Co-reporter:Enrique Iglesia;Do Kyoung Kim
The Journal of Physical Chemistry C November 6, 2008 Volume 112(Issue 44) pp:17235-17243
Publication Date(Web):2017-2-22
DOI:10.1021/jp8062178
This study provides kinetic and isotopic evidence for the identity and kinetic relevance of elementary steps in CH3OH−H2O reactions on monofunctional Cu-based catalysts. H2, CO2, and CO were the only products formed in these reactions, which proceed via kinetically relevant C−H bond activation in chemisorbed methoxides. All other elementary steps are in quasi-equilibrium, consistent with the effects of reactants and products on rates, with isotopic tracing and kinetic isotope evidence, and with the equilibrated nature of water gas shift reactions at all reaction conditions. Turnover rates increased with CH3OH pressure but did not depend on CO, CO2, or H2O concentrations. H2 inhibited CH3OH−H2O reactions by decreasing the concentration of surface methoxide intermediates via quasi-equilibrated CH3OH dissociation steps. Isotopic scrambling was complete among hydroxyl groups in methanol and water, but undetectable among hydroxyl and methyl groups in methanol, consistent with quasi-equilibrated O−H activation and irreversible C−H activation steps. Deuterium substitution at methyl groups in methanol gave normal kinetic isotope effects, whereas substitution at hydroxyl groups in methanol or water led to weaker isotope effects consistent with their thermodynamic origin from quasi-equilibrated O−H activation steps. These mechanistic conclusions are consistent with detailed kinetic data on both large and small Cu clusters and with reforming pathways requiring only Cu surfaces to complete catalytic turnovers. CH3OH−H2O turnover rates increased weakly with Cu dispersion as Cu cluster size decreased from 30 to 5 nm, suggesting that these reactions are insensitive to structure when CH3OH−H2O reaction occurs on Cu via monofunctional pathways limited by H-abstraction from methoxide intermediates. This structure insensitivity may reflect the titration of low-coordination surfaces with strongly held and unreactive forms of adsorbed intermediates, causing turnovers to occur preferentially on low-index surfaces for clusters of varying size.
Co-reporter:Jianwei Liu, David Hibbitts, and Enrique Iglesia
Journal of the American Chemical Society August 30, 2017 Volume 139(Issue 34) pp:11789-11789
Publication Date(Web):August 21, 2017
DOI:10.1021/jacs.7b04606
High CO* coverages lead to rates much higher than Langmuirian treatments predict because co-adsorbate interactions destabilize relevant transition states less than their bound precursors. This is shown here by kinetic and spectroscopic data—interpreted by rate equations modified for thermodynamically nonideal surfaces—and by DFT treatments of CO-covered Ru clusters and lattice models that mimic adlayer densification. At conditions (0.01–1 kPa CO; 500–600 K) which create low CO* coverages (0.3–0.8 ML from in situ infrared spectra), turnover rates are accurately described by Langmuirian models. Infrared bands indicate that adlayers nearly saturate and then gradually densify as pressure increases above 1 kPa CO, and rates become increasingly larger than those predicted from Langmuir treatments (15-fold at 25 kPa and 70-fold at 1 MPa CO). These strong rate enhancements are described here by adapting formalisms for reactions in nonideal and nearly incompressible media (liquids, ultrahigh-pressure gases) to handle the strong co-adsorbate interactions within the nearly incompressible CO* adlayer. These approaches show that rates are enhanced by densifying CO* adlayers because CO hydrogenation has a negative activation area (calculated by DFT), analogous to how increasing pressure enhances rates for liquid-phase reactions with negative activation volumes. Without these co-adsorbate effects and the negative activation area of CO activation, Fischer–Tropsch synthesis would not occur at practical rates. These findings and conceptual frameworks accurately treat dense surface adlayers and are relevant in the general treatment of surface catalysis as it is typically practiced at conditions leading to saturation coverages of reactants or products.
Co-reporter:Michele L. Sarazen, Enrique Iglesia
Journal of Catalysis 2017 Volume 354(Volume 354) pp:
Publication Date(Web):1 October 2017
DOI:10.1016/j.jcat.2017.08.002
•Alkane-alkene mixtures on solid acids lead to CC coupling & alkylation reactions.•Alkane incorporation is limited by hydride transfer (HT) to surface alkoxides.•HT rates increase with increasing substitution and chain length of donor alkane.•Molecular & catalyst descriptors are employed to describe reactivity differences.•Larger pore aluminosilicates better “fit” large, bimolecular HT transition states.Experimental and theoretical methods are used to probe the mechanism and site requirements for C–C bond formation and hydride transfer (HT) reactions of alkane-alkene mixtures on solid acids with diverse acid strength and confining voids. Such methods provide quantitative descriptors of reactivity in terms of the properties of molecules and solids for chemistries that enable the practice of alkylation and oligomerization catalysis. In these processes, chain growth is controlled by HT from alkanes to alkene-derived bound alkoxides formed via oligomerization or β-scission. Transition state (TS) treatments of the elementary steps that mediate these reactions show that HT rates depend on the energies required to desorb alkoxides as carbenium ions and to cleave the weakest C–H bond in gaseous alkanes. These energies serve as accurate molecular descriptors of hydride transfer reactivity and, taken together with the acid strength and van der Waals stabilization properties of catalytic solids, provide the kinetic details required to predict the relative rates at which alkoxides react with alkenes (to form C–C bonds and larger alkenes) or alkanes (to accept H-atoms and desorb as alkanes) for chains with a broad range of size and skeletal structure. Confinement effects reflect the size of the TS and its precursors for C–C coupling and HT relative to the dimensions of the confining voids, which determine how guest species form van der Waals contacts with the host without significant distortions. Smaller voids preferentially stabilize the smaller C–C bond formation TS over the larger structures that mediate HT. Acid strength, in turn, influences the stability of conjugate anions at the ion-pair TS: stronger acids lead to higher turnover rates for C–C coupling and HT, but to similar extents, because their TS structures contain fully-formed framework anions that benefit similarly from their more stable character in stronger acids.Download high-res image (112KB)Download full-size image
Co-reporter:Trenton Otto, Stacey I. Zones, Yongchun Hong, Enrique Iglesia
Journal of Catalysis 2017 Volume 356(Volume 356) pp:
Publication Date(Web):1 December 2017
DOI:10.1016/j.jcat.2017.10.017
•Procedure to prepare cobalt oxide nanoparticles encapsulated within LTA zeolite.•Cobalt oxide nanoparticles are highly dispersed, uniform in size, and thermally stable.•Size-selective catalysis and application to CO, NO, and alcohol oxidation.Small Co3O4 nanoparticles uniformly distributed in size were encapsulated within LTA zeolite crystals in a one-step process through hydrothermal self-assembly of crystalline frameworks around ligated Co2+ precursors. The use of bifunctional ligands containing a chelating bidentate amine functionality and an alkoxysilane moiety prevented the precipitation of Co2+ species as colloidal hydroxides in the highly alkaline synthesis gels, while also allowing the formation of linkages between precursors and the framework during the nucleation and growth of LTA crystals. Oxidative treatments of ligated compounds occluded within zeolite crystals removed ligand residues and formed small Co3O4 nanoparticles visible in transmission electron micrographs. These nanoparticles retained their small size (average diameter 1.5 nm) after oxidative treatment at 620–870 K, a reflection of their stabilization by confinement within zeolite voids. The infrared spectra of adsorbed CO on Co-LTA samples confirmed the absence of Co2+ as exchanged cations or aluminosilicates, indicating the presence of Co oxide clusters, with dynamics and stoichiometry of reduction in H2 corresponding to small Co3O4 clusters. Ethanol oxidation rates on Co-LTA samples, exchanged with K+ or Ca2+ cations to vary the diffusive properties of LTA crystals, indicated that more than 97% of the active surfaces on these Co3O4 clusters resided within zeolite crystals, where ethanol and O2 concentrations depend on the diffusive properties of the LTA framework. The Co3O4 clusters prepared by these methods, in contrast with Co2+ in exchanged or aluminosilicate forms, exhibit reactivity in CO and NO oxidation. Their turnover rates (per exposed Co atom), however, were lower than on bulk Co3O4 powders, because of the combined effects of diffusional constraints imposed by the confining framework and the small size of these clusters, which leads to lower intrinsic reactivities as a result of their more difficult reduction during catalytic redox cycles. These clusters would be attractive in catalytic applications requiring stability against sintering during reaction or regeneration, reactant or product shape selectivity, or protection from contact with large molecules that block active surfaces. Such oxide clusters cannot be formed by sequential ion exchange, detachment by reduction to Co0, and re-oxidation because the extremely high temperatures required for reduction destroy the aluminosilicate frameworks. The synthesis protocols and their mechanistic interpretations described herein represent a conceptual and practical platform for the encapsulation of nanoparticles of base elements within a broad range of confining crystalline environments through one-step hydrothermal self-assembly.Download high-res image (173KB)Download full-size image
Co-reporter:Shuai Wang;Iker Agirrezabal-Telleria;Aditya Bhan;Dante Simonetti;Kazuhiro Takanabe
Faraday Discussions 2017 (Volume 197) pp:9-39
Publication Date(Web):2017/05/02
DOI:10.1039/C7FD00018A
This account illustrates concepts in chemical kinetics underpinned by the formalism of transition state theory using catalytic processes that enable the synthesis of molecules suitable as fuels from C1 and oxygenate reactants. Such feedstocks provide an essential bridge towards a carbon-free energy future, but their volatility and low energy density require the formation of new C–C bonds and the removal of oxygen. These transformations are described here through recent advances in our understanding of the mechanisms and site requirements in catalysis by surfaces, with emphasis on enabling concepts that tackle ubiquitous reactivity and selectivity challenges. The hurdles in forming the first C–C bond from C1 molecules are illustrated by the oxidative coupling of methane, in which surface O-atoms form OH radicals from O2 and H2O molecules. These gaseous OH species act as strong H-abstractors and activate C–H bonds with earlier transition states than oxide surfaces, thus rendering activation rates less sensitive to the weaker C–H bonds in larger alkane products than in CH4 reactants. Anhydrous carbonylation of dimethyl ether forms a single C–C bond on protons residing within inorganic voids that preferentially stabilize the kinetically-relevant transition state through van der Waals interactions that compensate for the weak CO nucleophile. Similar solvation effects, but by intrapore liquids instead of inorganic hosts, also become evident as alkenes condense within MCM-41 channels containing isolated Ni2+ active sites during dimerization reactions. Intrapore liquids preferentially stabilize transition states for C–C bond formation and product desorption, leading to unprecedented reactivity and site stability at sub-ambient temperatures and to 1-alkene dimer selectivities previously achieved only on organometallic systems with co-catalysts or activators. C1 homologation selectively forms C4 and C7 chains with a specific backbone (isobutane, triptane) on solid acids, because of methylative growth and hydride transfer rates that reflect the stability of their carbenium ion transition states and are unperturbed by side reactions at low temperatures. Aldol condensation of carbonyl compounds and ketonization of carboxylic acids form new C–C bonds concurrently with O-removal. These reactions involve analogous elementary steps and occur on acid–base site pairs on TiO2 and ZrO2 catalysts. Condensations are limited by α-H abstraction to form enolates via concerted interactions with predominantly unoccupied acid–base pairs. Ketonization is mediated instead by C–C bond formation between hydroxy-enolates and monodentate carboxylates on site pairs nearly saturated by carboxylates. Both reactions are rendered practical through bifunctional strategies, in which H2 and a Cu catalyst function scavenge unreactive intermediates, prevent sequential reactions and concomitant deactivation, and remove thermodynamic bottlenecks. Alkanal–alkene Prins condensations on solid acids occur concurrently with alkene dimerization and form molecules with new C–C bonds as skeletal isomers unattainable by other routes. Their respective transition states are of similar size, leading to selectivities that cannot sense the presence of a confining host. Prins condensation reactions benefit from weaker acid sites because their transition states are less charged than those for oligomerization and consequently less sensitive to conjugate anions that become less stable as acids weaken.
Co-reporter:Iker Agirrezabal-Telleria, Enrique Iglesia
Journal of Catalysis 2017 Volume 352(Volume 352) pp:
Publication Date(Web):1 August 2017
DOI:10.1016/j.jcat.2017.06.025
•Intrapore liquid ethene leads to unprecedented Ni site stability and reactivity at sub-ambient temperatures.•Stability reflects solvation of late desorption transition states that prevent secondary growth and isomerization.•Stability allows elucidation of mechanism and of involvement of isolated grafted {Ni-OH}+ species as active sites.•Titrations during reaction demonstrate concerted involvement of protons and Ni centers in ethene dimerization events.•Transition state treatments of thermodynamic non-idealities account for reactivity enhancements by intrapore liquids.This study reports the high and stable ethene dimerization turnover rates conferred upon Ni-based active sites at subambient temperatures by the condensation of liquid ethene reactants within ordered mesopores in Al-MCM-41. Such active and stable catalysts do not require the activators or co-catalysts essential for organometallic Ni-based catalysts and their robust porous framework allows their full regeneration by thermal treatments in inert or oxidizing environments when deactivation occurs. Dimerization rates (per Ni) are independent of Ni content below Ni2+/H+ exchange ratios of unity, consistent with isolated (Ni-OH)+ cations as active moieties, and with the formation of inactive NiO oligomers at higher Ni contents. Both CO and 2,6 di-tertbutylpyridine, used as titrants of Ni centers and protons, respectively, fully suppressed reactivity, indicating that the Ni2+ and OH− centers in (Ni-OH)+ are involved in stabilizing the kinetically-relevant CC formation transition state. Deactivation constants decreased abruptly to undetectable values at those temperatures and ethene pressures that formed an extended liquid phase within the MCM-41 mesopores that confine the Ni active sites. This remarkable shift from rapid to undetectable deactivation reflects how intrapore liquids preferentially stabilize the late transition states that mediate the desorption of bound 1-butenes before subsequent isomerization and CC bond formation events during one surface sojourn. Their preferential desorption inhibits the formation of regioisomers and larger oligomers, thus inhibiting deactivation while also leading to very high C4 selectivities among products and to the predominant presence of 1-butene among linear C4 alkenes. Such unprecedented stability and high selectivities allow accurate kinetic measurements and their rigorous mechanistic interpretation in terms of transition state formalisms of chemical dynamics in thermodynamic non-ideal media. Dimerization turnovers involve kinetically-relevant CC bond formation between unbound ethene molecules and an ethene bound at low coverages on (Ni-OH)+ moieties. Such catalytic sequences lead to dimerization rates with a nearly second-order dependence in ethene fugacity, but with an effective rate constant that benefits from the stabilization of the CC bond formation transition state as intrapore liquids form. The essential elimination of deactivation events and secondary reactions by such liquids leads to dimerization rates and 1-butene selectivities that are much higher at 240–260 K with liquid-filled mesopores than at 448 K, conditions that maintain all species in their gaseous form. In addition to their significant practical impact, these results provide compelling evidence for the ability of an extended liquid to create a thermodynamically non-ideal environment that markedly alters reaction pathways through the selective stabilization of specific transition states based on their location along the reaction coordinate for a given elementary step.Download high-res image (73KB)Download full-size image
Co-reporter:Shuai Wang, Enrique Iglesia
Journal of Catalysis 2017 Volume 352(Volume 352) pp:
Publication Date(Web):1 August 2017
DOI:10.1016/j.jcat.2017.06.012
•CC bond formation steps limit Prins condensation and oligomerization reactions.•Confinement preferentially stabilizes transition states in voids of intermediate size.•Smaller voids lead to enthalpic/entropic penalties through host-guest structural distortions.•Larger voids cause ineffective van der Waals host-guest contacts.•MFI intersections show a unique selectivity to cyclization of 2,5-dimethyl-hexadiene.The effects of confinement of transition states and bound intermediates are described here based on kinetic and theoretical studies for Prins condensation and oligomerization reactions of isobutanal-isobutene reactant mixtures catalyzed by Brønsted acid sites of similar strength but confined within diverse microporous (TON, BEA, MFI, FAU) and mesoporous (Al-MCM-41) aluminosilicate structures. Kinetic data and density functional theory (DFT) show that smaller voids lead to more stable bound monomers derived from isobutene and isobutanal and to protons nearly saturated with H-bonded isobutanal. Prins condensation forms 2,5-dimethyl-hexadiene regioisomers (2,5-DMH) with rate constants that reflect the different stability of the kinetically-relevant CC bond formation transition states and their H-bonded isobutanal precursors. Their differences in size lead to reactivities that depend sensitively on the size and shape of the confining voids in each aluminosilicate and even on the location of the Al-atoms (and the protons) within a given framework. Specifically, FAU leads to the highest turnover rates, because larger voids in Al-MCM-41 form less effective van der Waals contacts with the transition state, while the smaller voids within TON and BEA lead to enthalpic and entropic penalties brought forth by host-guest distortions. MFI cage-like intersections provide more effective stabilization than BEA cylindrical channels of similar size and lead to higher turnover rates and smaller DFT-derived activation free energies; these subtle effects of void shape on reactivity are neglected in descriptions of the confining frameworks based on spherical constructs. Reactivity descriptors require instead theoretical treatments that rigorously account for van der Waals interactions and for the entropy losses and host-guest distortions required for confinement, as reported here from confinement free energies ensemble-averaged over all crystallographic locations in each aluminosilicate structure. The ratios of rate constants for these parallel isobutene dimerization and Prins condensation reactions depend only weakly on the nature of the aluminosilicate framework, because of the similar size and shape of their respective transition states and bound precursors. Transition states for secondary skeletal isomerization and cyclization of 2,5-DMH condensation products, however, are stabilized more effectively (relative to condensation transition states) within voids of intermediate size (BEA, MFI), leading to higher selectivity to secondary products. MFI cage-like structures specifically catalyze the conversion of 2,5-DMH to cyclic 1,4-dimethyl-cyclohexene isomers, direct precursors to p-xylene, with unique specificity.Download high-res image (140KB)Download full-size image
Co-reporter:Shuai Wang and Enrique Iglesia
ACS Catalysis 2016 Volume 6(Issue 11) pp:7664
Publication Date(Web):October 12, 2016
DOI:10.1021/acscatal.6b02171
The selectivity to 2,5-dimethyl-hexadiene isomers (2,5-DMH) via acid-catalyzed isobutanal–isobutene Prins condensation is limited by isobutene oligomerization reactions (to 2,4,4-trimethyl-pentene isomers) and by skeletal isomerization and cyclization of the primary 2,5-DMH products of Prins condensation. Experiment and theory are used here to assess and interpret acid strength effects on the reactivity and selectivity for isobutanal–isobutene Prins condensation routes to 2,5-DMH, useful as precursors to p-xylene. Non-coordinating 2,6-di-tert-butylpyridine titrants fully suppress reactivity on Keggin heteropolyacids, niobic acid, and mesoporous and microporous aluminosilicates, indicating that Prins condensation, parallel isobutene oligomerization, and secondary skeletal isomerization and cyclization of primary 2,5-DMH products occur exclusively on Brønsted acid sites. The number of titrants required to suppress rates allows site counts for active protons, a requirement for comparing reactivity among solid acids as turnover rates, as well as for the rigorous benchmarking of mechanistic proposals by theory and experiment. Kinetic and theoretical treatments show that both reactions involve kinetically relevant C–C bond formation elementary steps mediated by cationic C–C coupling transition states. Transition state charges increase with increasing acid strength for Prins condensation, becoming full carbenium-ions only on the stronger acids. Oligomerization transition state structures, in contrast, remain full ion-pairs, irrespective of acid strength. Turnover rates for both reactions increase with acid strength, but oligomerization transition states preferentially benefit from the greater stability of the conjugate anions in the stronger acids, leading to higher 2,5-DMH selectivities on weaker acids (niobic acid, aluminosilicates). These trends and findings are consistent with theoretical estimates of activation free energies for Prins condensation and oligomerization elementary steps on aluminosilicate slab and Keggin heteropolyacid cluster models. High 2,5-DMH selectivities require weak acids, which do not form a full ion-pair at transition states and thus benefit from significant stabilization by residual covalency. These trends demonstrate the previously unrecognized consequences of incomplete proton transfer at oxygen-containing transition states in dampening the effects of acid strength, which contrast the full ion-pair transition states and stronger acid strength effects in hydrocarbon rearrangements on solids acids of catalytic relevance. These mechanistic conclusions and the specific example used to illustrate them led us to conclude that reaction routes involving O-containing molecules become prevalent over hydrocarbon rearrangements on weak acids when parallel routes are accessible in mixtures of oxygenate and hydrocarbon reactants.Keywords: 2,5-dimethyl-hexadienes; acid strength; density functional theory; isobutanal; isobutene; oligomerization; Prins reaction; solid acid
Co-reporter:Alexandra M. Landry and Enrique Iglesia
Chemistry of Materials 2016 Volume 28(Issue 16) pp:5872
Publication Date(Web):July 22, 2016
DOI:10.1021/acs.chemmater.6b02346
We report the synthesis of bimetallic AuPt nanoparticles (3.3–4.3 nm) of uniform size and composition using colloidal methods and reagents containing only C, H, O, and N. These clusters were dispersed onto SiO2 and treated at low temperatures in the presence of reductants to remove all surface residues without concomitant agglomeration, thus leading to bimetallic structures suitable for mechanistic inquiries into bimetallic effects on surface reactivity. Synthesis protocols exploit and generalize galvanic displacement-reduction (GDR) processes previously used to prepare AuPd clusters; these routes promote bimetallic mixing but become more challenging for systems (e.g., AuPt) with smaller reduction potential differences and less favorable mixing enthalpies than AuPd. These hurdles are addressed here through procedural modifications that inhibit the formation of large Au-rich clusters, which compromise size and compositional uniformity. In doing so, we extend GDR techniques to endothermic alloys with elements of more similar redox properties. Higher temperatures and lower Au3+ precursor concentrations promoted metal mixing and inhibited homogeneous and heterogeneous nucleation. Cluster size and compositional uniformity were confirmed by UV–visible spectroscopy during and after colloid formation, transmission electron microscopy, and high-angle annular dark-field (HAADF) imaging with energy-dispersive X-ray spectroscopy (EDS). Particle-by-particle EDS analysis and HAADF imaging demonstrated the prevalence of GDR processes in AuPd bimetallic cluster assembly. These methods also showed that size-dependent intracluster diffusion during AuPt cluster formation, driven by unfavorable AuPt mixing thermodynamics, leads to Au surface enrichment, thus promoting autocatalytic Au deposition. This rigorous mechanistic comparison of AuPt and AuPd systems provides essential guidance and specific control variables and procedures for the synthesis of other bimetallic systems based on the redox potential differences and mixing thermodynamics of their two components.
