http://img.cochemist.com/ccimg/849200/849198-40-9.png)
![BENZENE, 1-METHOXY-4-[1-(PHENYLMETHOXY)ETHYL]-](http://img.cochemist.com/ccimg/849200/849198-40-9_b.png)
![Benzene, 1-methoxy-4-[(1-methylpropoxy)methyl]-](http://img.cochemist.com/ccimg/732300/732286-12-3.png)
![Benzene, 1-methoxy-4-[(1-methylpropoxy)methyl]-](http://img.cochemist.com/ccimg/732300/732286-12-3_b.png)




![Cyclopent[b]indole, 1,2,3,4-tetrahydro-6-methoxy-](http://img.cochemist.com/ccimg/327100/327021-89-6.png)
![Cyclopent[b]indole, 1,2,3,4-tetrahydro-6-methoxy-](http://img.cochemist.com/ccimg/327100/327021-89-6_b.png)
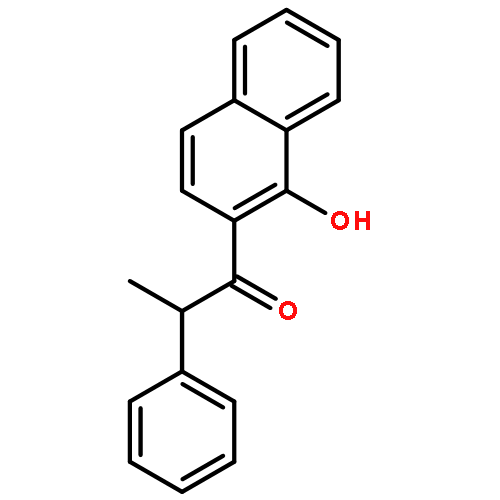
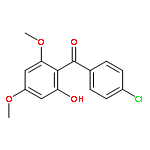
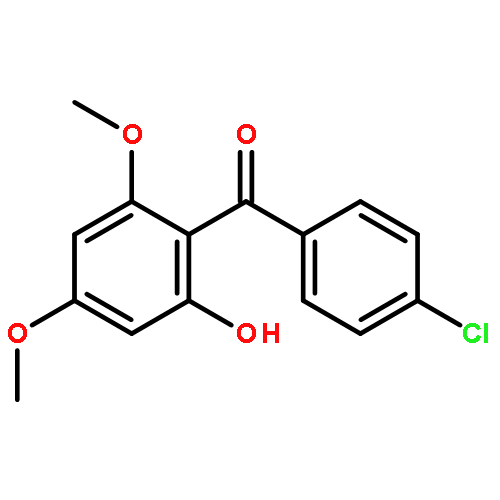


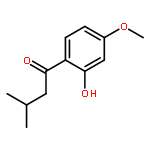
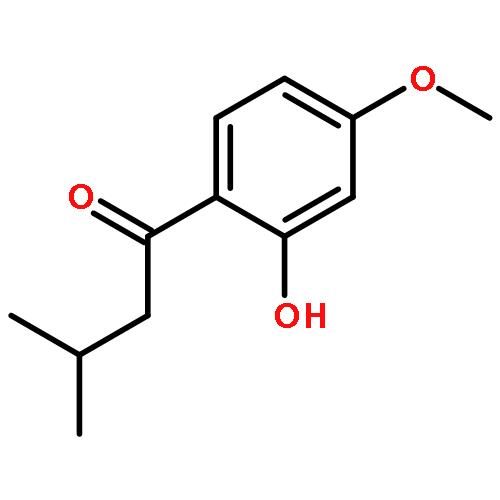
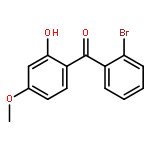
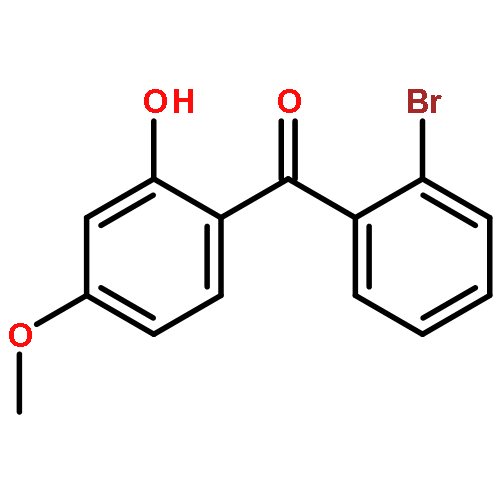
![Benzene, 1-[(cyclohexyloxy)methyl]-4-methoxy-](http://img.cochemist.com/ccimg/156200/156147-58-9.png)
![Benzene, 1-[(cyclohexyloxy)methyl]-4-methoxy-](http://img.cochemist.com/ccimg/156200/156147-58-9_b.png)




![Cyclohexane, [(1Z)-2-(triethylsilyl)ethenyl]-](/data/chemimg/3759000/148991-62-2.png)
![Cyclohexane, [(1Z)-2-(triethylsilyl)ethenyl]-](/data/chemimg/3759000/148991-62-2_b.png)




![Acetamide,2,2-dichloro-N-[1-(hydroxymethyl)-2-(4-nitrophenyl)ethyl]-, (S)- (9CI)](http://img.cochemist.com/ccimg/133200/133191-51-2.png)
![Acetamide,2,2-dichloro-N-[1-(hydroxymethyl)-2-(4-nitrophenyl)ethyl]-, (S)- (9CI)](http://img.cochemist.com/ccimg/133200/133191-51-2_b.png)
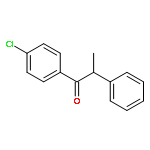
![Silane, triethyl[2-(trimethylsilyl)ethenyl]-, (E)-](/data/chemimg/1073100/125402-66-6.png)
![Silane, triethyl[2-(trimethylsilyl)ethenyl]-, (E)-](/data/chemimg/1073100/125402-66-6_b.png)
![1H-Benz[g]indole, 2,3-diphenyl-](/data/chemimg/3740700/116999-94-1.png)
![1H-Benz[g]indole, 2,3-diphenyl-](/data/chemimg/3740700/116999-94-1_b.png)
![1H-Benz[g]indole, 2-butyl-](http://img.cochemist.com/ccimg/107800/107784-26-9.png)
![1H-Benz[g]indole, 2-butyl-](http://img.cochemist.com/ccimg/107800/107784-26-9_b.png)
![Cyclohexane, [(1E)-2-(triethylsilyl)ethenyl]-](/data/chemimg/3740700/104014-94-0.png)
![Cyclohexane, [(1E)-2-(triethylsilyl)ethenyl]-](/data/chemimg/3740700/104014-94-0_b.png)
![Benzene, 1-methoxy-4-[(1-methylethoxy)methyl]-](http://img.cochemist.com/ccimg/98500/98446-83-4.png)
![Benzene, 1-methoxy-4-[(1-methylethoxy)methyl]-](http://img.cochemist.com/ccimg/98500/98446-83-4_b.png)
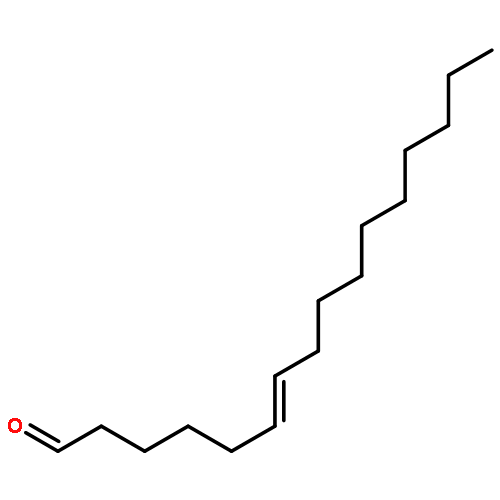
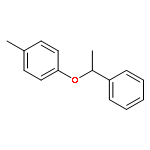
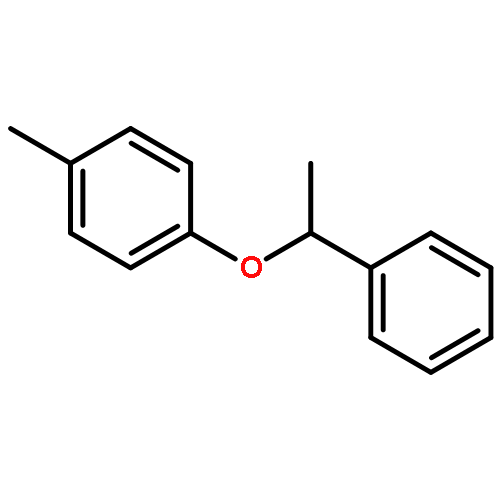
![Benzene, 1-methoxy-4-[1-(1-phenylethoxy)ethyl]-](http://img.cochemist.com/ccimg/88300/88288-59-9.png)
![Benzene, 1-methoxy-4-[1-(1-phenylethoxy)ethyl]-](http://img.cochemist.com/ccimg/88300/88288-59-9_b.png)
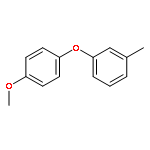
![SILANE, TRIETHYL[(1Z)-2-PHENYLETHENYL]-](http://img.cochemist.com/ccimg/75700/75645-33-9.png)
![SILANE, TRIETHYL[(1Z)-2-PHENYLETHENYL]-](http://img.cochemist.com/ccimg/75700/75645-33-9_b.png)
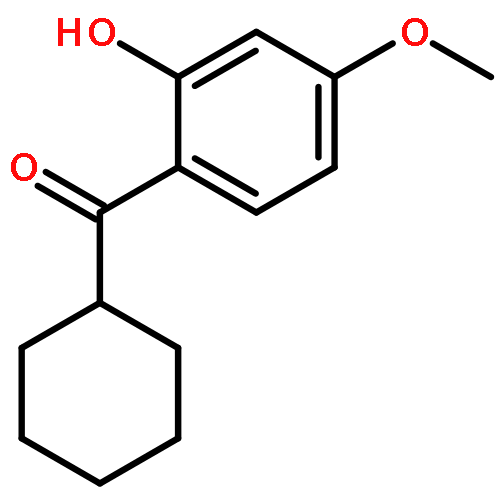
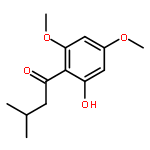
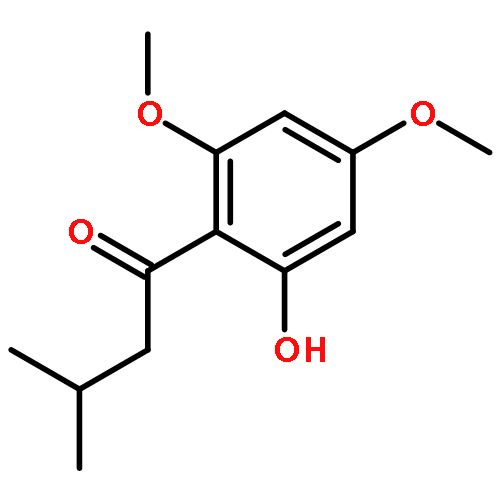