Co-reporter:David D. Hibbitts, David W. Flaherty, and Enrique Iglesia
ACS Catalysis 2016 Volume 6(Issue 1) pp:469
Publication Date(Web):December 18, 2015
DOI:10.1021/acscatal.5b01950
The kinetic relevance and rates of elementary steps involved in C–C bond hydrogenolysis for isobutane, neopentane, and 2,3-dimethylbutane reactants were systematically probed using activation enthalpies and free energies derived from density functional theory. Previous studies showed that C–C cleavage in alkanes occurs via unsaturated species formed in fast quasi-equilibrated C–H activation steps, leading to rates that decrease with increasing H2 pressure, because of a concomitant decrease in the concentration of the relevant transition states. This study, together with previous findings for n-alkanes, provides a general mechanistic construct for the analysis and prediction of C–C hydrogenolysis rates on metals. C–C cleavage in alkanes is preceded by the loss of two H atoms and the formation of two C-metal (C–M) bonds for each 1C and 2C atom involved in the C–C bond. Metal atoms transfer electrons into the 1C and 2C atoms as C–C bonds cleave and additional C–M bonds form. 3C and 4C atoms of isobutane, neopentane, and 2,3-dimethylbutane, however, do not lose H atoms before C–C cleavage, and thus, transition states cannot bind the 3C and 4C atoms in the C–C bond being cleaved to surface metal atoms. C–H activation occurs instead at 1C atoms vicinal to the C–C bond, which lose all H atoms and form three C–M bonds. These transition states involve electron transfer into the metal surface, leading to a net positive charge at the 3C and 4C atoms; these atoms exhibit sp2 geometry and resemble carbenium ions at the C–C cleavage transition state, in which they are not bound to the metal surface. These mechanistic features accurately describe measured H2 effects, activation enthalpies, and entropies, and furthermore, they provide the molecular details required to understand and predict the effects of temperature on hydrogenolysis rates and on the location of C–C bond cleavage within a given alkane reagent. The result shown and the conclusions reached are supported by rigorous theoretical assessments for C–C cleavage within about 200 intermediates on Ir surfaces, and the results appear to be applicable to other metals (Rh, Ru, and Pt), which show kinetic behavior similar to Ir.Keywords: alkane activation; density functional theory; hydrogenolysis; kinetics; metal catalysis
Co-reporter:Prashant Deshlahra and Enrique Iglesia
ACS Catalysis 2016 Volume 6(Issue 8) pp:5386
Publication Date(Web):July 20, 2016
DOI:10.1021/acscatal.6b01402
Density functional theory and classical electrostatics are used to develop reactivity descriptors for catalysis by solid acids. Acid strength, as deprotonation energies (DPE), reflects the charge reorganization required to disrupt covalent OH bonds in inorganic acids and the electrostatic forces that resist the separation of protons from conjugate anions. Both charge reorganization (covalent) and electrostatic (ionic) components vary monotonically with DPE on solid acids with different heteroatoms within a given type of oxide framework, but their relative contributions differ among different acid types. Ion-pair transition states recover predominantly the ionic part of the DPE, and the extent to which they recover each component is a unique property of a transition state and thus of an acid-catalyzed reaction, independent of the acid strength or type. These fractional recoveries, together with the ionic and covalent DPE components, a unique property of a solid acid, provide a general and complete descriptor of reactivity, which we illustrate here for diverse reactions (proton shuttling, H2O elimination, methyl shift, ring contraction) on several types of solid acids (Mo- and W-based polyoxometalate clusters with S, P, Si, Al, and Co central atoms and MFI type heterosilicates with Al, Ga, Fe, and B heteroatoms). For protons confined within small voids of heterosilicates, the transition state stabilization and reactivity depend additionally on van der Waals interactions that are unrelated to acid strength.Keywords: Brønsted acid catalysis; dehydration; isomerization; noncovalent interactions; proton shuttling; protonation; thermochemical cycles
Co-reporter:Michele L. Sarazen, Eric Doskocil, and Enrique Iglesia
ACS Catalysis 2016 Volume 6(Issue 10) pp:7059
Publication Date(Web):September 21, 2016
DOI:10.1021/acscatal.6b02128
The effects of channel connectivity, void environment, and acid strength on the relative rates of oligomerization, β-scission, and isomerization reactions during light alkene conversion (ethene, propene, isobutene; 2–400 kPa alkene; 473–533 K) were examined on microporous (TON, MFI, MOR, BEA, FAU) and mesoporous (amorphous silica–alumina (SiAl), MCM-41, Keggin POM) Brønsted acids with a broad range of confining voids and acid strength. Skeletal and regioisomers equilibrate under all conditions of pressure and conversion and on all catalysts, irrespective of their acid strength, void size, or framework connectivity, consistent with rapid hydride and methyl shifts of alkoxides intermediates and with their fast adsorption–desorption steps. Such equilibration is evident from detailed chemical speciation of the products and also from intramolecular isotopic scrambling in all oligomers formed from 2-13C-propene on TON, MFI, SiAl, and POM clusters. Previous claims of kinetic control of skeletal isomers in oligomerization catalysis through shape-selective effects conferred by void environments may have used inaccurate tabulated thermodynamics, as we show in this study. The void environment, however, influences the size distribution of the chains formed in these acid-catalyzed alkene reactions. One-dimensional microporous aluminosilicates predominantly form true oligomers, those expected from dimerization and subsequent oligomerization events for a given reactant alkene; such chains are preserved because they cannot grow to sizes that would inhibit their diffusion through essentially cylindrical channels in these frameworks. Amorphous SiAl and colloidal silica-supported POM clusters contain acid sites of very different strength; both exhibit size variations across the void space, but at length scales much larger than molecular diameters, thus preserving true oligomers by allowing them to egress the void before β-scission events. Mesoporous acids of very different strength (POM, SiAl) give similar true isomer selectivities, as also observed on MFI structures with different heteroatoms (X-MFI, where X = Al, Ga, Fe, B), which also differ in acid strength; this insensitivity reflects oligomerization and β-scission reactions that involve similar ion-pair transition states and therefore depend similarly on the stability of the conjugate anion. Three-dimensional microporous frameworks contain voids larger than their interconnecting paths, an inherent consequence of intersecting channels and cage–window structures. As a result, oligomers can reach sizes that restrict their diffusion through the interconnections, until β-scission events form smaller and faster diffusing chains. These undulations are of molecular dimensions and their magnitude, which is defined here as the ratio of the largest scale to the smallest scale along intracrystal diffusion paths, determines the extent to which oligomerization–scission cycles contribute to the size distribution of products. These contributions are evident in the extent to which chain size and the number of 13C atoms in each molecule formed from 2-13C-propene approach their binomial distributions, as they do on microporous acids with significant undulations. The general nature of these conclusions is evident from the similar effects of void shape and connectivity and of acid strength on selectivity for ethene, propene, and isobutene reactants.Keywords: Brønsted acid catalysis; oligomerization; skeletal isomerization; zeolites; β-scission
Co-reporter:Ya-Huei (Cathy) Chin
The Journal of Physical Chemistry C 2016 Volume 120(Issue 3) pp:1446-1460
Publication Date(Web):January 12, 2016
DOI:10.1021/acs.jpcc.5b06677
Methane oxidation rates uncorrupted by nonchemical effects of transport, taken together with stoichiometric oxygen uptake (oxidation cycle) and evolution (decomposition cycle) data, are used to establish for the first time a set of conditions required for true thermodynamic equilibrium during metal-to-oxide interconversions in small Pd clusters (1.8–8.8 nm). These conditions allow us to assess the intrinsic thermodynamics of small Pd clusters and their catalytic effects in CH4 oxidation. PdO decomposition in the absence of CH4 deviates from equilibrium, as this step is limited by the nucleation of an oxygen vacancy ensemble on oxide domains. The nucleation bottleneck is removed by CH4 during its catalytic sojourns, when CH4 pressure and the related rates exceed a critical value, because CH4 effectively removes the oxygen adatoms near an oxygen vacancy site via C–H bond activation on an oxygen–oxygen vacancy site pair that converts the O* adatom to a hydroxyl intermediate, which desorbs as H2O in sequential steps. CH4 oxidation turnovers promote the nucleation of oxygen vacancy ensembles at conditions that maintain the global oxygen equilibration, as confirmed from the absence of CH4 oxidation rate hysteresis in both Pd oxidation and PdO decomposition cycles and from coincidence of rate and oxygen content profiles during Pd oxidation. A theoretical construction decoupling the inherent cluster size variance from cluster diameter effects shows marked effects of size on bulk phase transition. The bulk phase transition occurs at lower oxygen chemical potentials for the smaller clusters, which confirm their more negative Gibbs free energy for PdO formation than the large structures. The bulk phase transition converts O*–O* adatom sites to Pd2+–O2– ion pairs that are more effective for the kinetically relevant C–H bond activation in CH4. These effects of size on the thermodynamics and reactivities of small clusters illustrated in this study are general and extend beyond the Pd–PdO system.
Co-reporter:William Knaeble
The Journal of Physical Chemistry C 2016 Volume 120(Issue 6) pp:3371-3389
Publication Date(Web):January 21, 2016
DOI:10.1021/acs.jpcc.5b11127
Elementary steps that mediate ethanol dehydration to alkenes and ethers are determined here from rate and selectivity data on solid acids of diverse acid strength and known structure and free energies derived from density functional theory (DFT). Measured ethene and ether formation rates that differed from those expected from accepted monomolecular and bimolecular routes led to our systematic enumeration of plausible dehydration routes and to a rigorous assessment of their contributions to the products formed. H-bonded monomers, protonated alkanol dimers, and alkoxides are the prevalent bound intermediates at conditions relevant to the practice of dehydration catalysis. We conclude that direct and sequential (alkoxide-mediated) routes contribute to ether formation via SN2-type reactions; alkenes form preferentially from sequential routes via monomolecular and bimolecular syn-E2-type eliminations; and alkoxides form via bimolecular SN2-type substitutions. The prevalence of these elementary steps and their kinetic relevance are consistent with measured kinetic and thermodynamic parameters, which agree with values from DFT-derived free energies and with the effects of acid strength on rates, selectivities, and rate constants; such effects reflect the relative charges in transition states and their relevant precursors. Dehydration turnover rates, but not selectivities, depend on acid strength because transition states are more highly charged than their relevant precursors, but similar in charge for transition states that mediate the competing pathways responsible for selectivity.
Co-reporter:David D. Hibbitts
The Journal of Physical Chemistry C 2016 Volume 120(Issue 15) pp:8125-8138
Publication Date(Web):April 12, 2016
DOI:10.1021/acs.jpcc.6b00323
C–C cleavage in C2–C10 n-alkanes involves quasi-equilibrated C–H activation steps to form dehydrogenated intermediates on surfaces saturated with H atoms. These reactions are inhibited by H2 to similar extents for C–C bonds of similar substitution in all acyclic and cyclic alkanes and, thus, show similar kinetic dependences on H2 pressure. Yet, turnover rates depend sensitively on chain length because of differences in activation enthalpies (ΔH⧧) and entropies (ΔS⧧) whose mechanistic origins remain unclear. Density functional theory (DFT) estimates of ΔH⧧ and ΔG⧧ for C–C cleavage via >150 plausible elementary steps for propane and n-butane reactants on Ir show that hydrogenolysis occurs via α,β-bound RC*–C*R′⧧ transition states (R = H, CxH2x+1) in which two H atoms are removed from each C*. Calculated ΔH⧧ values decrease with increasing alkane chain length (C2–C8), consistent with experiment, because attractive van der Waals interactions with surfaces preferentially stabilize larger transition states. A concomitant increase in ΔS⧧, evident from experiments, is not captured by periodic DFT methods, which treat low-frequency vibrational modes inaccurately, but statistical mechanics treatments describe such effects well for RC*–C*R⧧ species, as previously reported. These findings, together with parallel studies of the cleavage of more substituted C–C bonds in branched and cyclic alkanes, account for the reasons that chain length and substitution influence ΔH⧧ and ΔS⧧ values and the dependence of rates on H2 pressure and consequently explain differences in hydrogenolysis reactivities and selectivities across all alkanes.
Co-reporter:Prashant Deshlahra
The Journal of Physical Chemistry C 2016 Volume 120(Issue 30) pp:16741-16760
Publication Date(Web):July 20, 2016
DOI:10.1021/acs.jpcc.6b04604
C–H bond activation at lattice O atoms on oxides mediates some of the most important chemical transformations of small organic molecules. The relations between molecular and catalyst properties and C–H activation energies are discerned in this study for the diverse C–H bonds prevalent in C1–C4 hydrocarbons and oxygenates using lattice O atoms with a broad range of H atom abstraction properties. These activation energies determine, in turn, attainable selectivities and yields of desired oxidation products, which differ from reactants in their C–H bond strength. Brønsted-Evans–Polanyi (BEP) linear scaling relations predict that C–H activation energies depend solely and linearly on the C–H bond dissociation energies (BDE) in molecules and on the H-atom addition energies (HAE) of the lattice oxygen abstractors. These relations omit critical interactions between organic radicals and surface OH groups that form at transition states that mediate the H atom transfer, which depend on both molecular and catalyst properties; they also neglect deviations from linear relations caused by the lateness of transition states. Thus, HAE and BDE values, properties that are specific to a catalyst and a molecule in isolation, represent incomplete descriptors of reactivity and selectivity in oxidation catalysis. These effects are included here through crossing potential formalisms that account for the lateness in transition states in estimates of activation energies from HAE and BDE and by estimates of molecule-dependent but catalyst-independent parameters that account for diradical interactions that differ markedly for allylic and nonallylic C–H bonds. The systematic ensemble-averaging of activation energies for all C–H bonds in a given molecule show how strong abstractors and high temperatures decrease an otherwise ubiquitous preference for activating the weakest C–H bonds in molecules, thus allowing higher yields of products with C–H bonds weaker than in reactants than predicted from linear scaling relations based on molecule and abstractor properties. Such conclusions contradict the prevailing guidance to improve such yields by softer oxidants and lower temperatures, a self-contradictory strategy, given the lower reactivity of such weaker H-abstractors. The diradical-type interactions, not previously considered as essential reactivity descriptors in catalytic oxidations, may expand the narrow yield limits imposed by linear free energy relations by guiding the design of solids with surfaces that preferentially destabilize allylic radicals relative to those formed from saturated reactants at C–H activation transition states.
Co-reporter:Shuai Wang and Enrique Iglesia
The Journal of Physical Chemistry C 2016 Volume 120(Issue 38) pp:21589-21616
Publication Date(Web):August 26, 2016
DOI:10.1021/acs.jpcc.6b07304
Co-reporter:Elif I. Gürbüz; David D. Hibbitts
Journal of the American Chemical Society 2015 Volume 137(Issue 37) pp:11984-11995
Publication Date(Web):September 10, 2015
DOI:10.1021/jacs.5b05361
This study combines theory and experiment to determine the kinetically relevant steps and site requirements for deoxygenation of alkanols and alkanals. These reactants deoxygenate predominantly via decarbonylation (C–C cleavage) instead of C–O hydrogenolysis on Ir, Pt, and Ru, leading to strong inhibition effects by chemisorbed CO (CO*). C–C cleavage occurs via unsaturated species formed in sequential quasi-equilibrated dehydrogenation steps, which replace C–H with C–metal bonds, resulting in strong inhibition by H2, also observed in alkane hydrogenolysis. C–C cleavage occurs in oxygenates only at locations vicinal to the C═O group in RCCO* intermediates, because such adjacency weakens C–C bonds, which also leads to much lower activation enthalpies for oxygenates than hydrocarbons. C–O hydrogenolysis rates are independent of H2 pressure and limited by H*-assisted C–O cleavage in RCHOH* intermediates on surfaces with significant coverages of CO* formed in decarbonylation events. The ratio of C–O hydrogenolysis to decarbonylation rates increased almost 100-fold as the Ir cluster size increased from 0.7 to 7 nm; these trends reflect C–O hydrogenolysis reactions favored on terrace sites, while C–C hydrogenolysis prefers sites with lower coordination, because of the relative size of their transition states and the crowded nature of CO*-covered surfaces. C–O hydrogenolysis becomes the preferred deoxygenation route on Cu-based catalysts, thus avoiding CO inhibition effects. The relative rates of C–O and C–C cleavage on these metals depend on their relative ability to bind C atoms, because C–C cleavage transitions states require an additional M–C attachment.
Co-reporter:Prashant Deshlahra, Robert T. Carr, Song-Hai Chai, and Enrique Iglesia
ACS Catalysis 2015 Volume 5(Issue 2) pp:666
Publication Date(Web):December 24, 2014
DOI:10.1021/cs501599y
Acid and redox reaction rates of CH3OH-O2 mixtures on polyoxometalate (POM) clusters, together with isotopic, spectroscopic, and theoretical assessments of catalyst properties and reaction pathways, were used to define rigorous descriptors of reactivity and to probe the compositional effects for oxidative dehydrogenation (ODH) and dehydration reactions. 31P-MAS NMR, transmission electron microscopy and titrations of protons with di-tert-butylpyridine during catalysis showed that POM clusters retained their Keggin structure upon dispersion on SiO2 and after use in CH3OH reactions. The effects of CH3OH and O2 pressures and of D-substitution on ODH rates show that C–H activation in molecularly adsorbed CH3OH is the sole kinetically relevant step and leads to reduced centers as intermediates present at low coverages; their concentrations, measured from UV–vis spectra obtained during catalysis, are consistent with the effects of CH3OH/O2 ratios predicted from the elementary steps proposed. First-order ODH rate constants depend strongly on the addenda atoms (Mo vs W) but weakly on the central atom (P vs Si) in POM clusters, because C–H activation steps inject electrons into the lowest unoccupied molecular orbitals (LUMO) of the clusters, which are the d-orbitals at Mo6+ and W6+ centers. H-atom addition energies (HAE) at O-atoms in POM clusters represent the relevant theoretical probe of the LUMO energies and of ODH reactivity. The calculated energies of ODH transition states at each O-atom depend linearly on their HAE values with slopes near unity, as predicted for late transition states in which electron transfer and C–H cleavage are essentially complete. HAE values averaged over all accessible O-atoms in POM clusters provide the appropriate reactivity descriptor for oxides whose known structures allow accurate HAE calculations. CH3OH dehydration proceeds via parallel pathways mediated by late carbenium-ion transition states; effects of composition on dehydration reactivity reflect changes in charge reorganizations and electrostatic forces that stabilize protons at Brønsted acid sites.Keywords: C−H activation; electron transfer; H-atom addition energy; in situ UV−vis spectroscopy; redox cycle; time-dependent DFT
Co-reporter:Andrew J. Jones and Enrique Iglesia
ACS Catalysis 2015 Volume 5(Issue 10) pp:5741
Publication Date(Web):August 31, 2015
DOI:10.1021/acscatal.5b01133
Ensemble-averaged deprotonation energies (DPE) derived from periodic density functional theory models are insensitive to the location of isolated Al atoms and associated protons and similar among microporous aluminosilicates (i.e., zeolites) with different crystalline frameworks (MFI, BEA, FER, MOR, CHA, FAU). These DPE values are 1201 ± 11 kJ mol–1 after correcting for systematic artifacts of periodic DFT methods, which vary with framework density, and averaging over the four distinct proton locations at each Al atom. These energies rigorously reflect the strength of the acid sites in these important catalytic solids. Thus, the stability of the conjugate anions and the acid strength of these materials merely reflect the presence of Al atoms within the silicate framework, and not their specific siting or local confining environment. DPE values did not show any systematic trends with the vibrational frequency or length of O–H bonds, with Si–O–Al bond angles, or with NH3 adsorption enthalpies, properties that are frequently but inaccurately used as experimental indicators of acid strength. Such properties may reflect or bring forth confinement effects that do not influence acid strength, but which can stabilize the relevant ion-pair transition states and adsorbed intermediates through dispersion forces. These findings confirm that the different shape and size of the confining voids near Al atoms and their associated protons, instead of any differences in their acid strength, give rise to the remarkable diversity of acid forms of zeolites in the practice of catalysis.Keywords: acid strength; Brønsted acid; deprotonation energy; DFT; statistical mechanics
Co-reporter:Sarika Goel, Stacey I. Zones, and Enrique Iglesia
Chemistry of Materials 2015 Volume 27(Issue 6) pp:2056
Publication Date(Web):March 4, 2015
DOI:10.1021/cm504510f
We report synthetic protocols and guiding principles inspired by mechanistic considerations for the synthesis of crystalline microporous solids via interzeolite transformations that avoid direct intervention by organic structure-directing agents. These protocols are specifically implemented to synthesize high-silica MFI (ZSM-5), CHA (chabazite), STF (SSZ-35), and MTW (ZSM-12) zeolites from FAU (faujasite) or BEA (beta) parent materials. These transformations succeed when they lead to daughter structures with higher framework densities, and their nucleation and growth become possible by the presence of seeds or of structural building units common to the parent and target structures, leading, in the latter case, to spontaneous transformations by choosing appropriate synthesis conditions. These protocols allow the synthesis of high-silica frameworks without the use of organic templates otherwise required. The NaOH/SiO2 ratio and Al content in reagents are used to enforce synchronization between the swelling and local restructuring within parent zeolite domains with the spalling of fragments or building units from seeds of the target structure. Seed-mediated interconversions preserve the habit and volume of the parent crystals because of the incipient nucleation of the target structure at the outer regions of the parent domains. The pseudomorphic nature of these transformations requires the concurrent nucleation of mesopores within daughter zeolite crystals because their framework density is larger than that for the parent zeolites. The approach and evidence described shows, for the first time, that a broad range of zeolites rich in silica, and thus more useful as catalysts, can be made without the organic templates originally used to discover them.
Co-reporter:David W. Flaherty
The Journal of Physical Chemistry C 2015 Volume 119(Issue 5) pp:2597-2613
Publication Date(Web):January 26, 2015
DOI:10.1021/jp511688x
Rates and locations of C–C cleavage during the hydrogenolysis of alkyl-cyclohexanes determine the isomeric products of ring opening and the yield losses from dealkylation. Kinetically relevant transition states for C–C rupture form by sequential quasi-equilibrated dehydrogenation steps that break C–H bonds, form C–metal bonds, and desorb chemisorbed H atoms (H*) from H*-covered surfaces. Activation enthalpies (ΔH⧧), entropies (ΔS⧧), and the number of H2(g) formed with transition states are larger for 3C–xC rupture than for 2C–2C or 2C–1C cleavage for all cycloalkane reactants and Ir cluster sizes. 3C–xC rupture transition states bind to surfaces through three or more C atoms, whereas those for less-substituted 2C–2C bonds cleave via α,β species bound by two C atoms. 3C–xC rupture involves larger ΔH⧧ than 2C–2C and 2C–1C because the former requires that more C–H bonds cleave and H* desorb than for the latter two. These endothermic steps are partially compensated by C–metal bond formation, whereas the formation of additional H2(g) gives larger ΔS⧧. C–C rupture transition states for cycloalkanes have less entropy than those for C–C bonds in acyclic alkanes of similar size because C6 rings decrease the rotational and conformational freedom. ΔH⧧ values for all C–C bonds in a given reactant decrease with increasing Ir cluster size because the coordination of exposed metal atoms influences the stabilities of the H* atoms that desorb more than those of the transition states. ΔH⧧ for 3C–xC cleavage is more sensitive to cluster size because their transition states displace more H* than those for 2C–2C or 2C–1C bonds. These data and their mechanistic interpretation provide guidance for how surface coordination, reaction temperatures, and H2 pressures can be used to control ring-opening selectivities toward desirable products while minimizing yield losses. These findings are consistent with trends for the hydrogenolysis of acyclic isoalkanes and seem likely to extend to C–X bond cleavage (where X = O, S, and N atoms) reactions during hydrotreating processes.