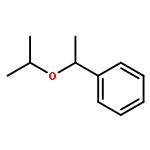
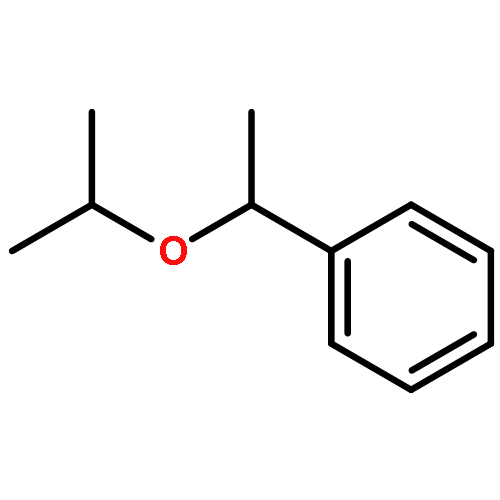


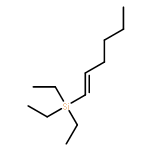
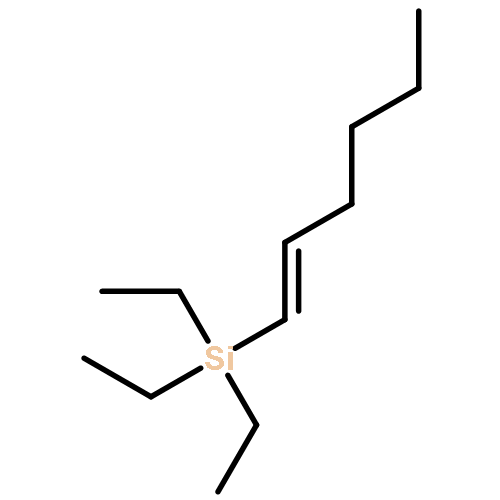
![2H-Phenanthro[9,10-b]pyran, 2,2-diphenyl-](/data/chemimg/3286000/61708-78-9.png)
![2H-Phenanthro[9,10-b]pyran, 2,2-diphenyl-](/data/chemimg/3286000/61708-78-9_b.png)
![Benzene, [(hexyloxy)methyl]-](http://img.cochemist.com/ccimg/61200/61103-84-2.png)
![Benzene, [(hexyloxy)methyl]-](http://img.cochemist.com/ccimg/61200/61103-84-2_b.png)
![2-[6-AMINO-3,5-DICYANO-4-[4-(CYCLOPROPYLMETHOXY)PHENYL]PYRIDIN-2-YL]SULFANYLACETAMIDE](http://img.cochemist.com/ccimg/61100/61001-99-8.png)
![2-[6-AMINO-3,5-DICYANO-4-[4-(CYCLOPROPYLMETHOXY)PHENYL]PYRIDIN-2-YL]SULFANYLACETAMIDE](http://img.cochemist.com/ccimg/61100/61001-99-8_b.png)
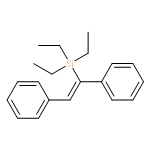