Co-reporter:David W. Flaherty ; David D. Hibbitts
Journal of the American Chemical Society 2014 Volume 136(Issue 27) pp:9664-9676
Publication Date(Web):June 25, 2014
DOI:10.1021/ja5037429
Methyl substituents at C–C bonds influence hydrogenolysis rates and selectivities of acyclic and cyclic C2–C8 alkanes on Ir, Rh, Ru, and Pt catalysts. C–C cleavage transition states form via equilibrated dehydrogenation steps that replace several C–H bonds with C-metal bonds, desorb H atoms (H*) from saturated surfaces, and form λ H2(g) molecules. Activation enthalpies (ΔH⧧) and entropies (ΔS⧧) and λ values for 3C–xC cleavage are larger than for 2C–2C or 2C–1C bonds, irrespective of the composition of metal clusters or the cyclic/acyclic structure of the reactants. 3C–xC bonds cleave through α,β,γ- or α,β,γ,δ-bound transition states, as indicated by the agreement between measured activation entropies and those estimated for such structures using statistical mechanics. In contrast, less substituted C–C bonds involve α,β-bound species with each C atom bound to several surface atoms. These α,β configurations weaken C–C bonds through back-donation to antibonding orbitals, but such configurations cannot form with 3C atoms, which have one C–H bond and thus can form only one C–M bond. 3C–xC cleavage involves attachment of other C atoms, which requires endothermic C–H activation and H* desorption steps that lead to larger ΔH⧧ values but also larger ΔS⧧ values (by forming more H2(g)) than for 2C–2C and 2C–1C bonds, irrespective of alkane size (C2–C8) or cyclic/acyclic structure. These data and their mechanistic interpretation indicate that low temperatures and high H2 pressures favor cleavage of less substituted C–C bonds and form more highly branched products from cyclic and acyclic alkanes. Such interpretations and catalytic consequences of substitution seem also relevant to C–X cleavage (X = S, N, O) in desulfurization, denitrogenation, and deoxygenation reactions.
Co-reporter:Sarika Goel ; Stacey I. Zones
Journal of the American Chemical Society 2014 Volume 136(Issue 43) pp:15280-15290
Publication Date(Web):October 14, 2014
DOI:10.1021/ja507956m
The encapsulation of metal clusters (Pt, Ru, Rh) within MFI was achieved by exchanging cationic metal precursors into a parent zeolite (BEA, FAU), reducing them with H2 to form metal clusters, and transforming these zeolites into daughter structures of higher framework density (MFI) under hydrothermal conditions. These transformations required MFI seeds or organic templates for FAU parent zeolites, but not for BEA, and occurred with the retention of encapsulated clusters. Clusters uniform in size (1.3–1.7 nm) and exposing clean and accessible surfaces formed in BEA and FAU zeolites; their size remained essentially unchanged upon transformation into MFI. Encapsulation selectivities, determined from the relative hydrogenation rates of small (toluene) and large (alkyl arenes) molecules and defined as the ratio of the surface areas of all the clusters in the sample to that of external clusters, were very high (8.1–40.9) for both parent and daughter zeolites. Encapsulation into MFI via direct hydrothermal syntheses was unsuccessful because metal precursors precipitated prematurely at the pH and temperatures required for MFI synthesis. Delayed introduction of metal precursors and F– (instead of OH–) as the mineralizing agent in hydrothermal syntheses increased encapsulation selectivities, but they remained lower than those achieved via interzeolite transformations. These interconversions provide a general and robust strategy for encapsulation of metals when precursors can be introduced via exchange into a zeolite that can be transformed into target daughter zeolites with higher framework densities, whether spontaneously or by using seeds or structure-directing agents (SDA).
Co-reporter:Prashant Deshlahra ; Robert T. Carr
Journal of the American Chemical Society 2014 Volume 136(Issue 43) pp:15229-15247
Publication Date(Web):October 21, 2014
DOI:10.1021/ja506149c
Reactivity descriptors describe catalyst properties that determine the stability of kinetically relevant transition states and adsorbed intermediates. Theoretical descriptors, such as deprotonation energies (DPE), rigorously account for Brønsted acid strength for catalytic solids with known structure. Here, mechanistic interpretations of methanol dehydration turnover rates are used to assess how charge reorganization (covalency) and electrostatic interactions determine DPE and how such interactions are recovered when intermediates and transition states interact with the conjugate anion in W and Mo polyoxometalate (POM) clusters and gaseous mineral acids. Turnover rates are lower and kinetically relevant species are less stable on Mo than W POM clusters with similar acid strength, and such species are more stable on mineral acids than that predicted from W-POM DPE–reactivity trends, indicating that DPE and acid strength are essential but incomplete reactivity descriptors. Born–Haber thermochemical cycles indicate that these differences reflect more effective charge reorganization upon deprotonation of Mo than W POM clusters and the much weaker reorganization in mineral acids. Such covalency is disrupted upon deprotonation but cannot be recovered fully upon formation of ion pairs at transition states. Predictive descriptors of reactivity for general classes of acids thus require separate assessments of the covalent and ionic DPE components. Here, we describe methods to estimate electrostatic interactions, which, taken together with energies derived from density functional theory, give the covalent and ionic energy components of protons, intermediates, and transition states. In doing so, we provide a framework to predict the reactive properties of protons for chemical reactions mediated by ion-pair transition states.
Co-reporter:Andrew J. Jones ; Enrique Iglesia
Angewandte Chemie 2014 Volume 126( Issue 45) pp:12373-12377
Publication Date(Web):
DOI:10.1002/ange.201406823
Abstract
Mechanistic interpretations of rates and in situ IR spectra combined with density functionals that account for van der Waals interactions of intermediates and transition states within confining voids show that associative routes mediate the formation of dimethyl ether from methanol on zeolitic acids at the temperatures and pressures of practical dehydration catalysis. Methoxy-mediated dissociative routes become prevalent at higher temperatures and lower pressures, because they involve smaller transition states with higher enthalpy, but also higher entropy, than those in associative routes. These enthalpy–entropy trade-offs merely reflect the intervening role of temperature in activation free energies and the prevalence of more complex transition states at low temperatures and high pressures. This work provides a foundation for further inquiry into the contributions of H-bonded methanol and methoxy species in homologation and hydrocarbon synthesis reactions from methanol.
Co-reporter:Sebastian Kunz
The Journal of Physical Chemistry C 2014 Volume 118(Issue 14) pp:7468-7479
Publication Date(Web):March 27, 2014
DOI:10.1021/jp500537v
Bimetallic Pd–Au clusters with (Pd/Au)at compositions of 0.5, 1.0, and 2.0 narrowly distributed in size were prepared using colloidal methods with reagents containing only C, H, and O atoms, specifically polyvinyl alcohol (PVA) as protecting species and ethanol as the organic reductant. Synthesis protocols involved contacting a solution of Au precursors with nearly monodisperse Pd clusters. The formation of Pd–Au clusters was inferred from the monotonic growth of clusters with increasing Au content and confirmed by the in situ detection of Au plasmon bands in their UV–visible spectra during synthesis. Specifically, transmission electron microscopy (TEM) showed that growth rates were proportional to the surface area of the clusters, and rigorous deconvolution and background subtraction allowed for determination of the intensity and energy of Au-derived plasmon bands. This feature emerged during initial contact between Au precursors and Pd clusters apparently because Au3+ species deposit as Au0 using Pd0 as the reductant in a fast galvanic displacement process consistent with their respective redox potentials. The plasmon band ultimately disappeared as a result of the subsequent slower reduction of the displaced Pd2+ species by ethanol and of their deposition onto the bimetallic clusters. Such displacement–reduction pathways are consistent with the thermodynamic redox tendencies of Au, Pd, and ethanol and lead to the conclusion that such triads (two metals and an organic reductant) can be chosen from thermodynamic data and applied generally to the synthesis of bimetallic clusters with other compositions. These bimetallic clusters were dispersed on mesoporous γ-Al2O3 supports, and PVA was removed by treatment in ozone at near-ambient temperature without any detectable changes in cluster size. The absence of strongly bound heteroatoms, ubiquitous in many other colloidal synthesis protocols, led to Al2O3-dispersed clusters with chemisorption uptakes consistent with their TEM-derived cluster size, thus demonstrating that cluster surfaces are accessible and free of synthetic debris. The infrared spectra of chemisorbed CO indicated that both Pd and Au were present at such clean surfaces but that any core–shell intracluster structure conferred by synthesis was rapidly destroyed by adsorption of catalytically relevant species, even at ambient temperature; this merely reflects the thermodynamic tendency and kinetic ability of an element to segregate and to decrease surface energies when it binds an adsorbate more strongly than another element in bimetallic particles.
Co-reporter:Andrew J. Jones ; Stacey I. Zones
The Journal of Physical Chemistry C 2014 Volume 118(Issue 31) pp:17787-17800
Publication Date(Web):July 18, 2014
DOI:10.1021/jp5050095
The catalytic diversity of microporous aluminosilicates reflects their unique ability to confine transition states within intracrystalline voids of molecular dimensions and the number (but not the strength) of the protons that act as Brønsted acids. First-order rate constants for CH3OH conversion to dimethyl ether (DME) reflect the energy of transition states relative to those for gaseous and H-bonded CH3OH molecules; on zeolites, these constants depend exponentially on n-hexane physisorption energies for different void size and shape and proton location, indicating that van der Waals stabilization of transition states causes their different reactivity, without concomitant effects of void structure or proton location on acid strength. The dispersive contribution to adsorption enthalpies of DME, a proxy in shape and size for relevant transition states, was calculated using density functional theory and Lennard-Jones interactions on FAU, SFH, BEA, MOR, MTW, MFI, and MTT zeolites and averaged over all proton locations; first-order rate constants also depended exponentially on these enthalpies. In contrast, zero-order rate constants, which reflect the stability of transition states relative to protonated CH3OH dimers similar in size, depended weakly on dispersive stabilization, whether measured from experiment or simulations, because dispersive forces influence species similar in size to the same extent. These results, taken together, demonstrate the preeminent effects of confinement on zeolite reactivity and the manner by which the local voids around protons held within diverse intracrystalline environments give rise to the unique behaviors that have made zeolites ubiquitous in the practice of catalysis. Enthalpic stabilization of relevant transition states prevail over entropic losses caused by confinement at low temperatures in a manner reminiscent of how catalytic pockets and solvents do so in catalysis by molecules or enzymes.
Co-reporter:Prashant Deshlahra
The Journal of Physical Chemistry C 2014 Volume 118(Issue 45) pp:26115-26129
Publication Date(Web):November 5, 2014
DOI:10.1021/jp507922u
The oxidative dehydrogenation (ODH) of alkanols on oxide catalysts is generally described as involving H-abstraction from alkoxy species formed via O–H dissociation. Kinetic and isotopic data cannot discern between such routes and those involving kinetically-relevant H-abstraction from undissociated alkanols. Here, we combine such experiments with theoretical estimates of activation energies and entropies to show that the latter molecular routes prevail over dissociative routes for methanol reactions on polyoxometalate (POM) clusters at all practical reaction temperatures. The stability of the late transition states that mediate H-abstraction depend predominantly on the stability of the O–H bond formed, making H-addition energies (HAE) accurate and single-valued descriptors of reactivity. Density functional theory-derived activation energies depend linearly on HAE values at each O-atom location on clusters with a range of composition (H3PMo12, H4SiMo12, H3PW12, H4PV1Mo11, and H4PV1W11); both barriers and HAE values reflect the lowest unoccupied molecular orbital energy of metal centers that accept the electron and the protonation energy of O-atoms that accept the proton involved in the H-atom transfer. Bridging O-atoms form O–H bonds that are stronger than those of terminal atoms and therefore exhibit more negative HAE values and higher ODH reactivity on all POM clusters. For each cluster composition, ODH turnover rates reflect the reactivity-averaged HAE of all accessible O-atoms, which can be evaluated for each cluster composition to provide a rigorous and accurate predictor of ODH reactivity for catalysts with known structure. These relations together with oxidation reactivity measurements can then be used to estimate HAE values and to infer plausible structures for catalysts with uncertain active site structures.
Co-reporter:Andrew J. Jones ; Enrique Iglesia
Angewandte Chemie International Edition 2014 Volume 53( Issue 45) pp:12177-12181
Publication Date(Web):
DOI:10.1002/anie.201406823
Abstract
Mechanistic interpretations of rates and in situ IR spectra combined with density functionals that account for van der Waals interactions of intermediates and transition states within confining voids show that associative routes mediate the formation of dimethyl ether from methanol on zeolitic acids at the temperatures and pressures of practical dehydration catalysis. Methoxy-mediated dissociative routes become prevalent at higher temperatures and lower pressures, because they involve smaller transition states with higher enthalpy, but also higher entropy, than those in associative routes. These enthalpy–entropy trade-offs merely reflect the intervening role of temperature in activation free energies and the prevalence of more complex transition states at low temperatures and high pressures. This work provides a foundation for further inquiry into the contributions of H-bonded methanol and methoxy species in homologation and hydrocarbon synthesis reactions from methanol.
Co-reporter:David W. Flaherty
Journal of the American Chemical Society 2013 Volume 135(Issue 49) pp:18586-18599
Publication Date(Web):November 22, 2013
DOI:10.1021/ja4093743
Statistical mechanics and transition state (TS) theory describe rates and selectivities of C–C bond cleavage in C2–C10 n-alkanes on metal catalysts and provide a general description for the hydrogenolysis of hydrocarbons. Mechanistic interpretation shows the dominant role of entropy, over enthalpy, in determining the location and rate of C–C bond cleavage. Ir, Rh, and Pt clusters cleave C–C bonds at rates proportional to coverages of intermediates derived by removing 3–4 H-atoms from n-alkanes. Rate constants for C–C cleavage reflect large activation enthalpies (ΔH⧧, 217–257 kJ mol–1) that are independent of chain length and C–C bond location in C4+ n-alkanes. C–C bonds cleave because of large, positive activation entropies (ΔS⧧, 164–259 J mol–1 K–1) provided by H2 that forms with TS. Kinetic and independent spectroscopic evidence for the composition and structure of these TS give accurate estimates of ΔS⧧ for cleavage at each C–C bond. Large differences between rate constants for ethane and n-decane (∼108) reflect an increase in the entropy of gaseous alkanes retained at the TS. The location of C–C bond cleavage depends solely on the rotational entropies of alkyl chains attached to the cleaved C–C bond, which depend on their chain length. Such entropy considerations account for the ubiquitous, but previously unexplained, preference for cleaving nonterminal C–C bonds in n-alkanes. This mechanistic analysis and thermodynamic treatment illustrates the continued utility of such approaches even for hydrogenolysis reactions, with complexity seemingly beyond the reach of classical treatments, and applies to catalytic clusters beyond those reported here (0.6–2.7 nm; Ir, Rh, Pt).
Co-reporter:Brett T. Loveless ; Corneliu Buda ; Matthew Neurock
Journal of the American Chemical Society 2013 Volume 135(Issue 16) pp:6107-6121
Publication Date(Web):March 12, 2013
DOI:10.1021/ja311848e
Density functional theory (DFT) and infrared spectroscopy results are combined with mechanism-based rate equations to assess the structure and thermodynamics of chemisorbed CO (CO*) and its activation during Fischer–Tropsch synthesis (FTS). CO* binding becomes weaker with increasing coverage on Ru(0001) and Ru201 clusters, but such decreases in binding energy occur at higher coverages on Ru201 clusters than on Ru(0001) surfaces (CO*/Ru = 1.55 to 0.75); such differences appear to reflect weaker repulsive interactions on the curved surfaces prevalent on small Ru201 clusters. Ru201 clusters achieve stable supramonolayer coverages (CO*/Ru > 1) by forming geminal dicarbonyls at low-coordination corner/edge atoms. CO* infrared spectra on Ru/SiO2 (∼7 nm diameter) detect mobile adlayers that anneal into denser structures at saturation. Mechanism-based FTS rate equations give activation energies that reflect the CO*-saturated surfaces prevalent during catalysis. DFT-derived barriers show that CO* predominantly reacts at (111) terraces via H-assisted reactions, consistent with measured effects of H2 and CO pressures and cluster size effects on rates and O-rejection selectivities. Barriers are much higher for unassisted CO* dissociation on (111) terraces and low-coordination atoms, including step-edge sites previously proposed as active sites for CO* dissociation during FTS. DFT-derived barriers indicate that unassisted CO* dissociation is irreversible, making such steps inconsistent with measured rates. The modest activation barriers of H-assisted CO* dissociation paths remove a requirement for special low-coordination sites for unassisted CO* activation, which is inconsistent with higher rates on larger clusters. These conclusions seem generally applicable to Co, Fe, and Ru catalysts, which show similar FTS rate equations and cluster size effects. This study also demonstrates the feasibility and relevance of DFT treatments on the curved and crowded cluster surfaces where catalysis occurs.
Co-reporter:Ya-Huei (Cathy) Chin ; Corneliu Buda ; Matthew Neurock
Journal of the American Chemical Society 2013 Volume 135(Issue 41) pp:15425-15442
Publication Date(Web):October 1, 2013
DOI:10.1021/ja405004m
Mechanistic assessments based on kinetic and isotopic methods combined with density functional theory are used to probe the diverse pathways by which C–H bonds in CH4 react on bare Pd clusters, Pd cluster surfaces saturated with chemisorbed oxygen (O*), and PdO clusters. C–H activation routes change from oxidative addition to H-abstraction and then to σ-bond metathesis with increasing O-content, as active sites evolve from metal atom pairs (*–*) to oxygen atom (O*–O*) pairs and ultimately to Pd cation-lattice oxygen pairs (Pd2+–O2–) in PdO. The charges in the CH3 and H moieties along the reaction coordinate depend on the accessibility and chemical state of the Pd and O centers involved. Homolytic C–H dissociation prevails on bare (*–*) and O*-covered surfaces (O*–O*), while C–H bonds cleave heterolytically on Pd2+–O2– pairs at PdO surfaces. On bare surfaces, C–H bonds cleave via oxidative addition, involving Pd atom insertion into the C–H bond with electron backdonation from Pd to C–H antibonding states and the formation of tight three-center (H3C···Pd···H)⧧ transition states. On O*-saturated Pd surfaces, C–H bonds cleave homolytically on O*–O* pairs to form radical-like CH3 species and nearly formed O–H bonds at a transition state (O*···CH3•···*OH)⧧ that is looser and higher in enthalpy than on bare Pd surfaces. On PdO surfaces, site pairs consisting of exposed Pd2+ and vicinal O2–, Pdox–Oox , cleave C–H bonds heterolytically via σ-bond metathesis, with Pd2+ adding to the C–H bond, while O2– abstracts the H-atom to form a four-center (H3Cδ−···Pdox···Hδ+···Oox)⧧ transition state without detectable Pdox reduction. The latter is much more stable than transition states on *–* and O*–O* pairs and give rise to a large increase in CH4 oxidation turnover rates at oxygen chemical potentials leading to Pd to PdO transitions. These distinct mechanistic pathways for C–H bond activation, inferred from theory and experiment, resemble those prevalent on organometallic complexes. Metal centers present on surfaces as well as in homogeneous complexes act as both nucleophile and electrophile in oxidative additions, ligands (e.g., O* on surfaces) abstract H-atoms via reductive deprotonation of C–H bonds, and metal–ligand pairs, with the pair as electrophile and the metal as nucleophile, mediate σ-bond metathesis pathways.
Co-reporter:Rajamani Gounder and Enrique Iglesia
Chemical Communications 2013 vol. 49(Issue 34) pp:3491-3509
Publication Date(Web):04 Mar 2013
DOI:10.1039/C3CC40731D
The ability of molecular sieves to control the access and egress of certain reactants and products and to preferentially contain certain transition states while excluding others based on size were captured as shape selectivity concepts early in the history of zeolite catalysis. The marked consequences for reactivity and selectivity, specifically in acid catalysis, have since inspired and sustained many discoveries of novel silicate frameworks and driven the engineering of hierarchical structures and void size to influence catalysis. The catalytic diversity of microporous voids is explored and extended here in the context of their solvating environments, wherein voids act as hosts and stabilize guests, whether reactive intermediates or transition states, by van der Waals forces. We use specific examples from acid catalysis, including activation of C–C and C–H bonds in alkanes, alkylation and hydrogenation of alkenes, carbonylation of dimethyl ether, and elimination and homologation reactions of alkanols and ethers, which involve transition states and adsorbed precursors of varying size and composition. Mechanistic interpretations of measured turnover rates enable us to assign precise chemical origins to kinetic and thermodynamic constants in rate equations and, in turn, to identify specific steps and intermediates that determine the free energy differences responsible for chemical reactivity and selectivity. These free energy differences reflect the stabilization of transition states and their relevant precursors via electrostatic interactions that depend on acid strength and van der Waals interactions that depend on confinement within voids. Their respective contributions to activation free energies are examined by Born–Haber thermochemical cycles by considering plausible transition states and the relevant precursors. These examples show that zeolite voids solvate transition states and precursors differently, and markedly so for guest moieties of different size and chemical composition, thus enabling voids of a given size and shape to provide the “right fit” for a given elementary step, defined as that which minimizes Gibbs free energies of activation. Tighter confinement is preferred at low temperatures because enthalpic gains prevail over concomitant entropic losses, while looser fits are favored at high temperatures because entropy gains offset losses in enthalpic stabilization. Confinement and solvation by van der Waals forces are not directly involved in the making or breaking of strong chemical bonds; yet, they confer remarkable diversity to zeolites, in spite of their structural rigidity and their common aluminosilicate composition. A single zeolite can itself contain a range of local void environments, each with distinct reactivity and selectivity; as a result, varying the distribution of protons among these locations within a given framework or modifying a given location by partial occlusion of the void space can extend the range of catalytic opportunities for zeolites. Taken together with theoretical tools that accurately describe van der Waals interactions between zeolite voids and confined guests and with synthetic protocols that place protons or space-filling moieties at specific locations, these concepts promise to broaden the significant impact and catalytic diversity already shown by microporous solids.
Co-reporter:Andrew J. Jones, Christopher Ostrouchov, Maciej Haranczyk, Enrique Iglesia
Microporous and Mesoporous Materials 2013 Volume 181() pp:208-216
Publication Date(Web):15 November 2013
DOI:10.1016/j.micromeso.2013.07.033
•Accessible voids are compared through stochastic distributions of ray lengths.•Ray histograms encode the shape, topology, distribution and size of voids.•Void similarity determined with Euclidean distance metrics.•Potential catalytically relevant zeolites are presented using similarity searches.Voids within crystalline microporous solids are represented here using stochastic distributions of rays placed and oriented randomly within the accessible void space, represented using Voronoi decompositions. This algorithm is provided in the Zeo++ software for open use. In this method, ray lengths are depicted as two-dimensional histograms that complement alternate descriptors, such as free and included sphere diameters. We illustrate the specific use of these methods as a tool to narrow the range of zeolites useful for a given catalytic application because of the shape and size of voids. DAC, AFS, AFY, SFO and EON zeolites contain void spaces similar, as suggested by Euclidean distance values between histograms, to those within MOR 8-MR side pockets, which stabilize the transition states that mediate dimethyl ether carbonylation to methyl acetate; these alternate structures offer different connecting void environments, which can enhance or restrict molecular access and influence the effectiveness of the 8-MR protons. NES, EON and USI zeolites exhibit histogram features similar to those of 12-MR MOR channels, where protons selectively catalyze alkylation of biphenyl and naphthalene to 4,4′-diisopropylbiphenyl and 2,6-diisopropylnaphthalene, respectively, with propene. SBT, FAU and SBS contain voids similar in topology to the 12-MR channels of LTL zeolites, within which Pt clusters remain active and stable during the dehydrocyclization of light alkanes, but without the one-dimensional nature of LTL channels. The approach and implementation of these methods are applicable to any microporous or mesoporous solids and to adsorption processes driven by van der Waals contacts between hosts and guest molecules.