![Benzo[h]quinoline, 3-methyl-](http://img.cochemist.com/ccimg/37100/37062-82-1.png)
![Benzo[h]quinoline, 3-methyl-](http://img.cochemist.com/ccimg/37100/37062-82-1_b.png)
![2,3-DIMETHYL-1H-BENZO[G]INDOLE](http://img.cochemist.com/ccimg/36800/36729-21-2.png)
![2,3-DIMETHYL-1H-BENZO[G]INDOLE](http://img.cochemist.com/ccimg/36800/36729-21-2_b.png)
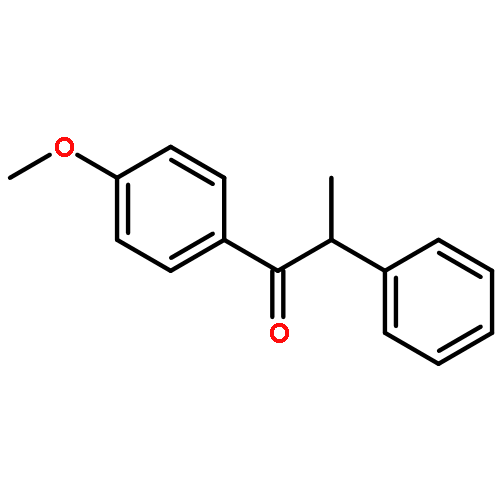
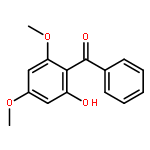
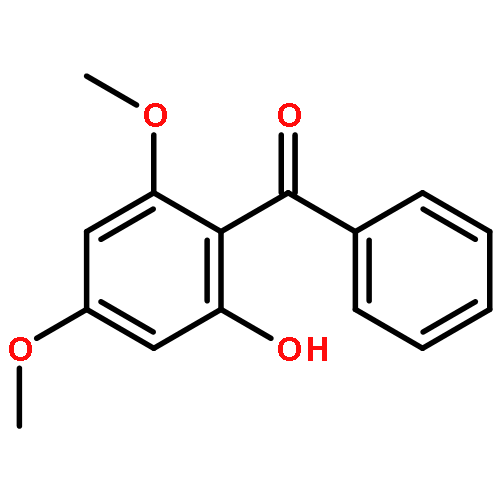
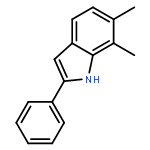
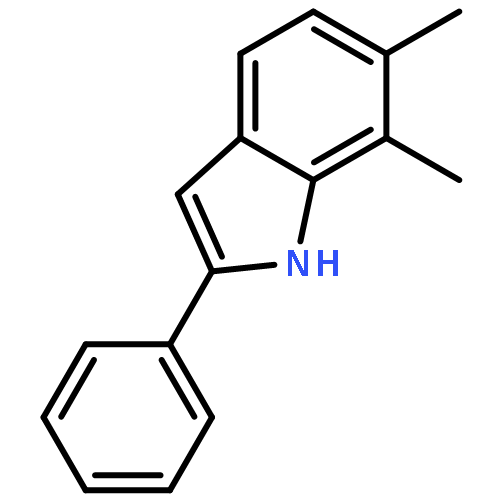
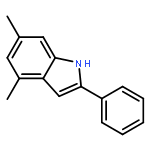
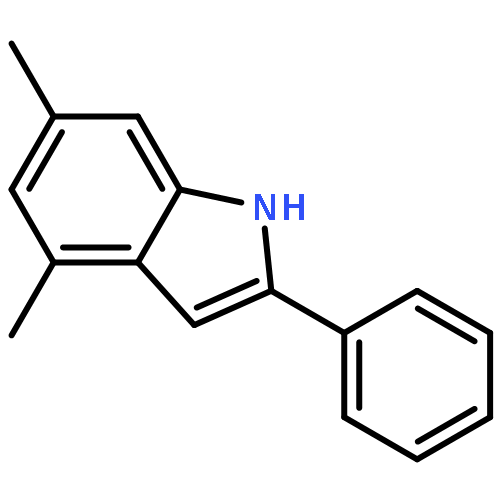
![2-PHENYL-1H-BENZO[G]INDOLE](http://img.cochemist.com/ccimg/33600/33555-17-8.png)
![2-PHENYL-1H-BENZO[G]INDOLE](http://img.cochemist.com/ccimg/33600/33555-17-8_b.png)
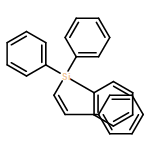
