Co-reporter:Rajamani Gounder and Enrique Iglesia
Accounts of Chemical Research 2012 Volume 45(Issue 2) pp:229
Publication Date(Web):August 26, 2011
DOI:10.1021/ar200138n
Acidic zeolites are indispensable catalysts in the petrochemical industry because they select reactants and their chemical pathways based on size and shape. Voids of molecular dimensions confine reactive intermediates and transition states that mediate chemical reactions, stabilizing them by van der Waals interactions. This behavior is reminiscent of the solvation effects prevalent within enzyme pockets and has analogous consequences for catalytic specificity. Voids provide the “right fit” for certain transition states, reflected in their lower free energies, thus extending the catalytic diversity of zeolites well beyond simple size discrimination. This catalytic diversity is even more remarkable because acid strength is essentially unaffected by confinement among known crystalline aluminosilicates. In this Account, we discuss factors that determine the “right fit” for a specific chemical reaction, exploring predictive criteria that extend the prevailing discourse based on size and shape. We link the structures of reactants, transition states, and confining voids to chemical reactivity and selectivity.Confinement mediates enthalpy–entropy compromises that determine the Gibbs free energies of transition states and relevant reactants; these activation free energies determine turnover rates via transition state theory. At low temperatures (400–500 K), dimethyl ether carbonylation occurs with high specificity within small eight-membered ring (8-MR) voids in FER and MOR zeolite structures, but at undetectable rates within larger voids (MFI, BEA, FAU, and SiO2–Al2O3). More effective van der Waals stabilization within 8-MR voids leads to lower ion-pair enthalpies but also lower entropies; taken together, carbonylation activation free energies are lower within 8-MR voids. The “right fit” is a “tight fit” at low temperatures, a consequence of how temperature appears in the defining equation for Gibbs free energy.In contrast, entropy effects dominate in high-temperature alkane activation (700–800 K), for which the “right fit” becomes a “loose fit”. Alkane activation turnovers are still faster on 8-MR MOR protons because these transition states are confined only partially within shallow 8-MR pockets; they retain higher entropies than ion-pairs fully confined within 12-MR channels at the expense of enthalpic stability. Selectivities for n-alkane dehydrogenation (relative to cracking) and isoalkane cracking (relative to dehydrogenation) are higher on 8-MR than 12-MR sites because partial confinement preferentially stabilizes looser ion-pair structures; these structures occur later along reaction coordinates and are higher in energy, consistent with Marcus theory for charge-transfer reactions. Enthalpy differences between cracking and dehydrogenation ion-pairs for a given reactant are independent of zeolite structure (FAU, FER, MFI, or MOR) and predominantly reflect the different gas-phase proton affinities of alkane C–C and C–H bonds, as expected from Born–Haber thermochemical cycles. These thermochemical relations, together with statistical mechanics-based treatments, predict that rotational entropy differences between intact reactants and ion-pair transition states cause intrinsic cracking rates to increase with n-alkane size.Through these illustrative examples, we highlight the effects of reactant and catalyst structures on ion-pair transition state enthalpies and entropies. Our discussion underscores the role of temperature in mediating enthalpic and entropic contributions to free energies and, in turn, to rates and selectivities in zeolite acid catalysis.
Co-reporter:Sarika Goel ; Zhijie Wu ; Stacey I. Zones
Journal of the American Chemical Society 2012 Volume 134(Issue 42) pp:17688-17695
Publication Date(Web):September 27, 2012
DOI:10.1021/ja307370z
The synthesis protocols for encapsulation of metal clusters reported here expand the diversity in catalytic chemistries made possible by the ability of microporous solids to select reactants, transition states, and products on the basis of their molecular size. We report a synthesis strategy for the encapsulation of noble metals and their oxides within SOD (Sodalite, 0.28 nm × 0.28 nm), GIS (Gismondine, 0.45 nm × 0.31 nm), and ANA (Analcime, 0.42 nm × 0.16 nm) zeolites. Encapsulation was achieved via direct hydrothermal synthesis for SOD and GIS using metal precursors stabilized by ammonia or organic amine ligands, which prevent their decomposition or precipitation as colloidal hydroxides at the conditions of hydrothermal synthesis (<380 K) and favor interactions between metal precursors and incipient aluminosilicate nuclei during self-assembly of microporous frameworks. The synthesis of ANA requires higher crystallization temperatures (∼415 K) and high pH (>12), thereby causing precipitation of even ligand-stabilized metal precursors as hydroxides. As a result, encapsulation was achieved by the recrystallization of metal clusters containing GIS into ANA, which retained these metal clusters within voids throughout the GIS–ANA transformation.
Co-reporter:Dr. Brian M. Weiss;Dr. Nancy Artioli; Enrique Iglesia
ChemCatChem 2012 Volume 4( Issue 9) pp:1397-1404
Publication Date(Web):
DOI:10.1002/cctc.201200050
Abstract
The elementary steps and site requirements for the oxidation of NO on Rh and Co and the oxidation state of the catalysts were probed by isotopic tracers, chemisorption methods, and kinetic measurements of the effects of the pressures of NO, O2, and NO2 on turnover rates. On both catalysts, NO oxidation rates were first order in NO and O2 and were inversely proportional to NO2 pressure, as observed on Pt and PdO. These data implied that O2 activation on an isolated vacancy (*) on the catalyst surfaces that were saturated with oxygen (O*) was the kinetically relevant step. Quasi-equilibrated NO–NO2 interconversion steps established the coverage of * and O* and the chemical potential of oxygen during the catalysis. These chemical potentials set the oxidation state of Rh and Co clusters and were described by an O2 virtual pressure, which was determined from the formalism of non-equilibrium thermodynamics. RhO2 and Co3O4 were the phases that were present during NO oxidation, which had several consequences for catalysis. Turnover rates increased with increasing cluster size because the vacancies that were needed for O2 activation were more abundant on large oxide clusters, which delocalized electrons better than small clusters. NO oxidation turnover rates on RhO2 and Co3O4 were higher than expected from the oxygen-binding energy on Rh and Co metal surfaces and from the reduction potentials of Rh3+ and Co2+. These NO oxidation rates were consistent with the rates on Pt and PdO when one-electron-reduction processes, which were accessible for Rh4+ and Co3+ but not for Pt2+ and Pd2+, were used to describe the reactivity of RhO2 and Co3O4. One-electron redox cycles caused the 16O2–18O2 exchange rates to be higher than the NO oxidation rates, in contrast with their analogous values on Pt and PdO, although O2 activation on the vacancies limited NO oxidation and O2 exchange on all of the catalysts. One-electron redox cycles allowed electron sharing between metal cations and a facile route to form vacancies on RhO2 and Co3O4. This interpretation of the data highlighted the role of vacancies in kinetically relevant O2-activation steps to explain the higher reactivity of larger metal and oxide clusters and to provide a common framework to describe NO oxidation and the active species on catalysts of practical interest.
Co-reporter:Ya-Huei(Cathy) Chin ; Corneliu Buda ; Matthew Neurock
Journal of the American Chemical Society 2011 Volume 133(Issue 40) pp:15958-15978
Publication Date(Web):September 15, 2011
DOI:10.1021/ja202411v
Kinetic and isotopic data and density functional theory treatments provide evidence for the elementary steps and the active site requirements involved in the four distinct kinetic regimes observed during CH4 oxidation reactions using O2, H2O, or CO2 as oxidants on Pt clusters. These four regimes exhibit distinct rate equations because of the involvement of different kinetically relevant steps, predominant adsorbed species, and rate and equilibrium constants for different elementary steps. Transitions among regimes occur as chemisorbed oxygen (O*) coverages change on Pt clusters. O* coverages are given, in turn, by a virtual O2 pressure, which represents the pressure that would give the prevalent steady-state O* coverages if their adsorption–desorption equilibrium was maintained. The virtual O2 pressure acts as a surrogate for oxygen chemical potentials at catalytic surfaces and reflects the kinetic coupling between C–H and O═O activation steps. O* coverages and virtual pressures depend on O2 pressure when O2 activation is equilibrated and on O2/CH4 ratios when this step becomes irreversible as a result of fast scavenging of O* by CH4-derived intermediates. In three of these kinetic regimes, C–H bond activation is the sole kinetically relevant step, but occurs on different active sites, which evolve from oxygen–oxygen (O*–O*), to oxygen–oxygen vacancy (O*–*), and to vacancy–vacancy (*–*) site pairs as O* coverages decrease. On O*-saturated cluster surfaces, O*–O* site pairs activate C–H bonds in CH4 via homolytic hydrogen abstraction steps that form CH3 groups with significant radical character and weak interactions with the surface at the transition state. In this regime, rates depend linearly on CH4 pressure but are independent of O2 pressure. The observed normal CH4/CD4 kinetic isotope effects are consistent with the kinetic-relevance of C–H bond activation; identical 16O2–18O2 isotopic exchange rates in the presence or absence of CH4 show that O2 activation steps are quasi-equilibrated during catalysis. Measured and DFT-derived C–H bond activation barriers are large, because of the weak stabilization of the CH3 fragments at transition states, but are compensated by the high entropy of these radical-like species. Turnover rates in this regime decrease with increasing Pt dispersion, because low-coordination exposed Pt atoms on small clusters bind O* more strongly than those that reside at low-index facets on large clusters, thus making O* less effective in H-abstraction. As vacancies (*, also exposed Pt atoms) become available on O*-covered surfaces, O*–* site pairs activate C–H bonds via concerted oxidative addition and H-abstraction in transition states effectively stabilized by CH3 interactions with the vacancies, which lead to much higher turnover rates than on O*–O* pairs. In this regime, O2 activation becomes irreversible, because fast C–H bond activation steps scavenge O* as it forms. Thus, O* coverages are set by the prevalent O2/CH4 ratios instead of the O2 pressures. CH4/CD4 kinetic isotope effects are much larger for turnovers mediated by O*–* than by O*–O* site pairs, because C–H (and C–D) activation steps are required to form the * sites involved in C–H bond activation. Turnover rates for CH4–O2 reactions mediated by O*–* pairs decrease with increasing Pt dispersion, as in the case of O*–O* active structures, because stronger O* binding on small clusters leads not only to less reactive O* atoms, but also to lower vacancy concentrations at cluster surfaces. As O2/CH4 ratios and O* coverages become smaller, O2 activation on bare Pt clusters becomes the sole kinetically relevant step; turnover rates are proportional to O2 pressures and independent of CH4 pressure and no CH4/CD4 kinetic isotope effects are observed. In this regime, turnover rates become nearly independent of Pt dispersion, because the O2 activation step is essentially barrierless. In the absence of O2, alternate weaker oxidants, such as H2O or CO2, lead to a final kinetic regime in which C–H bond dissociation on *–* pairs at bare cluster surfaces limit CH4 conversion rates. Rates become first-order in CH4 and independent of coreactant and normal CH4/CD4 kinetic isotope effects are observed. In this case, turnover rates increase with increasing dispersion, because low-coordination Pt atoms stabilize the C–H bond activation transition states more effectively via stronger binding to CH3 and H fragments. These findings and their mechanistic interpretations are consistent with all rate and isotopic data and with theoretical estimates of activation barriers and of cluster size effects on transition states. They serve to demonstrate the essential role of the coverage and reactivity of chemisorbed oxygen in determining the type and effectiveness of surface structures in CH4 oxidation reactions using O2, H2O, or CO2 as oxidants, as well as the diversity of rate dependencies, activation energies and entropies, and cluster size effects that prevail in these reactions. These results also show how theory and experiments can unravel complex surface chemistries on realistic catalysts under practical conditions and provide through the resulting mechanistic insights specific predictions for the effects of cluster size and surface coordination on turnover rates, the trends and magnitude of which depend sensitively on the nature of the predominant adsorbed intermediates and the kinetically relevant steps.
Co-reporter:María E. Sad ; Matthew Neurock
Journal of the American Chemical Society 2011 Volume 133(Issue 50) pp:20384-20398
Publication Date(Web):October 25, 2011
DOI:10.1021/ja207551f
This study reports evidence for catalytic deoxygenation of alkanols, alkanals, and alkanediols on dispersed Cu clusters with minimal use of external H2 and with the concurrent formation of new C–C and C–O bonds. These catalysts selectively remove O-atoms from these oxygenates as CO or CO2 through decarbonylation or decarboxylation routes, respectively, that use C-atoms present within reactants or as H2O using H2 added or formed in situ from CO/H2O mixtures via water-gas shift. Cu catalysts fully convert 1,3-propanediol to equilibrated propanol–propanal intermediates that subsequently form larger oxygenates via aldol-type condensation and esterification routes without detectable involvement of the oxide supports. Propanal–propanol–H2 equilibration is mediated by their chemisorption and interconversion at surfaces via C–H and O–H activation and propoxide intermediates. The kinetic effects of H2, propanal, and propanol pressures on turnover rates, taken together with measured selectivities and the established chemical events for base-catalyzed condensation and esterification reactions, indicate that both reactions involve kinetically relevant bimolecular steps in which propoxide species, acting as the base, abstract the α-hydrogen in adsorbed propanal (condensation) or attack the electrophilic C-atom at its carbonyl group (esterification). These weakly held basic alkoxides render Cu surfaces able to mediate C–C and C–O formation reactions typically catalyzed by basic sites inherent in the catalyst, instead of provided by coadsorbed organic moieties. Turnover rates for condensation and esterification reactions decrease with increasing Cu dispersion, because low-coordination corner and edge atoms prevalent on small clusters stabilize adsorbed intermediates and increase the activation barriers for the bimolecular kinetically relevant steps required for both reactions.
Co-reporter:Ayman D. Allian ; Kazuhiro Takanabe ; Kyle L. Fujdala ; Xianghong Hao ; Timothy J. Truex ; Juan Cai ; Corneliu Buda ; Matthew Neurock
Journal of the American Chemical Society 2011 Volume 133(Issue 12) pp:4498-4517
Publication Date(Web):March 2, 2011
DOI:10.1021/ja110073u
Kinetic, isotopic, and infrared studies on well-defined dispersed Pt clusters are combined here with first-principle theoretical methods on model cluster surfaces to probe the mechanism and structural requirements for CO oxidation catalysis at conditions typical of its industrial practice. CO oxidation turnover rates and the dynamics and thermodynamics of adsorption−desorption processes on cluster surfaces saturated with chemisorbed CO were measured on 1−20 nm Pt clusters under conditions of strict kinetic control. Turnover rates are proportional to O2 pressure and inversely proportional to CO pressure, consistent with kinetically relevant irreversible O2 activation steps on vacant sites present within saturated CO monolayers. These conclusions are consistent with the lack of isotopic scrambling in C16O−18O2−16O2 reactions, and with infrared bands for chemisorbed CO that did not change within a CO pressure range that strongly influenced CO oxidation turnover rates. Density functional theory estimates of rate and equilibrium constants show that the kinetically relevant O2 activation steps involve direct O2* (or O2) reactions with CO* to form reactive O*−O−C*═O intermediates that decompose to form CO2 and chemisorbed O*, instead of unassisted activation steps involving molecular adsorption and subsequent dissociation of O2. These CO-assisted O2 dissociation pathways avoid the higher barriers imposed by the spin-forbidden transitions required for unassisted O2 dissociation on surfaces saturated with chemisorbed CO. Measured rate parameters for CO oxidation were independent of Pt cluster size; these parameters depend on the ratio of rate constants for O2 reactions with CO* and CO adsorption equilibrium constants, which reflect the respective activation barriers and reaction enthalpies for these two steps. Infrared spectra during isotopic displacement and thermal desorption with 12CO−13CO mixtures showed that the binding, dynamics, and thermodynamics of CO chemisorbed at saturation coverages do not depend on Pt cluster size in a range that strongly affects the coordination of Pt atoms exposed at cluster surfaces. These data and their theoretical and mechanistic interpretations indicate that the remarkable structure insensitivity observed for CO oxidation reactions reflects average CO binding properties that are essentially independent of cluster size. Theoretical estimates of rate and equilibrium constants for surface reactions and CO adsorption show that both parameters increase as the coordination of exposed Pt atoms decreases in Pt201 cluster surfaces; such compensation dampens but does not eliminate coordination and cluster size effects on measured rate constants. The structural features and intrinsic non-uniformity of cluster surfaces weaken when CO forms saturated monolayers on such surfaces, apparently because surfaces and adsorbates restructure to balance CO surface binding and CO−CO interaction energies.
Co-reporter:Tatiana Luts, Enrique Iglesia and Alexander Katz
Journal of Materials Chemistry A 2011 vol. 21(Issue 4) pp:982-990
Publication Date(Web):24 Nov 2010
DOI:10.1039/C0JM02826F
The design of new materials for gaseous NOx (NO and NO2) removal at ambient temperature using organic active sites is reported. The materials consist of unfunctionalized silica and silica modified by immobilized aminoxyls and function via sequential processes consisting of (i) NO oxidation to NO2 and (ii) NO2 storage. NOx removal by physical mixtures of immobilized PTIO (2-phenyl-4,4,5,5-tetramethyl-imidazoline-3-oxide-1-oxyl) sites on silica as the NO oxidant and hydrated silica as the NO2 trap occurs with significant degradation of the PTIO oxidant via undesired side reactions with NO2 when NO2 adsorption sites are fewer than required for its complete removal along the packed bed. The use of packed beds with sufficient NO2 adsorption sites requires a large excess of unfunctionalized silica, because of its low surface density of geminal silanols, which are shown to be the relevant sites for NO2 storage on silica at ambient temperature based on density functional theory calculations. This PTIO degradation is circumvented by the design of NOx traps consisting of immobilized PTIO on silica as the NO oxidant and immobilized TEMPO (2,2,6,6-tetramethylpiperidine-1-oxyl) on silica as an adsorbent with a high density of strong NO2 binding sites. Packed beds consisting of a 2:1 molar mixture of PTIO and TEMPO sites consume NOx as predicted by stoichiometry without detectable PTIO degradation and also without a contribution from geminal silanols as NO2 storage sites. This result requires that PTIO and TEMPO sites on silica render geminal silanols as inactive towards NO2 storage presumably because of the titration of these silanols by immobilized aminoxyls.
Co-reporter: Dante A. Simonetti;John H. Ahn ;Dr. Enrique Iglesia
ChemCatChem 2011 Volume 3( Issue 4) pp:704-718
Publication Date(Web):
DOI:10.1002/cctc.201000383
Abstract
We provide kinetic and isotopic evidence for the co-homologation of linear and branched alkanes with dimethyl ether (DME) to form larger branched alkanes and isobutane on H-BEA zeolites, and for the role of adamantane as a hydride transfer co-catalyst that allows the activation of CH bonds in alkanes at low temperatures (<500 K) on Brønsted acid sites. Branched alkanes (isobutane, isopentane, and 2,3-dimethylbutane) present in equimolar mixtures with DME form the corresponding alkenes via hydride transfer to bound alkoxides (formed in DME homologation steps) and subsequent deprotonation; these alkenes, derived from the added alkanes, are then methylated to lengthen their chain by using DME-derived C1 species, as shown by the isotopologues formed in reactions of 13C-DME with 12C-alkanes. Linear alkanes are much less reactive than branched alkanes, because of their stronger CH bonds and larger carbenium ion formation energies, which determines hydride-transfer rates to a given acceptor molecule. Adamantane increased the hydride-transfer rates to bound alkoxides from branched alkanes, and even from unreactive linear alkanes, while also increasing their extent of incorporation into DME homologation pathways; adamantane acts as a reversible hydrogen donor that mediates dehydrogenation of alkanes at low temperatures on acid sites. The co-homologation of alkanes with DME avoids the need for carbon rejection in the form of arenes to satisfy the hydrogen balance in the DME conversion to alkanes, provides a robust strategy for increasing the chain length and extent of branching in light alkanes through the selective addition of C1 species, and mitigates the formation of unsaturated by-products ubiquitous in the homologation of DME or methanol on Brønsted acids.
Co-reporter:Rajamani Gounder; Enrique Iglesia
ChemCatChem 2011 Volume 3( Issue 7) pp:1134-1138
Publication Date(Web):
DOI:10.1002/cctc.201100051
Co-reporter:Dr. Huamin Wang ; Enrique Iglesia
ChemCatChem 2011 Volume 3( Issue 7) pp:1166-1175
Publication Date(Web):
DOI:10.1002/cctc.201100027
Abstract
Kinetic, isotopic, and chemical analysis methods are used to examine the identity and kinetic relevance of elementary steps and the effects of Pt cluster size on thiophene hydrodesulfurization (HDS) turnover rates. Quasi-equilibrated H2 and H2S heterolytic dissociation steps lead to sulfur chemical potentials given by the prevalent H2S/H2 ratio and to cluster surfaces with a metallic bulk, but near-saturation sulfur coverages, during steady-state catalysis. Sulfur-vacancies on such surfaces are required for η1(S) or η4 thiophene adsorption modes and for H2 and H2S dissociation steps. H-assisted CS bond cleavage of η1(S) thiophene and H-addition to η4 thiophene limits rates of direct desulfurization and hydrogenation sulfur removal pathways, respectively. These steps, their kinetic relevance, and the prevalent sulfur-saturated surfaces resemble those on Ru clusters; they are also consistent with the observed kinetic effects of reactants and products on rates, with the rapid isotopic exchange in H2/D2/H2S mixtures during HDS catalysis, and with measured H2/D2 kinetic isotope effects. Small Pt clusters exhibit lower turnover rates, stronger inhibition by H2S, and a greater preference for desulfurization pathways than those of large clusters. These effects reflect the prevalence of coordinatively unsaturated corner and edge sites on small clusters, which bind sulfur atoms more strongly and lead to lower densities of vacancies and to a preference for η1(S)-bound thiophene species. Sulfur binding energies and their concomitant effects on the number of available vacancies also account for the higher turnover rates measured on Pt clusters compared with Ru clusters of similar size. These data and their mechanistic interpretation suggest that the concepts and steps proposed here apply generally to hydrogenation and direct desulfurization of organosulfur compounds. Taken together with similar observed effects of oxygen binding strength, metal identity, and cluster size for oxidation reactions of NO, hydrocarbons, and oxygenates, which also require vacancies in their respective kinetically relevant steps, these data also indicate that low reactivity of small clusters may reflect in most instances their coordinative unsaturation and the concomitant kinetic and thermodynamic preference for low vacancy concentrations on nearly saturated surfaces.
Co-reporter:Ya-Huei (Cathy) Chin
The Journal of Physical Chemistry C 2011 Volume 115(Issue 36) pp:17845-17855
Publication Date(Web):July 21, 2011
DOI:10.1021/jp203324y
Kinetic and isotopic data and effects of cluster size are used to probe elementary steps and their kinetic relevance in CH4–O2 reactions on Pd clusters that retain a metallic bulk during catalysis. CO2 and H2O were the only products detected, except when O2 was nearly depleted, during which trace CO amounts were formed. 13CH4–12CO–O2 reactions showed that CO reacts with chemisorbed oxygen (O*) much faster than CH4 with reactive collision probability ratios for CO and CH4 proportional to O2/CO ratios via a constant exceeding 500. Thus, even if CO desorbed before forming CO2, it would oxidize via reactions with O* at any reactor residence time required for detectable CH4 conversion, making direct partial oxidation impractical as a molecular route to H2–CO mixtures on Pd. CH4 turnover rates and effective first-order rate constants initially decreased and then reached constant values as O2 pressure and O* coverage increased as a result of a transition in the surface species involved in kinetically relevant C–H bond activation steps from O*–* to O*–O* site pairs (*, vacancy site). On O*–O* site pairs, C–H bonds are cleaved via H-abstraction mediated by O* and radical-like CH3 fragments weakly stabilized by the vicinal O* are formed at the transition state. These reactions show large activation barriers (158 kJ mol–1) but involve high entropy transition states that lead to larger pre-exponential factors (1.48 × 109 kPa–1 s–1) than for tighter transition states involved in C–H bond activation by *–* site pairs for CH4 reactions with H2O or CO2 (barriers: 82.5 kJ mol–1 and pre-exponential factors: 3.5 × 105 kPa–1 s–1). CH3 fragments at the transition state are effectively stabilized by interactions with vacancy sites on O*–* site pairs, which lead to higher turnover rates, as vacancies become available with decreasing O2 pressure. CH4–O2 turnover rates and C–H bond activation rate constants on O*–O* site pairs decreased with decreasing Pd cluster size, because coordinatively unsaturated exposed atoms on small clusters bind O* more strongly and decrease its reactivity for H-abstraction. The stronger O* binding on small Pd clusters also causes the kinetic involvement of O*–* sites to become evident at lower O2 pressures than on large clusters. These effects of metal–oxygen bond strength on O* reactivity also lead to the smaller turnover rates observed on Pd clusters compared with Pt clusters of similar size. These effects of cluster size and metal identity and their O* binding energy are the root cause for reactivity differences and appear to be general for reactions involving vacancies in kinetically relevant steps, as is the case for CH4, C2H6, NO, and CH3OCH3 oxidation on O*-covered surfaces and for hydrogenation of organosulfur compounds on surfaces nearly saturated with chemisorbed sulfur.