![2-[2-(hydroxymethyl)phenyl]ethanol](http://img.cochemist.com/ccimg/6400/6346-00-5.png)
![2-[2-(hydroxymethyl)phenyl]ethanol](http://img.cochemist.com/ccimg/6400/6346-00-5_b.png)



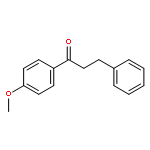





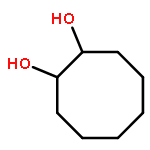



![Benzene,1-methoxy-4-[(phenylmethoxy)methyl]-](http://img.cochemist.com/ccimg/3700/3613-53-4.png)
![Benzene,1-methoxy-4-[(phenylmethoxy)methyl]-](http://img.cochemist.com/ccimg/3700/3613-53-4_b.png)
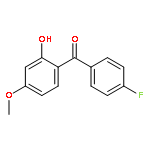
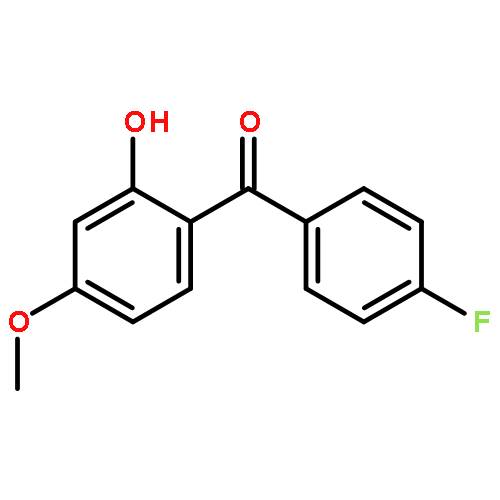











![2,4-dimethylbenzo[h]quinoline](http://img.cochemist.com/ccimg/700/605-67-4.png)
![2,4-dimethylbenzo[h]quinoline](http://img.cochemist.com/ccimg/700/605-67-4_b.png)