Co-reporter:Minkee Choi ; Zhijie Wu
Journal of the American Chemical Society 2010 Volume 132(Issue 26) pp:9129-9137
Publication Date(Web):June 10, 2010
DOI:10.1021/ja102778e
We report here a general synthetic strategy to encapsulate metal clusters within zeolites during their hydrothermal crystallization. Precursors to metal clusters are stabilized against their premature colloidal precipitation as hydroxides during zeolite crystallization using bifunctional (3-mercaptopropyl)trimethoxysilane ligands. Mercapto (−SH) groups in these ligands interact with cationic metal centers, while alkoxysilane moieties form covalent Si−O−Si or Si−O−Al linkages that promote zeolite nucleation around ligated metal precursors. These protocols led to the successful encapsulation of Pt, Pd, Ir, Rh, and Ag clusters within the NaA zeolite, for which small channel apertures (0.41 nm) preclude postsynthesis deposition of metal clusters. Sequential treatments in O2 and H2 formed small (∼1 nm) clusters with uniform diameter. Titration of exposed atoms with H2 or O2 gave metal dispersions that agree well with mean cluster sizes measured from electron microscopy and X-ray absorption spectroscopy, consistent with accessible cluster surfaces free of mercaptosilane residues. NaA micropore apertures restrict access to encapsulated clusters by reactants based on their molecular size. The ratio of the rates of hydrogenation of ethene and isobutene is much higher on clusters encapsulated within NaA than those dispersed on SiO2, as also found for the relative rates of methanol and isobutanol oxidation. These data confirm the high encapsulation selectivity achieved by these synthetic protocols and the ability of NaA micropores to sieve reactants based on molecular size. Containment within small micropores also protects clusters against thermal sintering and prevents poisoning of active sites by organosulfur species, thus allowing alkene hydrogenation to persist even in the presence of thiophene. The bifunctional nature and remarkable specificity of the mercapto and alkoxysilane functions for metal and zeolite precursors, respectively, render these protocols extendable to diverse metal−zeolite systems useful as shape-selective catalysts in demanding chemical environments.
Co-reporter:Manuel Ojeda ; Anwu Li ; Rahul Nabar ; Anand U. Nilekar ; Manos Mavrikakis
The Journal of Physical Chemistry C 2010 Volume 114(Issue 46) pp:19761-19770
Publication Date(Web):October 29, 2010
DOI:10.1021/jp1073076
H2/D2 isotope effects on Fischer−Tropsch synthesis (FTS) rate and selectivity are examined here by combining measured values on Fe and Co at conditions leading to high C5+ yields with theoretical estimates on model Fe(110) and Co(0001) surfaces with high coverages of chemisorbed CO (CO*). Inverse isotope effects (rH/rD < 1) are observed on Co and Fe catalysts as a result of compensating thermodynamic (H2 dissociation to H*; H* addition to CO* species to form HCO*) and kinetic (H* reaction with HCO*) isotope effects. These isotopic effects and their rigorous mechanistic interpretation confirm the prevalence of H-assisted CO dissociation routes on both Fe and Co catalysts, instead of unassisted pathways that would lead to similar rates with H2 and D2 reactants. The small contributions from unassisted pathways to CO conversion rates on Fe are indeed independent of the dihydrogen isotope, as is also the case for the rates of primary reactions that form CO2 as the sole oxygen rejection route in unassisted CO dissociation paths. Isotopic effects on the selectivity to C5+ and CH4 products are small, and D2 leads to a more paraffinic product than does H2, apparently because it leads to preference for chain termination via hydrogen addition over abstraction. These results are consistent with FTS pathways limited by H-assisted CO dissociation on both Fe and Co and illustrate the importance of thermodynamic contributions to inverse isotope effects for reactions involving quasi-equilibrated H2 dissociation and the subsequent addition of H* in hydrogenation catalysis, as illustrated here by theory and experiment for the specific case of CO hydrogenation.
Co-reporter:Dr. Eva Díaz;Dr. María Eugenia Sad; Enrique Iglesia
ChemSusChem 2010 Volume 3( Issue 9) pp:1063-1070
Publication Date(Web):
DOI:10.1002/cssc.201000142
Abstract
O2 reacts with propanediols via homogeneous pathways at 400–500 K. 1,2-Propanediol forms CH3CHO, HCHO, and CO2 via oxidative CC cleavage and acetone via dehydration routes, while symmetrical 1,3-propanediol undergoes dehydration and oxidative dehydrogenation to form, almost exclusively, acrolein (ca. 90 % selectivity). The products formed and their kinetic dependence on reactant concentrations are consistent with radical-mediated pathways initiated by O2 insertion into CH bonds in a β position relative to oxygen atoms in diol reactants. Propagation involves β-scission reactions that form hydroxyl and hydroxyalkyl radicals. Acrolein/O2/H2O mixtures from the homogeneous oxidation of 1,3-propanediol form acrylic acid (with 90 % yield) in tandem reactors containing molybdenum-vanadium oxide catalysts. These data reveal the unique reactivity of diols, compared with triols and alkanols, in homogeneous oxidations, while also providing useful insight into the molecular basis for reactivity in biomass-derived oxygenates.
Co-reporter:Manuel Ojeda and Enrique Iglesia
Chemical Communications 2009 (Issue 3) pp:352-354
Publication Date(Web):03 Dec 2008
DOI:10.1039/B813589D
Au/TiO2catalysts form hydroperoxy species from H2O–O2 mixtures at near-ambient temperatures; these species can be used in the selective epoxidation of propene to propylene oxide.
Co-reporter:Kazuhiro Takanabe and Enrique Iglesia
The Journal of Physical Chemistry C 2009 Volume 113(Issue 23) pp:10131-10145
Publication Date(Web):May 13, 2009
DOI:10.1021/jp9001302
Kinetic and isotopic methods were used to determine the identity, rate constants, and reversibility of elementary steps for primary and secondary reactions involved in the oxidative coupling of methane (OCM) on Mn/Na2WO4/SiO2. We provide evidence in this study for parallel C−H bond activation pathways, in which H-abstraction is mediated by either oxygen species on surfaces or by OH radicals formed via H2O/O2 equilibration on catalyst surfaces. OCM rates and C2+ yields are higher when H2O is present and OH-mediated pathways prevail, because of the high reactivity of OH radicals and of their lesser sensitivity to the energy of the C−H bond containing the hydrogen abstracted. These coupled homogeneous-catalytic sequences account for all observed kinetic effects of O2, CH4, and H2O on rates and selectivities for both CH4 conversion and for subsequent reactions of C2H6, C2H4 and C3 products; they are also consistent with measured kinetic and thermodynamic isotope effects for C−H bond activation mediated by surface and OH radicals. Kinetic isotope effects and isotopic scrambling studies (CD4/CH4; D2O/H2O; 18O2/16O2) indicate that C−H bond activation is irreversible and kinetically-relevant. O2 dissociation is quasi-equilibrated, but becomes irreversible as H2O/O2 ratios increase with increasing conversion and residence time. Competitive reactions of 13CH4/O2 with 12C2H6, 12C2H4, and 12C3H6 with and without added H2O show that H-abstraction from hydrocarbons is much less sensitive to C−H bond strength when OH radicals are used to abstract hydrogen instead of oxide surfaces. Maximum C2+ yields require conditions that favor OH-mediated pathways while maintaining equilibrium oxygen surface coverages and OH radical concentrations. OH-mediated pathways are more sensitive to O2 pressure than surface-mediated pathways; thus, low O2 pressures and staging strategies that maintain stoichiometric O2 requirements and low local O2 pressures can improve C2+ selectivities but only when OH radicals are maintained at equilibrium concentrations via catalytic H2O−O2 reactions. These findings and interpretations indicate that intermediate O2 pressures give maximum C2+ yields, but that their optimal value depends sensitively on prevalent H2O concentrations as they vary with conversion along the reactor. These predictions about the consequences of various operating strategies have become feasible because of the detailed and quantitative nature of the mechanism-based kinetic networks reported here for the first time.
Co-reporter:Michael Zboray, Alexis T. Bell and Enrique Iglesia
The Journal of Physical Chemistry C 2009 Volume 113(Issue 28) pp:12380-12386
Publication Date(Web):June 19, 2009
DOI:10.1021/jp901595k
The oxidative dehydrogenation of alkanes (C2H6, C3H8, i-C4H10, and n-C4H10) was investigated on VOx supported on Al2O3. Rate constants for alkane dehydrogenation (k1), alkane combustion (k2), and alkene combustion (k3) were measured, and a model was developed to describe the effects of alkane composition on these rate constants. The proposed model accounts for the effects of the number of C−H bonds available for activation and the relative strengths of these bonds in both the reactant and the product molecules. The Brønsted−Evans−Polanyi (BEP) relationship is used to relate activation energies of secondary and tertiary C−H bonds to that of primary C−H bonds. The model gives a reasonable approximation of the relative order of alkane reactivity, expressed by k1 + k2, and the relative ranking of alkanes with respct to combustion versus oxidative dehydrogenation, expressed by k2/k1. The ratio of k2/k1 is described by the product of two components: one that depends on the nature, number, and relative strength of C−H bonds of surface alkoxides, and a second one that is independent of the alkoxide composition and structure but depends on the difference in the entropy of activation for COx precursor versus alkene formation. The model also explains the observed variation of k3 with alkene composition by considering two precursor states for alkenes. One is strongly bound through π-orbital interactions with Lewis acid centers, and the second weakly binds via H bonding and van der Waals interactions, similar to the binding of alkanes. As a result, the rate of alkene combustion depends strongly on the large heats of adsorption of alkenes and only slightly on the presence of weak allylic C−H bonds. The high rate of C2H4 combustion is thus a consequence of its high heat of adsorption.
Co-reporter:JohnH. Ahn;Burcin Temel Dr.
Angewandte Chemie 2009 Volume 121( Issue 21) pp:3872-3874
Publication Date(Web):
DOI:10.1002/ange.200900541
Co-reporter:Manuel Ojeda Dr.
Angewandte Chemie 2009 Volume 121( Issue 26) pp:4894-4897
Publication Date(Web):
DOI:10.1002/ange.200805723
Co-reporter:Brian M. Weiss and Enrique Iglesia
The Journal of Physical Chemistry C 2009 Volume 113(Issue 30) pp:13331-13340
Publication Date(Web):July 1, 2009
DOI:10.1021/jp902209f
Kinetic and isotopic methods show that NO oxidation on supported Pt clusters involves kinetically relevant reaction of O2 with vacancy sites on surfaces nearly saturated with oxygen adatoms (O*). The oxygen chemical potential at Pt surfaces that determines the O* coverage is rigorously described by an O2 virtual pressure and determined by the thermodynamics of NO2−NO interconversion reactions. NO oxidation and oxygen isotopic exchange processes are described by the same rate constant, consistent with similar kinetically relevant O2 dissociation steps for both reactions. NO oxidation, NO2 decomposition, and 16O2−18O2 exchange rates increased markedly with increasing Pt cluster size (1−8 nm); these clusters remain metallic at all O2 virtual pressures prevalent during NO oxidation. These effects of cluster size reflect the higher vacancy concentrations and more facile oxygen desorption on larger Pt clusters, which bind oxygen adatoms weaker than more coordinatively unsaturated surface Pt atoms on smaller clusters. These trends are similar to those found for methane and dimethyl ether combustion on Pt and Pd catalysts, which also require vacancy sites on O*-saturated cluster surfaces in their respective kinetically relevant steps. Inhibition of NO oxidation by NO2 persists to undetectable NO2 concentrations; thus, NO oxidation turnover rates increase significantly when NO2 adsorption sites present on BaCO3/Al2O3 are placed within diffusion distances of Pt clusters. NO oxidation rates on intrapellet catalyst-adsorbent mixtures are described accurately by a simple reaction-adsorption model in which NO2 adsorbs via displacement of CO2 on BaCO3 surfaces.
Co-reporter:Manuel Ojeda Dr.
Angewandte Chemie International Edition 2009 Volume 48( Issue 26) pp:4800-4803
Publication Date(Web):
DOI:10.1002/anie.200805723
Co-reporter:Beata Kilos, Alexis T. Bell and Enrique Iglesia
The Journal of Physical Chemistry C 2009 Volume 113(Issue 7) pp:2830-2836
Publication Date(Web):2017-2-22
DOI:10.1021/jp8078056
The mechanism and structural requirements for ethanol oxidation to acetaldehyde were examined on VOx domains supported on γ-Al2O3 at surface densities of 1.7−11.8 VOx/nm2. Raman and UV−visible spectra showed that VOx species evolve from monovanadate to polyvanadate structures with increasing surface density with only traces of crystalline V2O5. Oxidative dehydrogenation (ODH) of ethanol to acetaldehyde occurs at low temperatures (473−523 K) with high primary selectivities of CH3CHO (∼80%) on a catalyst with one theoretical polyvanadate monolayer. ODH turnover rates (per V-atom) increased with increasing VOx surface density for surface densities up to 7.2 V/nm2, indicating that polyvanadate domain surfaces are more reactive than monovanadate structures. Similar trends were evident for alkane ODH reactions that also involve kinetically relevant H-abstraction steps within reduction−oxidation catalytic sequences. Turnover rates ultimately decreased at higher surface densities because of the incipient formation of three-dimensional structures. VOx domains of intermediate size therefore provide a compromise between site reactivity and accessibility during ethanol ODH. The effects of O2 and C2H5OH pressures on ethanol ODH rates and the kinetic isotope effects for C2H5OD and C2D5OD confirmed the kinetic relevance of H-abstraction from ethoxide species formed in quasiequilibrated ethanol dissociation steps; taken together with in situ infrared spectra, these data also show that ethoxide species are present at near saturation coverages on fully oxidized VOx domains that undergo reduction−oxidation cycles during each ethanol oxidation turnover.
Co-reporter:JohnH. Ahn;Burcin Temel Dr.
Angewandte Chemie International Edition 2009 Volume 48( Issue 21) pp:3814-3816
Publication Date(Web):
DOI:10.1002/anie.200900541
Co-reporter:Aditya Bhan and Enrique Iglesia
Accounts of Chemical Research 2008 Volume 41(Issue 4) pp:559
Publication Date(Web):February 16, 2008
DOI:10.1021/ar700181t
The extent to which spatial constraints influence rates and pathways in catalysis depends on the structure of intermediates, transition states, and active sites involved. We aim to answer, as we seek insights into catalytic mechanisms and site requirements, persistent questions about the potential for controlling rates and selectivities by rational design of spatial constraints around active sites within inorganic structures useful as catalysts. This Account addresses these matters for the specific case of reactions on zeolites that contain Brønsted acid sites encapsulated within subnanometer channels. We compare and contrast here the effects of local zeolite structure on the dynamics of the carbonylation of surface methyl groups and of the isotopic exchange of CD4 with surface OH groups on zeolites. Methyl and hydroxyl groups are the smallest monovalent cations relevant in catalysis by zeolites. Their small size, taken together with their inability to desorb except via reactions with other species, allowed us to discriminate between stabilization of cationic transition states and stabilization of adsorbed reactants and products by spatial constraints. We show that apparent effects of proton density and of zeolite channel structure on dimethyl ether carbonylation turnover rates reflect instead the remarkable specificity of eight-membered ring zeolite channels in accelerating kinetically relevant steps that form *COCH3 species via CO insertion into methyl groups. This specificity reflects the selective stabilization of cationic transition states via interactions with framework oxygen anions. These findings for carbonylation catalysts contrast sharply the weak effects of channel structure on the rate of exchange of CD4 with OH groups. This latter reaction involves concerted symmetric transition states with much lower charge than that required for CH3 carbonylation. Our Account extends the scope of shape selectivity concepts beyond those reflecting size exclusion and preferential adsorption. Our ability to discriminate among various effects of spatial constraints depends critically on dissecting chemical conversions into elementary steps of kinetic relevance and on eliminating secondary reactions and accounting for the concomitant effects of zeolite structure on the stability of adsorbed reactants and intermediates.
Co-reporter:Xuebing Li and Enrique Iglesia
Chemical Communications 2008 (Issue 5) pp:594-596
Publication Date(Web):27 Nov 2007
DOI:10.1039/B715543C
Pt clusters within [Fe]ZSM-5 channels provide active and stable sites for the selective catalytic dehydrogenation of n-alkanes to n-alkenes. Cs and Na cations titrate acid sites and inhibit skeletal isomerization and cracking side reactions.
Co-reporter:Josef Macht and Enrique Iglesia
Physical Chemistry Chemical Physics 2008 vol. 10(Issue 35) pp:5331-5343
Publication Date(Web):09 Jul 2008
DOI:10.1039/B805251D
Redox and acid–base properties of dispersed oxide nanostructures change markedly as their local structure and electronic properties vary with domain size. These changes give rise to catalytic behavior, site structures, and reaction chemistries often unavailable on bulk crystalline oxides. Turnover rates for redox and acid catalysis vary as oxide domains evolve from isolated monomers to two-dimensional oligomers, and ultimately into clusters with bulk-like properties. These reactivity changes reflect the ability of oxide domains to accept or redistribute electron density in kinetically-relevant reduction steps, in the formation of temporary acid sites via reductive processes, and in the stabilization of cationic transition states. Reduction steps are favored by low-lying empty orbitals prevalent in larger clusters, which also favor electron delocalization, stable anions, and strong Brønsted acidity. Isomerization of xylenes and alkanes, elimination reactions of alkanols, and oxidation of alkanes to alkenes on V, Mo, Nb, and W oxide domains are used here to demonstrate the remarkable catalytic diversity made available by changes in domain size. The reactive and disordered nature of small catalytic domains introduces significant challenges in their synthesis and their structural and mechanistic characterization, which require in situprobes and detailed kinetic analysis. The local structure and electronic properties of these materials must be probed during catalysis and their catalytic function be related to specific kinetically-relevant steps. Structural uniformity can be imposed on oxide clusters by the use of polyoxometalate clusters with thermodynamically stable and well-defined size and connectivity. These clusters provide the compositional diversity and the structural fidelity required to develop composition-function relations from synergistic use of experiments and theory. In these clusters, the valence and electronegativity of the central atom affects the acid strength of the polyoxometalate clusters and the rate constants for acid catalyzed elementary steps via the specific stabilization of cationic transition states in isomerization and elimination reactions.
Co-reporter:Xuebing Li
The Journal of Physical Chemistry C 2008 Volume 112(Issue 38) pp:15001-15008
Publication Date(Web):August 29, 2008
DOI:10.1021/jp801488y
Kinetic and isotopic studies showed that C−H bond activation in ethane by surfaces essentially saturated with lattice oxygens is the sole kinetically relevant step in ethane oxidation on Mo−V−NbOx mixed oxides. These conclusions are consistent with the dependence of oxidation rates on O2 and C2H6 pressures, with H/D exchange and kinetic isotope effects, and with the preferential initial incorporation of 16O atoms from the oxide lattice into products formed from 18O2−C2H6 mixtures. The precipitation of active components (Mo0.61V0.31Nb0.08Ox) in the presence of colloidal TiO2 led to 10-fold increases in all rate constants (per active component), consistent with higher dispersion of active components resembling in structure and surface reactivity those prevalent in bulk powders. The concurrent presence of PdOx cocatalyst, even as a separate solid, markedly increased all rate constants for oxidation of ethene intermediates and specifically that for ethene oxidation to acetaldehyde molecules, which are rapidly converted to acetic acid on active Mo−V−NbOx sites. Water, whether formed as a byproduct or added with C2H6−O2 reactants, increases acetic acid selectivities by promoting the desorption of adsorbed acetate species as acetic acid. Ethene molecules, formed as reactive intermediates, inhibit ethane oxidation rates by depleting surface lattice oxygen atoms in fast oxidation reactions, thus decreasing the number of sites available at steady state for the kinetically relevant C−H bond activation step required for ethane conversion.
Co-reporter:Kazuhiro Takanabe Dr.
Angewandte Chemie International Edition 2008 Volume 47( Issue 40) pp:7689-7693
Publication Date(Web):
DOI:10.1002/anie.200802608
Co-reporter:Kazuhiro Takanabe Dr.
Angewandte Chemie 2008 Volume 120( Issue 40) pp:7803-7807
Publication Date(Web):
DOI:10.1002/ange.200802608
Co-reporter:Akio Ishikawa and Enrique Iglesia
Chemical Communications 2007 (Issue 28) pp:2992-2993
Publication Date(Web):11 May 2007
DOI:10.1039/B702693E
Mixtures of Pt clusters dispersed on γ-Al2O3 and additional γ-Al2O3 led to much higher DME combustion turnover rates than on the individual components or on Pt clusters supported on non-acidic oxides.
Co-reporter:Janine Lichtenberger, Doohwan Lee and Enrique Iglesia
Physical Chemistry Chemical Physics 2007 vol. 9(Issue 35) pp:4902-4906
Publication Date(Web):27 Jul 2007
DOI:10.1039/B707465D
Supported Pd clusters catalyze methanol oxidation to methyl formate with high turnover rates and >90% selectivity at near ambient temperatures (313 K). Metal clusters are much more reactive than PdO clusters and rates are inhibited by the reactant O2. These data suggest that ensembles of Pd metal atoms on surfaces nearly saturated with chemisorbed oxygen are required for kinetically-relevant C–H bond activation in chemisorbed methoxide intermediates. Pd metal surfaces become more reactive with increasing metal particle size. The higher coordination of surface atoms on larger clusters leads to more weakly-bound chemisorbed species and to a larger number of Pd metal ensembles available during steady-state catalysis. Chemisorbed oxygen removes H-atoms formed in C–H bond activation steps and inhibits methoxide decomposition and CO2 formation, two functions essential for the high turnover rates and methyl formate selectivities reported here.
Co-reporter:Josef Macht;Michael J. Janik ;Matthew Neurock
Angewandte Chemie 2007 Volume 119(Issue 41) pp:
Publication Date(Web):7 SEP 2007
DOI:10.1002/ange.200701292
Die Geschwindigkeitskonstanten der säurekatalysierten Butanoldehydratisierung an Keggin-Polyoxometallatclustern H8−nXn+W12O40 (X=P, Si, Al, Co) steigen mit zunehmendem Oxidationszustand des Zentralatoms und, damit einhergehend, abnehmender Zahl von Protonen im Cluster (siehe Diagramm). Diese Trends spiegeln die niedrigeren Deprotonierungsenthalpien von Clustern mit hochvalentem Zentralatom und ihre Stabilität als anionische konjugierte Base in ionischen Übergangszuständen bei der katalytischen Dehydratisierung wider.
Co-reporter:Bi-Zeng Zhan Dr.
Angewandte Chemie 2007 Volume 119(Issue 20) pp:
Publication Date(Web):5 APR 2007
DOI:10.1002/ange.200700128
Gefangen! Das Titelsystem (ca. 1 nm Durchmesser; links) katalysiert die Oxidation von Methanol mit höheren Umsatzzahlen als Cluster auf SiO2-Trägern. In dem beschränkten Raum wird Methanol gegenüber größeren Alkoholen bevorzugt oxidiert. Der eingeschränkte Zugang zu aktiven Stellen schützt auch eingeschlossene Ru-Cluster (rechts) gegen die Inhibierung der Ethenhydrierung durch Organoschwefelverbindungen.
Co-reporter:Xuebing Li Dr.
Angewandte Chemie International Edition 2007 Volume 46(Issue 45) pp:
Publication Date(Web):9 OCT 2007
DOI:10.1002/anie.200700593
A great show at the Palladium: Precipitation of Mo–V–Nb oxides in the presence of colloidal TiO2 leads to more dispersed active structures and much higher rates of ethene oxidation to acetic acid. The presence of trace amounts of palladium as supported clusters in physical mixtures (<0.05 % w/w) catalyzes the formation of acetaldehyde intermediates and leads to unprecedented rates and selectivities in the synthesis of acetic acid from ethene and O2.
Co-reporter:Josef Macht;Michael J. Janik ;Matthew Neurock
Angewandte Chemie International Edition 2007 Volume 46(Issue 41) pp:
Publication Date(Web):7 SEP 2007
DOI:10.1002/anie.200701292
Rate constants for acid-catalyzed butanol dehydration on Keggin-type polyoxometalate clusters H8−nXn+W12O40 (X=P, Si, Al, Co) increase as the oxidation state of the central atom increases and the number of cluster protons concurrently decreases (see diagram). These trends reflect the lower deprotonation enthalpies of clusters with high valent central atoms and their stability as the anionic conjugate base in ionic transition states involved in dehydration catalysis.
Co-reporter:Bi-Zeng Zhan Dr.
Angewandte Chemie International Edition 2007 Volume 46(Issue 20) pp:
Publication Date(Web):5 APR 2007
DOI:10.1002/anie.200700128
Trapped! The title system (ca. 1 nm diameter; left) catalyzes methanol oxidation with higher turnover rates than clusters on SiO2 supports. Spatial constraints lead to the preferential oxidation of methanol over larger alcohols. Restricted access to active sites also protects encapsulated Ru clusters (right) against inhibition of ethene hydrogenation by organosulfur compounds.
Co-reporter:Xuebing Li Dr. Dr.
Chemistry - A European Journal 2007 Volume 13(Issue 33) pp:
Publication Date(Web):2 OCT 2007
DOI:10.1002/chem.200700579
The direct oxidation of ethanol to acetic acid is catalyzed by multicomponent metal oxides (Mo-V-NbOx) prepared by precipitation in the presence of colloidal TiO2 (Mo0.61V0.31Nb0.08Ox/TiO2). Acetic acid synthesis rates and selectivities (∼95 % even at 100 % ethanol conversion) were much higher than in previous reports. The presence of TiO2 during synthesis led to more highly active surface areas without detectable changes in the reactivity or selectivity of exposed active oxide surfaces. Ethanol oxidation proceeds via acetaldehyde intermediates that are converted to acetic acid. Water increases acetic acid selectivity by inhibiting acetaldehyde synthesis more strongly than its oxidation to acetic acid, thus minimizing prevalent acetaldehyde concentrations and its intervening conversion to COx. Kinetic and isotopic effects indicate that CH bond activation in chemisorbed ethoxide species limits acetaldehyde synthesis rates and overall rates of ethanol conversion to acetic acid. The VOx component in Mo-V-Nb is responsible for the high reactivity of these materials. Mo and Nb oxide components increase the accessibility and reducibility of VOx domains, while concurrently decreasing the number of unselective V-O-Ti linkages in VOx domains dispersed on TiO2.
Co-reporter:Xuebing Li Dr.
Angewandte Chemie 2007 Volume 119(Issue 45) pp:
Publication Date(Web):9 OCT 2007
DOI:10.1002/ange.200700593
Palladium gibt Gas: Die Fällung von Mo-V-Nb-Oxiden in Gegenwart von kolloidalem TiO2 führt zu fein verteilten, aktiven Strukturen für die Oxidation von Ethen zu Essigsäure. Zugemischte Palladiumcluster auf einem Träger (<0.05 Gew.-%) katalysieren die Bildung von Acetaldehyd-Zwischenstufen. Mit diesem System gelingt die beispiellos schnelle und selektive Synthese von Essigsäure aus Ethen und O2.
Co-reporter:Trenton Otto, José M. Ramallo-López, Lisandro J. Giovanetti, Félix G. Requejo, Stacey I. Zones, Enrique Iglesia
Journal of Catalysis (October 2016) Volume 342() pp:125-137
Publication Date(Web):1 October 2016
DOI:10.1016/j.jcat.2016.07.017
•Synthesis of AuPd, AuPt, and PdPt bimetallic clusters encapsulated in LTA zeolite.•Small bimetallic clusters uniform in size and composition.•Thermal sinter stability through small-pore zeolite encapsulation.•Encapsulated metals are size selective and sulfur-tolerant catalysts.AuPd, AuPt, and PdPt bimetallic clusters uniform in size and composition were prepared using hydrothermal assembly of LTA crystals around cationic precursors stabilized by protecting mercaptosilane ligands. The sulfur moiety in these bifunctional ligands forms adducts that prevent premature reduction or precipitation of metal precursors during crystallization. The silane groups can form bridges with silicate oligomers as they form, thus enforcing homogeneous distributions of precursors throughout crystals and ensuring that subsequent reductive treatments lead to the two elements residing within small and nearly monodisperse clusters. Their confinement within LTA crystals, evident from microscopy and titrations with large poisons, renders them stable against sintering during thermal treatments at high temperatures (820–870 K). Infrared spectra of chemisorbed CO show that bimetallic surfaces are free of synthetic debris after thermal treatments; these spectra also indicate that intracluster segregation occurs upon CO chemisorption, a demonstration of the presence of the two elements within the same clusters. The number and type of atoms coordinated to a given absorber atom, determined from the fine structure in X-ray absorption spectra, are consistent with bimetallic structures of uniform composition. The rates of ethanol oxidative dehydrogenation on these bimetallic clusters were essentially unaffected by exposure to dibenzothiophene, a large poison that suppresses rates on unconfined clusters, indicating that bimetallic clusters are protected within the confines of LTA crystals. These synthetic protocols seem generally applicable to other bimetallic compositions and zeolites, for which the monometallic counterparts have been successfully encapsulated within several microporous frameworks using ligand-stabilized precursors and hydrothermal crystallization methods.Download high-res image (289KB)Download full-size image
Co-reporter:Masaoki Iwasaki, Enrique Iglesia
Journal of Catalysis (October 2016) Volume 342() pp:84-97
Publication Date(Web):1 October 2016
DOI:10.1016/j.jcat.2016.07.011
•Kinetic and UV-visible methods assess elementary steps and the number of active sites.•NO oxidation involves kinetically-relevant O2 adsorption and O2∗ dissociation assisted by NO.•NO oxidation turnover rates depend weakly on W surface density (2.1–9.5 W/nm2).•WO3 domains in contact with CeO2 promote at CeO2-derived redox-active centers near their interface.The effects of NO, NO2 and O2 pressures on NO oxidation rates and UV-visible spectra are used here to assess the elementary steps and the number and type of redox-active sites involved in NO oxidation on CeO2 promoted by contact with WO3 domains. The reversible chemisorption of O2 on vacancies (∗) and the subsequent dissociation of O2∗ assisted by NO to form O∗ and NO2 are the kinetically-relevant steps on surfaces with O∗ coverage set by NO−NO2 equilibration. O2p → Ce4f ligand-to-metal charge transfer (LMCT) bands probe the rate constants for O2∗ formation and desorption at catalytic conditions; their comparison with those derived from rate data confirms the mechanistic conclusions and the involvement of CeO2 surfaces promoted by contact with WO3 domains. These data allow an accurate assessment of the number and type of redox-active sites, thus allowing reactivity comparisons among catalysts based on turnover rates. The number of redox-active sites increased with increasing W surface density (2.1–9.5 W/nm2), but NO oxidation turnover rates were essentially unchanged. These elementary steps and active structures differ markedly from those that mediate NO oxidation on Pt, PdO, RhO2 and Co3O4 catalysts. Turnover rates are similar on WO3/CeO2 and Pt-based catalysts at practical temperatures of diesel exhaust treatment (∼500 K), but WO3/CeO2 catalysts exhibit much higher rates based on catalyst mass (>10-fold), thus rendering useful as less costly and more resilient alternatives to noble metals. These findings illustrate a method to probe the number and type of redox-active sites and conceptual insights into the pathways that mediate the chemisorption and activation of O2 by isolated vacancies and the subsequent dissociation of OO bonds by assistance from co-reactants.Download high-res image (238KB)Download full-size image
Co-reporter:William Knaeble, Robert T. Carr, Enrique Iglesia
Journal of Catalysis (November 2014) Volume 319() pp:283-296
Publication Date(Web):1 November 2014
DOI:10.1016/j.jcat.2014.09.005
•Skeletal isomerization rates depend on alkene concentrations at acid sites.•Turnover rates (per proton) increase exponentially with deprotonation energy.•Ion-pair transition states mediate kinetically-relevant backbone rearrangements.•Transition states for hexene interconversions depend similarly on deprotonation energies.•Alkene isomerization product selectivity is independent of Brønsted acid strength.Acid strength effects on alkane isomerization turnover rates and selectivities are probed using hexene isomers as reactants on bifunctional catalysts containing tungsten Keggin polyoxometalates (POM) with different central atoms and exhibiting well-defined structures amenable to reliable estimates of deprotonation energies (DPE) as rigorous descriptors of acid strength. Titrations of protons with hindered bases during catalysis and mechanistic interpretations of rate data on POM acids in terms of a common sequence of elementary steps give isomerization rate constants that decrease exponentially with increasing DPE. The sensitivity to acid strength is the same for all interconversions among isomeric hexenes because their respective transition states are similar in the amount and localized character of their cationic charges, which determine, in turn, the extent to which the ionic and covalent interactions that determine DPE are recovered upon formation of ion pairs at transition states. The ratios of rate constants for such interconversions, and thus selectivities, are independent of acid strength and their magnitude merely reflects the stability of the gaseous analogs of their respective transition states on all acids.Download high-res image (266KB)Download full-size image
Co-reporter:David D. Hibbitts, Romel Jiménez, Masayuki Yoshimura, Brian Weiss, Enrique Iglesia
Journal of Catalysis (November 2014) Volume 319() pp:95-109
Publication Date(Web):1 November 2014
DOI:10.1016/j.jcat.2014.07.012
•NO–H2 turnover rates depend weakly on Pt cluster size (1.5–10 nm).•HD formation rates in NO–H2–D2 reactions show quasi-equilibrated H2 dissociation.•Isotopic effects are consistent with kinetically-relevant *HNOH* formation.•Theory indicates that N–O cleaves only after H*-additions to form NOH* and *HNOH*.Kinetic and isotopic data on Pt clusters and activation free energy barriers from density functional theory (DFT) on Pt(1 1 1) are used to assess the elementary steps involved in NO–H2 reactions. Pt clusters 1–10 nm in diameter gave similar turnover rates, indicating that these elementary steps are insensitive to surface-atom coordination. N–O cleavage occurs after sequential addition of two chemisorbed H-atoms (H*) to NO* which are quasi-equilibrated with H2 and NO co-reactants. The first step is equilibrated and forms HNO*, while the second addition is irreversible and forms *HNOH*; this latter step limits NO–H2 rates and forms OH* and NH* intermediates that undergo fast reactions to give H2O, N2O, NH3, and N2. These conclusions are consistent with (i) measured normal H/D kinetic isotope effects; (ii) rates proportional to H2 pressure, but reaching constant values at higher pressures; (iii) fast H2–D2 equilibration during catalysis; and (iv) DFT-derived activation barriers. These data and calculations, taken together, rule out N–O cleavage via NO* reactions with another NO* (forming O* and N2O) or with vicinal vacancies (forming N* and O*), which have much higher barriers than H*-assisted routes. The cleavage of N–O bonds via *HNOH* intermediates is reminiscent of C–O cleavage in CO–H2 reactions (via *HCOH*) and of O–O cleavage in O2–H2 reactions (via OOH* or *HOOH*). H*-addition weakens the multiple bonds in NO, CO, and O2 and allows coordination of each atom to metal surfaces; as a result, dissociation occurs via such assisted routes at all surface coverages relevant in the practice of catalysis.Download high-res image (201KB)Download full-size image
Co-reporter:Mónica García-Diéguez, Enrique Iglesia
Journal of Catalysis (May 2013) Volume 301() pp:198-209
Publication Date(Web):1 May 2013
DOI:10.1016/j.jcat.2013.02.014
The effects of CO and O2 concentrations on turnover rates and 18O2–16O2 exchange rates during catalysis are used to assess the relevant elementary steps and the consequences of Pt coordination for CO oxidation catalysis at moderate temperatures (700–800 K) on supported Pt clusters 1.8–25 nm in diameter. Turnover rates, measured under conditions of strict kinetic control, are proportional to O2 pressure and inhibited by CO; these data are consistent with kinetically-relevant O2 dissociation steps on cluster surfaces covered partially by chemisorbed CO (CO*). O2 dissociation also limits CO oxidation rates at higher temperatures, which lead to bare Pt surfaces, and at lower temperatures, where saturation CO* coverages require O2 dissociation to be assisted by CO* because of a dearth of vacant sites. At the intermediate temperatures used here, kinetic coupling between irreversible O2 activation and CO* reactions with O* causes edge and corner atoms to become decorated by unreactive O* species; consequently, turnovers occur predominantly on exposed low-index planes, which account for a decreasing fraction of exposed atoms with increasing metal dispersion. These decoration effects confer the appearance of structure sensitivity to the prototypical structure insensitive reaction by rendering only a fraction of exposed metal atoms able to turnover. These active sites, residing at exposed low-index planes, show similar CO* binding energies on large and small Pt clusters, but their relative abundance decreases as clusters become smaller, leading to a sharp decrease in turnover rates with increasing Pt dispersion. These trends stand in marked contrast with the absence of cluster size effects on CO oxidation rates at low temperatures, where high CO* coverages dampen the intrinsic site non-uniformity of metal clusters, and at high temperatures, where all Pt atoms remain accessible irrespective of coordination and active for catalytic turnovers.Graphical abstractCO oxidation turnovers occur predominantly on Pt atoms exposed at low-index planes, because strongly chemisorbed and unreactive oxygens chemisorb at low-coordination sites. As a result, turnover rates increase with Pt cluster size, while CO adsorption constants remain unchanged at those exposed atoms that remain accessible and are responsible for CO oxidation catalytic turnovers.Download high-res image (279KB)Download full-size imageHighlights► Pt surfaces are partially covered by CO* at 723–793 K during CO–O2 reactions. ► CO oxidation takes place predominantly on low-index planes. ► Low-coordination sites are decorated by strongly bound and less reactive oxygen. ► O* decoration confers structure sensitivity to CO oxidation. ► Mechanistic analyses are required to identify structure sensitiveness of reactions.
Co-reporter:David W. Flaherty, David D. Hibbitts, Elif I. Gürbüz, Enrique Iglesia
Journal of Catalysis (March 2014) Volume 311() pp:350-356
Publication Date(Web):1 March 2014
DOI:10.1016/j.jcat.2013.11.026
•Kinetic data and DFT used to study C–C rupture on Ir clusters.•C2-intermediates formed by quasi-equilibrated adsorption and dehydrogenation.•Close agreement between measured kinetic barrier and DFT-derived barriers.•C–C cleavage occurs via *CHCH* > 107 times faster than for other intermediates.Ethane hydrogenolysis involves C–C bond rupture in unsaturated species in quasi-equilibrium with gaseous reactants and H2 on metal clusters, because C–C bonds weaken as C-atoms replace hydrogen with exposed metal atoms from catalyst surfaces. The nature and reactivity of such adsorbed species are probed here using kinetic data and density functional theory (DFT) for the case of Ir surfaces, but with conclusions that appear to be general to hydrogenolysis on noble metals. On surfaces saturated with chemisorbed H-atoms (H*), theory and experiments indicate that C–C cleavage occurs predominantly via an α,β-bound *CHCH* species that forms via sequential dehydrogenation of adsorbed ethane; all other intermediates cleave C–C bonds at much lower rates (>107-fold). Measured activation energies (213 kJ mol−1) and free energies (130 kJ mol−1) reflect the combined values for quasi-equilibrated steps that desorb H*, adsorb C2H6, form C2-intermediates by dehydrogenation, and form the transition state from *CHCH* species. DFT-derived activation energies (218 kJ mol−1) and free energies estimated from these values and statistical mechanics treatments of reaction and activation entropies (137 kJ mol−1) are in excellent agreement with measured values. The removal of four H-atoms in forming the kinetically-relevant *CHCH* intermediates, taken together with measured effects of H2 pressure on hydrogenolysis rates, show that 2–3 H* must be removed to bind this intermediate and the transition state, as expected from the structure of the proposed adsorbed species and H* adsorption stoichiometries on Ir surface atoms that vary slightly with surface coordination on the non-uniform surfaces of metal clusters. Theory and experiments combine here to provide mechanistic insights inaccessible to direct observation and provide compelling evidence for reaction pathways long considered to be plausible for hydrogenolysis on noble metals. The extent of unsaturation in the single relevant intermediate and its C–C cleavage rates will depend on the identity of the metal, but the elementary steps and their kinetic relevance appear to be a general feature of metal-catalyzed hydrogenolysis.Download high-res image (252KB)Download full-size image
Co-reporter:Zhijie Wu, Sarika Goel, Minkee Choi, Enrique Iglesia
Journal of Catalysis (March 2014) Volume 311() pp:458-468
Publication Date(Web):1 March 2014
DOI:10.1016/j.jcat.2013.12.021
•A general strategy to encapsulate noble metal clusters within small-pore LTA zeolite.•Small and uniform metal clusters with good thermal stability against sintering.•Zeolite-encapsulated metals as shape-selective and sulfur-tolerant catalysts.Noble metal clusters (Pt, Pd, Rh, Ir, Re, and Ag) are selectively encapsulated within LTA voids via hydrothermal synthesis using metal precursors with ligands (NH3 for Pt and Ir; ethylenediamine for Pd, Rh, Re and Ag) that prevent their premature precipitation as colloidal oxyhydroxides. Such stability appears to be necessary and sufficient for successful encapsulation of cationic precursors during nucleation and growth of zeolite frameworks. Mean cluster diameters measured by titration of exposed metal atoms (H2 on Pt, Pd, Rh, Ir and Re; O2 on Ag; 1.1–1.8 nm) and by transmission electron microscopy (1.2–1.9 nm) were similar, indicating that cluster surfaces were clean and accessible to molecules used as titrants or reactants. Metal clusters were narrowly distributed in size and stable against sintering and coalescence during oxidative thermal treatments (573–873 K). Encapsulation selectivities were measured from turnover rates for reactions of small and large reactants, specifically hydrogenation of alkenes (ethene and isobutene) and oxidation of alkanols (methanol, ethanol, and isobutanol), which reflect the restricted access to encapsulated clusters by the larger molecules. These encapsulation selectivities, which reflect the ratio of metal surface areas within and outside LTA crystals ranged from 7.5 to 83 for all samples. Confinement within LTA crystals protects clusters from contact with thiophene and allows ethene hydrogenation to proceed at thiophene concentrations that fully suppressed reactivity for metal clusters dispersed on mesoporous SiO2. These protocols provide a general strategy for encapsulating clusters within small-pore zeolite voids, for which post-synthesis exchange is infeasible. Their successful encapsulation protects such clusters from coalescence and growth and allows them to select reactants and reject poisons based on their molecular size.Download high-res image (266KB)Download full-size image
Co-reporter:Shuai Wang, Enrique Iglesia
Journal of Catalysis (January 2017) Volume 345() pp:183-206
Publication Date(Web):1 January 2017
DOI:10.1016/j.jcat.2016.11.006
•Ketonization turnovers occur on TiO or ZrO pairs saturated with monodentate carboxylates.•Turnover rates benefit from intermediate strength of acid and base sites and distances that enforce orbital overlap.•Inactive bidentate carboxylates form slowly and cause site blocking.•Ketonization is limited by CC coupling of 1-hydroxy enolates with monodentate carboxylates.•The CC coupling transition state is preferentially stabilized by H-bonds at high coverages.Ketonization of carboxylic acids removes O-atoms and forms new CC bonds, thus providing routes from sustainable carbon feedstocks to fuels and chemicals. The elementary steps involved and their kinetic relevance, as well as the number and nature of the active sites on active TiO2 and ZrO2 catalysts, remain matters of active discourse. Here, site titrations demonstrate the requirement for coordinatively-unsaturated M-O-M sites (M = Ti, Zr) with specific geometry and intermediate acid-base strength. The measured site densities allow rigorous reactivity comparisons among catalysts based on turnover rates and activation free energies, as well as the benchmarking of mechanistic proposals against theoretical assessments. Kinetic, isotopic, spectroscopic, and theoretical methods show that C2C4 acids react on anatase TiO2 via kinetically-relevant CC coupling between 1-hydroxy enolate species and coadsorbed acids bound at vicinal acid-base pairs saturated with active monodentate carboxylates. Smaller TiTi distances on rutile TiO2 lead to the prevalence of unreactive bidentate carboxylates and lead to its much lower ketonization reactivity than anatase. The prevalent dense monolayers of chemisorbed acid reactants reflect their strong binding at acid-base pairs and their stabilization by H-bonding interactions with surface OH groups derived from the dissociation of the carboxylic acids or the formation of 1-hydroxy enolates; these interactions also stabilize CC coupling transition states preferentially over their carboxylate precursors; high coverages favor sequential dehydration routes of the α-hydroxy-γ-carboxy-alkoxide CC coupling products over previously unrecognized concerted six-membered-ring transition states. Infrared spectra show that ubiquitous deactivation, which has precluded broader deployment of ketonization in practice and unequivocal mechanistic inquiries, reflects the gradual formation of inactive bidentate carboxylates. Their dehydration to ketene-like gaseous species is faster on anatase TiO2 than on ZrO2 and allows the effective scavenging of bidentate carboxylates via ketene hydrogenation to alkanals/alkanols on a Cu function present within diffusion distances. These strategies make anatase TiO2, a more effective catalyst than ZrO2, in spite of its slightly lower initial turnover rates. This study provides details about the mechanism of ketonization of C2C4 carboxylic acids on TiO2 and a rigorous analysis of the sites required and of active and inactive bound species on TiO2 and ZrO2. The preference for specific distances and for intermediate acid-base strength in M-O-M species is consistent with the structure and energy of the proposed transition states and intermediates; their relative stabilities illustrate how densely-covered surfaces, prevalent during ketonization catalysis, represent an essential requirement for the achievement of practical turnover rates.Download high-res image (224KB)Download full-size image
Co-reporter:Manuel Ojeda, Bi-Zeng Zhan, Enrique Iglesia
Journal of Catalysis (January 2012) Volume 285(Issue 1) pp:92-102
Publication Date(Web):1 January 2012
DOI:10.1016/j.jcat.2011.09.015
Kinetic and isotopic data are used to interpret the mechanistic role of gaseous H2O molecules and of non-reducible (Al2O3) and reducible (TiO2, Fe2O3) supports on CO oxidation turnovers catalyzed by small Au clusters (<5 nm). H2O acts as a co-catalyst essential for O2 activation and for catalyst stability in CO oxidation at near-ambient temperatures, but also inhibits rates via competitive adsorption at higher H2O pressures. The effects of CO, O2, and H2O pressures on CO oxidation turnover rates, the absence of 16O2/18O2 and 16O2/H218O exchange, and the small H2O/D2O kinetic isotope effects are consistent with quasi-equilibrated molecular adsorption of CO, O2, and H2O on Au clusters with the kinetic relevance of H2O-mediated O2 activation via the formation of hydroperoxy intermediates (*OOH), which account for the remarkable reactivity and H2O effects on Au clusters. These elementary steps proceed on Au clusters without detectable requirements for support interface sites, which are no longer required when H2O is present and mediates O2 activation steps. Rate enhancements by H2O were also observed for CO oxidation on Pt clusters (1.3 nm), which is also limited by O2 activation steps, suggesting H2O-aided O2 activation and *OOH species in oxidations involving kinetically-relevant O2 activation. These intermediates have also been proposed to account for the ability of O2/H2O mixtures to act as reactants in alkene epoxidation on Au-based catalysts.Graphical abstractKinetic and isotopic data show that H2O acts as a co-catalyst for the activation of O2 and the retention of catalytic reactivity in CO oxidation on Au clusters. H2O mediates O2 activation steps, circumvents the kinetic bottlenecks prevalent under anhydrous conditions, and accounts for the remarkable reactivity of monofunctional Au metal clusters at near ambient temperatures.Download high-res image (107KB)Download full-size imageHighlights► H2O addition is essential in CO/O2 reactions to obtain high rates with Au clusters. ► CO oxidation on Au involves H2O-mediated O2 activation steps to form *OOH species. ► *OOH species are responsible for the high rates measured with CO/O2/H2O mixtures. ► Support identity is inconsequential for CO oxidation in the presence of H2O.
Co-reporter:Dante A. Simonetti, Robert T. Carr, Enrique Iglesia
Journal of Catalysis (January 2012) Volume 285(Issue 1) pp:19-30
Publication Date(Web):1 January 2012
DOI:10.1016/j.jcat.2011.09.007
Dimethyl ether (DME) homologation forms isobutane and triptane (2,2,3-trimethylbutane) with supra-equilibrium selectivities within C4 and C7 hydrocarbons on both mesoporous solid acids (SiO2–Al2O3, H3PW12O40/SiO2) and the acid forms of various zeolites (BEA, FAU, MFI) via methylation and hydride transfer steps that favor isobutane and triptane formation because of the relative stabilities of ion-pairs at transition states for chains along the preferred growth path. The stabilities of ion-pair transition states increase as acid sites become stronger and energies for charge separation decrease and as van der Waals interactions within pores become stronger, which respectively lead to higher rates on H3PW12O40/SiO2 and aluminosilicate zeolites than on amorphous SiO2–Al2O3. Solid acids with different strengths and abilities to solvate ion-pairs by confinement differ in selectivity because strength and solvation influence transition states for the hydride transfer, methylation, and isomerization steps to different extents. Stronger acid sites on H3PW12O40/SiO2 favor isomerization and hydride transfer over methylation; they lead to higher selectivities to n-butane and non-triptane C7 isomers than the weaker acid sites on BEA, FAU, and mesoporous SiO2–Al2O3. This preference for hydride transfer and isomerization on stronger acids reflects transition states with more diffuse cationic charge, which interact less effectively with conjugate anions than more localized cations at methylation transition states. The latter recover a larger fraction of the energy required to form the ion-pair, and their stabilities are less sensitive to acid strength than for diffuse cations. Large-pore zeolites (BEA, FAU) form triptane with higher selectivity than SiO2–Al2O3 because confinement within large pores preferentially solvates the larger transition states for hydride transfer and methylation, which preserve the four-carbon backbone in triptane, over smaller transition states for alkoxide isomerization steps, which disrupt this backbone and cause growth beyond C7 chains and subsequent facile β-scission to form isobutane. MFI forms isobutane and triptane with much lower selectivity than mesoporous acids or large-pore zeolites, because smaller pores restrict the formation of bimolecular methylation and hydride transfer transition states required for chain growth and termination steps to a greater extent than those for monomolecular alkoxide isomerization. These data and their mechanistic interpretations show that the selective formation of isobutane and triptane from C1 precursors like DME is favored on all acids as a result of the relative stability of methylation, hydride transfer, and isomerization transition states, but to a lesser extent when small confining voids and stronger acid sites preferentially stabilize monomolecular isomerization transition states. The observed effects of acid strength and confinement on rates and selectivities reflect the more effective stabilization of all ion-pairs on stronger acids and within solvating environments, but a preference for transition states with more diffuse charge on stronger acids and for ion-pairs with the appropriate solvation within voids of molecular dimensions.Graphical abstractFormation of isobutane and triptane from C1 precursors is favored on all solid acids, because methylation and hydride transfer steps selectively grow hydrocarbons without significant deviations via isomerization steps. Triptane and isobutane selectivities are maximized on BEA and FAU because weak acid sites and confinement within large channels preferentially stabilize hydride transfer and methylation transition states that preserve the structure of triptane in growing hydrocarbons over isomerization transition states.Download high-res image (65KB)Download full-size imageHighlights► Both mesoporous solid acids and zeolites selectively form triptane from C1 species. ► Solid acids with weak acid sites and large micropores maximize triptane formation. ► Stronger acids favor isomerization and hydride transfer over methylation. ► Large micropores selectively solvate methylation and hydride transfer transition states.
Co-reporter:Mónica García-Diéguez, Ya-Huei (Cathy) Chin, Enrique Iglesia
Journal of Catalysis (January 2012) Volume 285(Issue 1) pp:260-272
Publication Date(Web):1 January 2012
DOI:10.1016/j.jcat.2011.09.036
C2H6 reactions with O2 only form CO2 and H2O on dispersed Pt clusters at 0.2–28 O2/C2H6 reactant ratios and 723–913 K without detectable formation of partial oxidation products. Kinetic and isotopic data, measured under conditions of strict kinetic control, show that CH4 and C2H6 reactions involve similar elementary steps and kinetic regimes. These kinetic regimes exhibit different rate equations, kinetic isotope effects and structure sensitivity, and transitions among regimes are dictated by the prevalent coverages of chemisorbed oxygen (O*). At O2/C2H6 ratios that lead to O*-saturated surfaces, kinetically-relevant CH bond activation steps involve O*O* pairs and transition states with radical-like alkyls. As oxygen vacancies (∗) emerge with decreasing O2/alkane ratios, alkyl groups at transition states are effectively stabilized by vacancy sites and CH bond activation occurs preferentially at O** site pairs. Measured kinetic isotope effects and the catalytic consequences of Pt cluster size are consistent with a monotonic transition in the kinetically-relevant step from CH bond activation on O*O* site pairs, to CH bond activation on O** site pairs, to O2 dissociation on ** site pairs as O* coverage decrease for both C2H6 and CH4 reactants. When CH bond activation limits rates, turnover rates increase with increasing Pt cluster size for both alkanes because coordinatively unsaturated corner and edge atoms prevalent in small clusters lead to more strongly-bound and less-reactive O* species and lower densities of vacancy sites at nearly saturated cluster surfaces. In contrast, the highly exothermic and barrierless nature of O2 activation steps on uncovered clusters leads to similar turnover rates on Pt clusters with 1.8–8.5 nm diameter when this step becomes kinetically-relevant at low O2/alkane ratios. Turnover rates and the O2/alkane ratios required for transitions among kinetic regimes differ significantly between CH4 and C2H6 reactants, because of the different CH bond energies, strength of alkylO* interactions, and O2 consumption stoichiometries for these two molecules. Vacancies emerge at higher O2/alkane ratios for C2H6 than for CH4 reactants, because their weaker CH bonds lead to faster scavenging of O* and to lower O* coverages, which are set by the kinetic coupling between CH and OO activation steps. The elementary steps, kinetic regimes, and mechanistic analogies reported here for C2H6 and CH4 reactions with O2 are consistent with all rate and isotopic data, with their differences in CH bond energies and in alkyl binding, and with the catalytic consequences of surface coordination and cluster size. The rigorous mechanistic interpretation of these seemingly complex kinetic data and cluster size effects provides useful kinetic guidance for larger alkanes and other catalytic surfaces based on the thermodynamic properties of these molecules and on the effects of metal identity and surface coordination on oxygen binding and reactivity.Graphical abstractKinetic and isotopic data and Pt cluster size effects show that C2H6O2 and CH4O2 form CO2 and H2O via analogous elementary steps; turnover rates are higher for C2H6 in all kinetic regimes where CH bond cleavage limits rates because weaker CH bonds in C2H6 and stronger ethyl interactions with adsorbed oxygens (O*) at transition states lead to lower barriers for C2H6 than for CH4 activation. Reactivity differences cause transitions between kinetic regimes to occur at higher O2/alkane ratios for C2H6 because it scavenges O* species more effectively than CH4 and leads to lower O* coverages. These mechanistic analogies and insights can be rigorously extended to other alkanes and metal clusters.Download high-res image (129KB)Download full-size imageHighlights► C2H6O2 and CH4O2 form CO2 and H2O via similar elementary steps on Pt clusters. ► Elementary steps and rate equations depend on oxygen (O*) coverages. ► O* coverages are set by kinetic coupling between CH and OO activation steps. ► Rates reflect CH strength in alkanes and alkylO* interactions at transition states. ► Strongly bound O* at low-coordination surfaces are less reactive in CH activation.
Co-reporter:Stanley Herrmann, Enrique Iglesia
Journal of Catalysis (February 2017) Volume 346() pp:134-153
Publication Date(Web):1 February 2017
DOI:10.1016/j.jcat.2016.12.011
•H2 and Pt prevent deactivation and unselective β-scission routes.•Condensation rates are limited by CC formation at H+ saturated with H-bonded acetone.•Turnover rates are highest when voids and transition states optimize van der Waals contacts.•Lennard-Jones potentials give dispersion energies that accurately describe reactivity.The unselective nature and ubiquitous deactivation of Brønsted acids in aldol condensations have precluded their practical use and unequivocal mechanistic assessments. H2 and a Pt function are used here to confer stability by scavenging unsaturated intermediates to form stable products, thus allowing kinetic, spectroscopic, and isotopic assessments of elementary steps and their kinetic relevance, confirmed by density functional theory (DFT), for acetone condensation on microporous and mesoporous aluminosilicates (FER, TON, MFI, BEA, FAU, MCM-41). The selective titration of protons with 2,6-di-tert-butyl pyridine during catalysis shows that condensations occur exclusively on protons; the number of titrants required to suppress reactivity measures accessible sites and allows reactivity to be rigorously reported as turnover rates. Infrared spectra show that H-bonded acetone is present at saturation coverages during condensation catalysis. Taken together with rates that depend linearly on acetone pressure and with the absence of H/D kinetic isotope effects, these data indicate that condensation turnovers are mediated by the kinetically-relevant formation of a CC bond via reactions of H-bonded acetone with another acetone molecule, a conclusion confirmed by DFT-derived free energies along the reaction coordinate. Measured rate constants reflect free energy differences between this transition state and its relevant precursors, a H-bonded and a gaseous acetone. These rate constants (per H+) depend sensitively on size and shape of the confining voids among aluminosilicates of similar acid strength but diverse framework structure. Confinement effects are mediated by van der Waals contacts and are accurately described by energies derived from Lennard-Jones potentials of DFT-derived transition state structures; these energies are ensemble-averaged over all accessible configurations and T-site locations in each aluminosilicate framework. These energy descriptors replace incomplete metrics based solely on the sizes of voids and transition states, which fail to capture differences in reactivity among different confining frameworks.Download high-res image (83KB)Download full-size image
Co-reporter:Rajamani Gounder, Andrew J. Jones, Robert T. Carr, Enrique Iglesia
Journal of Catalysis (February 2012) Volume 286() pp:214-223
Publication Date(Web):1 February 2012
DOI:10.1016/j.jcat.2011.11.002
Kinetic, spectroscopic, and chemical titration data indicate that differences in monomolecular isobutane cracking and dehydrogenation and methanol dehydration turnover rates (per H+) among FAU zeolites treated thermally with steam (H-USY) and then chemically with ammonium hexafluorosilicate (CD-HUSY) predominantly reflect differences in the size and solvating properties of their supercage voids rather than differences in acid strength. The number of protons on a given sample was measured consistently by titrations with Na+, with CH3 groups via reactions of dimethyl ether, and with 2,6-di-tert-butylpyridine during methanol dehydration catalysis; these titration values were also supported by commensurate changes in acidic OH infrared band areas upon exposure to titrant molecules. The number of protons, taken as the average of the three titration methods, was significantly smaller than the number of framework Al atoms (Alf) obtained from X-ray diffraction and 27Al magic angle spinning nuclear magnetic resonance spectroscopy on H-USY (0.35 H+/Alf) and CD-HUSY (0.69 H+/Alf). These data demonstrate that the ubiquitous use of Alf sites as structural proxies for active H+ sites in zeolites can be imprecise, apparently because distorted Al structures that are not associated with acidic protons are sometimes detected as Alf sites. Monomolecular isobutane cracking and dehydrogenation rate constants, normalized non-rigorously by the number of Alf species, decreased with increasing Na+ content on both H-USY and CD-HUSY samples and became undetectable at sub-stoichiometric exchange levels (0.32 and 0.72 Na+/Alf ratios, respectively), an unexpected finding attributed incorrectly in previous studies to the presence of minority “super-acidic” sites. These rate constants, when normalized rigorously by the number of residual H+ sites were independent of Na+ content on both H-USY and CD-HUSY samples, reflecting the stoichiometric replacement of protons that are uniform in reactivity by Na+ cations. Monomolecular isobutane cracking and dehydrogenation rate constants (per H+; 763 K), however, were higher on H-USY than CD-HUSY (by a factor of 1.4). Equilibrium constants for the formation of protonated methanol dimers via adsorption of gaseous methanol onto adsorbed methanol monomers, determined from kinetic studies of methanol dehydration to dimethyl ether (433 K), were also higher on H-USY than CD-HUSY (by a factor of 2.1). These larger constants predominantly reflect stronger dispersive interactions in H-USY, consistent with its smaller supercage voids that result from the occlusion of void space by extraframework Al (Alex) residues. These findings appear to clarify enduring controversies about the mechanistic interpretation of the effects of Na+ and Alex species on the catalytic reactivity of FAU zeolites. They also illustrate the need to normalize rates by the number of active sites instead of more convenient but less accurate structural proxies for such sites.Graphical abstractDemystifying faujasite: Isobutane cracking (C) and dehydrogenation (D) rate constants (per framework Al; Alf), but not their ratio, decreased monotonically with Na+ content in ultrastable Y (USY) zeolite. Similar Na+ and 2,6-di-tert-butylpyridine titrant uptakes completely suppressed catalytic rates, indicating that there are fewer H+ sites than Alf atoms and that Na+ stoichiometrically replaces H+ sites that are uniform in reactivity and acid strength.Download high-res image (128KB)Download full-size imageHighlights► Protons are counted directly by titration with Na+, CH3 groups via dimethyl ether, and 2,6-di-tert-butylpyridine. ► There are fewer protons than framework aluminum atoms on FAU zeolites. ► Na+ stoichiometrically replaces H+ sites that are similar in reactivity and acid strength. ► Thermal treatments do not create Brønsted acid sites of increased strength. ► Thermal and chemical treatments influence supercage void size and solvation properties.
Co-reporter:Alexandra M. Landry, Enrique Iglesia
Journal of Catalysis (December 2016) Volume 344() pp:389-400
Publication Date(Web):1 December 2016
DOI:10.1016/j.jcat.2016.10.007
•Colloidal synthesis of PtPd catalysts uniform in composition and size.•PtPd clusters form via galvanic displacement-reduction processes.•Compare nanoparticle formation pathways for AuPt, PtPd and AuPd systems.•Compare synthesis strategies for AuPt, PtPd and AuPd systems.•Low-temperature reductive treatment cleans Pd and PtPd cluster surfaces.Bimetallic PtPd clusters (2.1–2.9 nm) dispersed on SiO2 and uniform in composition and size were prepared using colloidal methods with reagents containing only C, O, H, and N atoms. These synthetic protocols extend galvanic displacement-reduction (GDR) processes previously used to prepare AuPd and AuPt clusters. Such processes exploit the different redox potentials of two elements to encourage their deposition within the same cluster. The size, composition, and formation mechanism of PtPd clusters were probed using transmission electron microscopy, UV–visible spectroscopy, energy-dispersive X-ray spectroscopy, and high-angle annular dark-field imaging. Taken together with previous data for AuPd and AuPt systems, these findings highlight key general features, properties, and protocols required to form uniform bimetallic clusters. Exothermic alloys, such as PtPd and AuPd, form predominantly via selective GDR routes; in contrast, alternate routes become significant for endothermic alloys, such as AuPt. Bimetallic clusters grow via GDR processes (PtPd, AuPd) at rates proportional to the surface area of each cluster; therefore, compositional uniformity is dictated by the size distribution of the seed clusters. The rate of GDR processes reflects the difference in reduction potentials of the two components, as shown by more facile formation of AuPd than PtPd clusters. These considerations and experimental evidence provide useful guidance for conditions and protocols likely to succeed for other bimetallic pairs. Low-temperature (≤423 K) reductive treatments (in H2 or EtOH) successfully removed all synthetic detritus from Pd and PtPd clusters dispersed on SiO2, without significant coalescence. Such removal strategies are more challenging for Pd than for Pt clusters because of stronger Pd-polymer bonds and the greater sintering tendency of Pd clusters.Download high-res image (258KB)Download full-size image
Co-reporter:William Knaeble, Enrique Iglesia
Journal of Catalysis (December 2016) Volume 344() pp:817-830
Publication Date(Web):1 December 2016
DOI:10.1016/j.jcat.2016.08.007
•Exponential effects of deprotonation energy on intrinsic kinetic constants (per H+).•Kinetic constants for difficult and facile isomerization routes similarly affected by acid strength.•Ion-pair transition states mediate kinetically-relevant steps that form each isomer.•Diffusion-enhanced interconversions within acid domains influence measured selectivities.•Measured selectivities reflect characteristic diffusion and reaction times for each C5-ring isomer.Methylcyclohexane ring contraction is used here to assess the effects of acid strength and metal-acid site proximity on turnover rates and selectivities for bifunctional catalysts consisting of Keggin type polyoxometalates (POM) with different central atoms that act as Brønsted acids. Bifunctional catalysts with metal sites that fully equilibrate cycloalkanes and cycloalkenes give methylcyclohexene conversion rate constants that decreased exponentially with increasing deprotonation energy, a rigorous descriptor of acid strength, consistent with the ion-pair character of transition states that mediate the kinetically-relevant ring contraction of bound methylcyclohexoxide intermediates. The measured rates of formation of each alkylcyclopentane isomer, however, do not reflect their intrinsic formation kinetics, because of fast diffusion-enhanced interconversions within acid domains; thus, isomer selectivities cannot be used to infer, even indirectly, the strength of acid sites, as often proposed in previous studies. Selectivities reflect instead diffusional effects that become more severe as the number and strength of acid sites and the size and diffusive resistances increase within these acid domains, which shift, in turn, the products formed from relative abundances dictated by kinetics to those prescribed by thermodynamics. A rigorous accounting of these diffusional effects using a kinetic-transport model leads to ratios of intrinsic rate constants for the formation of the different alkylcyclopentane isomers that do not depend on acid strength, because all isomerization routes are mediated by bicyclo[3.1.0]hexyl cation transition states similar in the amount and location of charge and that therefore benefit to the same extent from the more stable conjugate anions characteristic of stronger acids.Download high-res image (209KB)Download full-size image
Co-reporter:Michele L. Sarazen, Eric Doskocil, Enrique Iglesia
Journal of Catalysis (December 2016) Volume 344() pp:553-569
Publication Date(Web):1 December 2016
DOI:10.1016/j.jcat.2016.10.010
•CC bond formation is mediated by bimolecular ion-pair transition states (TS).•Propene dimerization rate constants increase with decreasing zeolitic void size.•DFT dispersive energies show that the larger TS is stabilized over precursors.•Rate constants decrease exponentially with increasing deprotonation energies.•Al density does not influence the acid strength of MFI, TON and BEA samples.This study addresses fundamental descriptions of confinement and acid strength effects on stability for transition states and intermediates involved in alkene oligomerization on solid acids. Kinetic and infrared data and theoretical treatments that account for dispersive interactions show that turnover rates (per H+) on aluminosilicates and heterosilicates with microporous voids (TON, MFI, BEA, FAU) and on mesoporous acids (amorphous silica-alumina, dispersed polyoxometalates) reflect the free energy of CC bond formation transition states referenced to gaseous alkenes and bound alkene-derived precursors present at saturation coverages. These free energy barriers decrease as the size of confining voids decreases in aluminosilicates containing acid sites of similar acid strength and approaches bimolecular transition state (TS) sizes derived from density functional theory (DFT) for propene and isobutene reactants. Such TS structures are preferentially stabilized over smaller bound precursors via contacts with the confining framework. These effects of size, typically based on heuristic geometric analogies, are described here instead by the dispersive component of DFT-derived energies for TS and intermediates, which bring together the effects of size and the shape, for different framework voids and TS and precursor structures derived from alkenes of different size; these organic moieties differ in “fit” within voids but also in their proton affinity, as a result of the ion-pair character of TS structures. The larger charge in TS structures relative to their alkene-derived precursors causes free energy barriers to decrease as conjugate anions become more stable in stronger acids. Consequently, oligomerization rate constants decrease exponentially with increasing deprotonation energy on unconfined acid sites in polyoxometalates and silica-alumina and on confined sites within MFI frameworks with Al, Ga, Fe, or B heteroatoms. Reactivity descriptions based on geometry or acid strength are replaced by their more relevant energetic descriptors–van der Waals confinement energies, proton affinities of organic molecules, and deprotonation energies–to account for reactivity, here for different reactants on diverse solid acids, but in general for acid catalysis.Download high-res image (134KB)Download full-size image
Co-reporter:Andrew J. Jones, Robert T. Carr, Stacey I. Zones, Enrique Iglesia
Journal of Catalysis (April 2014) Volume 312() pp:58-68
Publication Date(Web):1 April 2014
DOI:10.1016/j.jcat.2014.01.007
•CH3OH dehydration rate constants are measured on MFI samples with Al, Ga, Fe and B heteroatoms.•Rate constants decrease exponentially with increasing deprotonation energies (DPE).•Polyoxometalates and MFI rate constants are influenced similarly by changes in DPE.•Al density does not influence the acid strength of Al-MFI samples.•High Al densities create H+ in the more confining channels of Al-MFI.The effects of heteroatom identity (Al3+, Ga3+, Fe3+, or B3+), concentration and location on catalysis by MFI zeolites are examined and interpreted mechanistically using methanol dehydration rate constants and density functional theory estimates of acid strength (deprotonation energies, DPE). In doing so, we shed light on the concomitant effects of confinement and acid strength on catalytic reactivity. Rate constants (per H+ from pyridine titrations during catalysis) in the first-order and zero-order kinetic regimes decreased exponentially as the DPE of MFI with different heteroatoms increased. These trends reflect a decrease in the stability of ion-pair transition states relative to the relevant precursors (H-bonded methanol and methanol dimers, respectively, for these two regimes) with decreasing acid strength and resemble those in mesoporous solid acids (e.g., polyoxometalates). Confinement effects, weaker in mesoporous solids, give larger rate constants on MFI than on POM clusters with similar DPE. Such reactivity enhancements reflect the effects of MFI voids that solvate transition states preferentially over smaller precursors via van der Waals interactions with the confining voids. Both dehydration rate constants on MFI with 0.7–2.4 H+ per unit cell volume (5.2 nm3) are independent of Al or H+ densities, indicating that neither H+ location nor acid strength depend on acid site concentration. Higher site densities (3.6 H+ per unit cell) lead to larger first-order rate constants, but do not influence their zero-order analogs. These data reflect, and in turn provide evidence for, the initial siting of H+ in less constrained channel intersections and their ultimate placement within the more solvating environments of the channels themselves. Thus, the higher reactivity of Al-rich samples, often attributed to the stronger acid sites, arises instead from the ubiquitous role of zeolites as inorganic solvents for the relevant transition state, taken together with H+ siting that depends on Al density. We find that heteroatom composition, but not Al density, influences acid strength in MFI, consistent with experiment and theoretical estimates of DPE, and that methanol dehydration rate constants, properly interpreted, provide relevant insights into the combined effects of acid strength and confinement on the reactivity of solid Brønsted acids.Graphical abstractDownload high-res image (167KB)Download full-size image
Co-reporter:Ya-Huei (Cathy) Chin, Corneliu Buda, Matthew Neurock, Enrique Iglesia
Journal of Catalysis (6 October 2011) Volume 283(Issue 1) pp:10-24
Publication Date(Web):6 October 2011
DOI:10.1016/j.jcat.2011.06.011
Rate measurements, density functional theory (DFT) within the framework of transition state theory, and ensemble-averaging methods are used to probe oxygen selectivities, defined as the reaction probability ratios for O* reactions with CO and CH4, during CH4–O2 catalysis on Pt and Rh clusters. CO2 and H2O are the predominant products, but small amounts of CO form as chemisorbed oxygen atoms (O*) are depleted from cluster surfaces. Oxygen selectivities, measured using 12CO–13CH4–O2 reactants, increase with O2/CO ratio and O* coverage and are much larger than unity at all conditions on Pt clusters. These results suggest that O* reacts much faster with CO than with CH4, causing any CO that forms and desorbs from metal cluster surfaces to react along the reactor bed with other O* to produce CO2 at any residence time required for detectable extents of CH4 conversion. O* selectivities were also calculated by averaging DFT-derived activation barriers for CO and CH4 oxidation reactions over all distinct surface sites on cubo-octahedral Pt clusters (1.8 nm diameter, 201 Pt atoms) at low O* coverages, which are prevalent at low O2 pressures during catalysis. CO oxidation involves non-activated molecular CO adsorption as the kinetically relevant step on exposed Pt atoms vicinal of chemisorbed O* atoms (on *–O* site pairs). CH4 oxidation occurs via kinetically relevant C–H bond activation on *–* site pairs involving oxidative insertion of a Pt atom into one of the C–H bonds in CH4, forming a three-centered HC3–Pt–H transition state. C–H bond activation barriers reflect the strength of Pt–CH3 and Pt–H interactions at the transition state, which correlates, in turn, with the Pt coordination and with CH3* binding energies. Ensemble-averaged O* selectivities increase linearly with O2/CO ratios, which define the O* coverages, via a proportionality constant. The proportionality constant is given by the ratio of rate constants for O2 dissociation and C–H bond activation elementary steps; the values for this constant are much larger than unity and are higher on larger Pt clusters (1.8–33 nm) at all temperatures (573–1273 K) relevant for CH4–O2 reactions. The barriers for the kinetically relevant C–H bond dissociation step increase, while those for CO oxidation remain unchanged as the Pt coordination number and cluster size increase, and lead, in turn, to higher O* selectivities on larger Pt clusters. Oxygen selectivities were much larger on Rh than Pt, because the limiting reactants for CO oxidation were completely consumed in 12CO–13CH4–O2 mixtures, consistent with lower CO/CO2 ratios measured by varying the residence time and O2/CH4 ratio independently in CH4–O2 reactions. These mechanistic assessments and theoretical treatments for O* selectivity provide rigorous evidence of low intrinsic limits of the maximum CO yields, thus confirming that direct catalytic partial oxidation of CH4 to CO (and H2) does not occur at the molecular scale on Pt and Rh clusters. CO (and H2) are predominantly formed upon complete O2 depletion from the sequential reforming steps.Graphical abstractChemisorbed oxygen (O*) preferentially reacts with CO instead of CH4 during CH4–O2 catalysis on Pt and Rh clusters. High O* selectivities (SO*) for CO reactions with O* lead to very low CO yields when O2 is present, indicating that direct catalytic partial oxidation to form CO (and H2) from CH4–O2 reactants is unlikely to occur at the molecular scale or at CH4 conversions of practical interest. Download high-res image (145KB)Download full-size imageHighlights► Chemisorbed oxygen selectively reacts with CO instead of CH4 on Pt and Rh clusters. ► O2 selectivity is described rigorously in terms of elementary step rate constants. ► Experiments and theory (DFT/TST) show much larger O* reactivity with CO than CH4. ► Preferential oxidation of CO limits the maximum CO yields from CH4–O2 reactions. ► Direct CH4 partial oxidation is unlikely to occur at practical CH4 conversions.
Co-reporter:Rajamani Gounder, Enrique Iglesia
Journal of Catalysis (3 January 2011) Volume 277(Issue 1) pp:36-45
Publication Date(Web):3 January 2011
DOI:10.1016/j.jcat.2010.10.013
Brønsted acid sites in zeolites (H-FER, H-MFI, H-MOR) selectively hydrogenate alkenes in excess H2 at high temperatures (>700 K) and at rates proportional to alkene and H2 pressures. This kinetic behavior and the De Donder equations for non-equilibrium thermodynamics show that, even away from equilibrium, alkene hydrogenation and monomolecular alkane dehydrogenation occur on predominantly uncovered surfaces via microscopically reverse elementary steps, which involve kinetically-relevant (C–H–H)+ carbonium-ion-like transition states in both directions. As a result, rate constants, activation energies and activation entropies for these two reactions are related by the thermodynamics of the overall stoichiometric gas-phase reaction. The ratios of rate constants for hydrogenation and dehydrogenation reactions do not depend on the identity or reactivity of active sites; thus, sites within different zeolite structures (or at different locations within a given zeolite) that favor alkane dehydrogenation reactions, because of their ability to stabilize the required transition states, also favor alkene hydrogenation reactions to the exact same extent. These concepts and conclusions also apply to monomolecular alkane cracking and bimolecular alkane–alkene reaction paths on Brønsted acids and, more generally, to any forward and reverse reactions that proceed via the same kinetically-relevant step on vacant surfaces in the two directions, even away from equilibrium. The evidence shown here for the sole involvement of Brønsted acids in the hydrogenation of alkoxides with H2 is unprecedented in its mechanistic clarity and thermodynamic rigor. The scavenging of alkoxides via direct H-transfer from H2 indicates that H2 can be used to control the growth of chains and the formation of unreactive deposits in alkylation, oligomerization, cracking and other acid-catalyzed reactions.Alkene Hydrogenation on Acidic Zeolites: Brønsted acid sites in FER, MFI, and MOR zeolites catalyze monomolecular alkane dehydrogenation and its microscopic reverse, alkene hydrogenation with H2, via the same kinetically-relevant (C–H–H)+ carbonium-ion-like transition states, even far from equilibrium. The measured rate constants, activation energies and activation entropies for dehydrogenation and hydrogenation are related by the equilibrium constant and the enthalpy and entropy for the stoichiometric gas-phase reaction, which are independent of the reactivity and structure of the active sites.Download high-res image (83KB)Download full-size image
Co-reporter:Dante A. Simonetti, John H. Ahn, Enrique Iglesia
Journal of Catalysis (24 January 2011) Volume 277(Issue 2) pp:173-195
Publication Date(Web):24 January 2011
DOI:10.1016/j.jcat.2010.11.004
We report here kinetic and isotopic evidence for the elementary steps involved in dimethyl ether (DME) homologation and for their role in the preferential synthesis of 2,2,3-trimethylbutane (triptane) and isobutane. Rates of methylation of alkenes and of hydrogen transfer, isomerization and β-scission reactions of the corresponding alkoxides formed along the homologation path to triptane were measured using mixtures of 13C-labeled dimethyl ether (13C-DME) and unlabeled alkenes on H-BEA. DME-derived C1 species react with these alkenes to form linear butyls from propene, isopentyls from n-butenes, 2,3-dimethylbutyls from isopentenes, and triptyls from 2,3-dimethylbutenes; these kinetic preferences reflect the selective formation of the more highly substituted carbenium ions and the retention of a four-carbon backbone along the path to triptane. Hydrogen transfer reactions terminate chains as alkanes; chain termination probabilities are low for species along the preferred methylation path, but reach a maximum at triptyl species, because tertiary carbenium ions involved in hydrogen transfer are much more stable than those with primary character required for triptene methylation. Alkenes and alkanes act as hydrogen donors and form unsaturated species as precursors to hexamethylbenzene, which forms to provide the hydrogen required for the DME-to-alkanes stoichiometry. Weak allylic C–H bonds in isoalkenes are particularly effective hydrogen donors, as shown by the higher termination probabilities and 12C content in hexamethylbenzene as 12C-2-methyl-2-butene and 12C-2,3-dimethyl-2-butene pressures increased in mixtures with 13C-DME. The resulting dienes and trienes can then undergo Diels–Alder cyclizations to form arenes as stable by-products. Isomerization and β-scission reactions of the alkoxides preferentially formed in methylation of alkenes are much slower than hydrogen transfer or methylation rates, thus preventing molecular disruptions along the path to triptane. Methylation at less preferred positions leads to species with lower termination probabilities, which tend to grow to C8+ molecules; these larger alkoxides undergo facile β-scission to form tert-butoxides that desorb preferentially as isobutane via hydrogen transfer; such pathways resolve methylation “missteps” by recycling the carbon atoms in such chains to the early stages of the homologation chain and account for the prevalence of isobutane among DME homologation products. These findings were motivated by an inquiry into the products formed via C1 homologation, but they provide rigorous insights about how the structure and stability of carbenium ions specifically influence the rates of methylation, hydrogen transfer, β-scission, and isomerization reactions catalyzed by solid acids.Graphical abstractThe selective homologation of C1 species to isobutane and 2,2,3-trimethylbutane on solid acids reflects the relative stability of the carbenium ions involved in methylation and hydrogen transfer, the formation of isobutane via facile β-scission of chains larger than C7, and the substantial absence of isomerization or cracking for smaller chains. These reaction rates and chain termination probabilities were rigorously measured using mixtures of 13C-dimethyl ether and 12C-alkenes on H-BEA; their mechanistic interpretations are broadly applicable to chain growth and rearrangements of alkenes and alkoxides via carbenium ion transition states.Download high-res image (84KB)Download full-size imageResearch highlights► Acid-catalyzed C1 homologation selectively forms isobutane and triptane. ► Carbenium ion structure and stability dictate C1 homologation chain growth paths. ► C1 homologation mechanism provides rigorous, general insights into acid catalysis.
Co-reporter:Aritomo Yamaguchi, Enrique Iglesia
Journal of Catalysis (19 August 2010) Volume 274(Issue 1) pp:52-63
Publication Date(Web):19 August 2010
DOI:10.1016/j.jcat.2010.06.001
The effects of reactant and product concentrations on turnover rates and isotopic tracing and kinetic isotope effects have led to a sequence of elementary steps for CH4 reactions with CO2 and H2O on supported Pd catalysts. Rate constants for kinetically-relevant C–H bond activation steps are much larger on Pd than on other metals (Ni, Ru, Rh, Ir, Pt). As a result, these steps become reversible during catalysis, because the products of CH4 dissociation rapidly deplete the required oxygen co-reactant formed from CO2 or H2O and co-reactant activation, and water–gas shift reactions remain irreversible in the time scale required for CH4 conversion. H2 and CO products inhibit CH4 reactions via their respective effects on CH4 and CO dissociation steps. These mechanistic conclusions are consistent with the kinetic effects of reactants and products on turnover rates, with the similar and normal CH4/CD4 kinetic isotope effects measured with H2O and CO2 co-reactants, with the absence of H2O/D2O isotope effects, and with the rate of isotopic scrambling between CH4 and CD4, 12C16O and 13C18O, and 13CO and 12CO during CH4 reforming catalysis. This catalytic sequence, but not the reversibility of its elementary steps, is identical to that reported on other Group VIII metals. Turnover rates are similar on Pd clusters on various supports (Al2O3, ZrO2, ZrO2−La2O3) and independent of Pd dispersion over the narrow range accessible at reforming conditions, because kinetically-relevant C–H bond activation steps occur predominantly on Pd surfaces. ZrO2 and ZrO2−La2O3 supports, with detectable reactivity for CO2 and H2O activation, can reverse the infrequent formation of carbon overlayers and inhibit deactivation, but do not contribute to steady-state catalytic reforming rates. The high reactivity of Pd surfaces in C–H bond activation reflects their strong binding for C and H and the concomitant stabilization of the transition state for kinetically-relevant C–H activation steps and causes the observed kinetic inhibition by chemisorbed carbon species formed in CH4 and CO dissociation steps.Pd cluster surfaces show much higher reactivity and rate constants for C–H bond activation than other Group VIII metals, irrespective of support or metal cluster size. This high reactivity leads to reversible C–H and C–O dissociation steps and to concomitant inhibition effects of H2 and CO on CH4 reactions with H2O and CO2.Download high-res image (51KB)Download full-size image
Co-reporter:Akio Ishikawa, Enrique Iglesia
Journal of Catalysis (15 November 2007) Volume 252(Issue 1) pp:49-56
Publication Date(Web):15 November 2007
DOI:10.1016/j.jcat.2007.08.012
Clusters of Pt and Pd catalyze dimethyl ether (DME) combustion to CO2 and H2O at 400–600 K without detectable formation of byproducts. Rh clusters are less active and also form CO, HCHO, and CH3OH in this temperature range. On Pd, isotopic and kinetic studies have shown that DMEO2 reactions proceed via redox cycles limited by hydrogen abstraction from chemisorbed DME molecules without the involvement of methoxide intermediates in kinetically relevant steps, as shown previously on Pt clusters. Kinetic inhibition by H2O is much stronger on Pd than on Pt clusters, apparently because of weaker binding of chemisorbed oxygen (O*) and hydroxyl (OH*) groups on Pt than on Pd. H2O titrates vacancies (*) required to chemisorb DME molecules involved in kinetically relevant H-abstraction steps. DME combustion turnover rates (per exposed metal atom) on Pt, Pd, and Rh increased with increasing cluster size, but were not affected by the identity of the support (Al2O3, ZrO2). These size effects reflect the stronger binding of O* and OH* on smaller clusters, which contain surface atoms with fewer neighbors and greater coordinative unsaturation. The higher reactivity of Pt compared with Pd and Rh also reflects the weaker binding of O* on Pt surfaces and the higher density of vacancies and of DME intermediates interacting with such vacancies. These trends resemble those reported for CH4O2 on Pt and Pd clusters. They represent a general feature of reactions that require vacancies and the abstraction of H-atoms by basic oxygens on surfaces covered predominately by O* or OH* during catalysis.
Co-reporter:Josef Macht, Robert T. Carr, Enrique Iglesia
Journal of Catalysis (15 May 2009) Volume 264(Issue 1) pp:54-66
Publication Date(Web):15 May 2009
DOI:10.1016/j.jcat.2009.03.005
We describe here a rigorous method to estimate the deprotonation energy (DPE) and acid strength for solid Brønsted acids with uncertain structure using rate constants for reactions involving cationic transition states. The approach exploits relations between turnover rates for dehydration and isomerization reactions and DPE values on Keggin polyoxometalates and H-BEA solids with known structures. These relations are used to estimate the strength of acid sites in SO4–ZrO2(SZr), WOx–ZrO2(WZr), and perfluorosulfonic resins (SAR) from their alkanol dehydration and alkane isomerization rate constants. Alkanol dehydration and alkane isomerization proceed via pathways independent of acid identity and are limited by steps involving late transition states. Turnover rates (per accessible acid sites measured by titration during catalysis) are related to the relevant rate constants and are used to estimate DPE values for SZr, WZr, and SAR. Isomerization data estimate DPE values of 1110 kJ mol−1 and 1120 kJ mol−1 for SZr and WZr, respectively, while dehydration rate data lead to slightly higher values (1165 kJ mol−1 and 1185 kJ mol−1). The DPE value for SAR was 1154 kJ mol−1 from dehydration reactions, but diffusional constraints during reactions of non-polar alkanes precluded isomerization rate measurements. SZr and SAR contain stronger acid sites than zeolites (1185 kJ mol−1), but weaker than those in H3PW12O40 and H4SiW12O40 (1087 kJ mol−1 and 1105 kJ mol−1). Acid sites present in WZr during alkane isomerization are stronger than those present in zeolites, but these become similar in strength in the polar environment prevalent during dehydration catalysis. These effects of reaction media (and treatment protocols) reflect differences in the extent of dehydroxylation of catalytic surfaces. OH groups remaining after dehydroxylation are stronger acid sites because of a concomitant decrease in electron density in the conjugate anion and the formation of Brønsted–Lewis acid conjugate pairs. The method proposed and used here probes acid strength (as DPE) on sites of uncertain structure and within solvating media inherent in their use as catalysts. It can be used for any Brønsted acid or reaction, but requires reactivity-DPE relations for acids of known structure, the mechanistic interpretations of rates, and the measurement of accessible protons during catalysis. The resulting DPE values provide a rigorous benchmark for the structural fidelity of sites proposed for acids with uncertain structure, a method to assess the consequences of the dynamic nature of active sites in acid catalysis, and a connection between theory and experiment previously unavailable.The acid strength of solid acids with uncertain structure was determined from elimination and isomerization rate constants using their sensitivity to deprotonation energies, established independently for catalysts with known structures.Download high-res image (149KB)Download full-size image
Co-reporter:Manuel Ojeda, Rahul Nabar, Anand U. Nilekar, Akio Ishikawa, Manos Mavrikakis, Enrique Iglesia
Journal of Catalysis (15 June 2010) Volume 272(Issue 2) pp:287-297
Publication Date(Web):15 June 2010
DOI:10.1016/j.jcat.2010.04.012
Unresolved mechanistic details of monomer formation in Fischer–Tropsch synthesis (FTS) and of its oxygen rejection routes are addressed here by combining kinetic and theoretical analyses of elementary steps on representative Fe and Co surfaces saturated with chemisorbed CO. These studies provide experimental and theoretical evidence for hydrogen-assisted CO activation as the predominant kinetically-relevant step on Fe and Co catalysts at conditions typical of FTS practice. H2 and CO kinetic effects on FTS rates and oxygen rejection selectivity (as H2O or CO2) and density functional theory estimates of activation barriers and binding energies are consistent with H-assisted CO dissociation, but not with the previously accepted kinetic relevance of direct CO dissociation and chemisorbed carbon hydrogenation elementary steps. H-assisted CO dissociation removes O-atoms as H2O, while direct dissociation forms chemisorbed oxygen atoms that desorb as CO2. Direct CO dissociation routes are minor contributors to monomer formation on Fe and may become favored at high temperatures on alkali-promoted catalysts, but not on Co catalysts, which remove oxygen predominantly as H2O because of the preponderance of H-assisted CO dissociation routes. The merging of experiment and theory led to the clarification of persistent mechanistic issues previously unresolved by separate experimental and theoretical inquiries.CO activation occurs predominantly by reaction with chemisorbed hydrogen before C–O bond cleavage with preferential rejection of oxygen as water on Fe and Co catalysts at conditions relevant to Fischer–Tropsch synthesis practice.Download high-res image (99KB)Download full-size image
Co-reporter:Robert T. Carr, Matthew Neurock, Enrique Iglesia
Journal of Catalysis (14 February 2011) Volume 278(Issue 1) pp:78-93
Publication Date(Web):14 February 2011
DOI:10.1016/j.jcat.2010.11.017
The effects of acid identity on CH3OH dehydration are examined here using density functional theory (DFT) estimates of acid strength (as deprotonation energies, DPE) and reaction energies, combined with rate data on Keggin polyoxometalate (POM) clusters and zeolite H-BEA. Measured first-order (kmono) and zero-order (kdimer) CH3OH dehydration rate constants depend exponentially on DPE for POM clusters; the value of kmono depends more strongly on DPE than kdimer does. The chemical significance of these rate parameters and the basis for their dependences on acid strength were established by using DFT to estimate the energies of intermediates and transition states involved in elementary steps that are consistent with measured rate equations. We conclude from this treatment that CH3OH dehydration proceeds via direct reactions of co-adsorbed CH3OH molecules for relevant solid acids and reaction conditions. Methyl cations formed at ion-pair transition states in these direct routes are solvated by H2O and CH3OH more effectively than those in alternate sequential routes involving methoxide formation and subsequent reaction with CH3OH. The stability of ion-pairs, prevalent as intermediates and transition states on solid acids, depends sensitively on DPE because of concomitant correlations between the stability of the conjugate anionic cluster and DPE. The chemical interpretation of kmono and kdimer from mechanism-based rate equations, together with thermochemical cycles of their respective transition state formations, show that similar charge distributions in the intermediate and transition state involved in kdimer cause its weaker dependence on DPE. Values of kmono involve uncharged reactants and the same ion-pair transition state as kdimer; these species sense acid strength differently and cause the larger effects of DPE on kmono. Confinement effects in H-BEA affect the value of kmono because the different sizes and number of molecules in reactants and transition states selectively stabilize the latter; however, they do not influence kdimer, for which reactants and transition states of similar size sense spatial constraints to the same extent. This combination of theory and experiment for solid acids of known structure sheds considerable light on the relative contributions from solvation, electrostatic and van der Waals interactions in stabilizing cationic transition states and provides predictive insights into the relative contributions of parallel routes based on the size and charge distributions of their relevant intermediates and transition states. These findings also demonstrate how the consequences of acid strength on measured turnover rates depend on reaction conditions and their concomitant changes in the chemical significance of the rate parameters measured. Moreover, the complementary use of experiment and theory in resolving mechanistic controversies has given predictive guidance about how rate and equilibrium constants, often inextricably combined as measured rate parameters, individually depend on acid strength based on the magnitude and spatial distributions of charges in reactants, products and transition states involved in relevant elementary steps. The unique relations between kmono, kdimer and DPE developed here for CH3OH dehydration can be applied in practice to assess the acid strength of any solid acid, many of which have unknown structures, preventing reliable calculations of their DPE by theory.Graphical abstractDimethyl ether synthesis on solid acids occurs via direct reactions of two co-adsorbed CH3OH molecules because methyl cations at transition states are effectively solvated by H2O and CH3OH molecules. On acids, activation barriers and their sensitivity to acid strength depend on differences in charge distribution and density in adsorbed reactants and their respective transition states.Download high-res image (98KB)Download full-size imageResearch highlights► CH3OH conversion to dimethyl ether on polyoxometalate and zeolite solid acids. ► Mechanism discrimination by combined kinetic and density functional theory analyses. ► Reaction sensitivity to acid strength determination by structure–function relations.
Co-reporter:Xuebing Li, Enrique Iglesia
Applied Catalysis A: General (1 January 2008) Volume 334(Issues 1–2) pp:339-347
Publication Date(Web):1 January 2008
DOI:10.1016/j.apcata.2007.10.021
Co-reporter:Aditya Bhan, Rajamani Gounder, Josef Macht, Enrique Iglesia
Journal of Catalysis (1 January 2008) Volume 253(Issue 1) pp:221-224
Publication Date(Web):1 January 2008
DOI:10.1016/j.jcat.2007.11.003
Compensation between adsorption entropies and enthalpies results in less than a two-fold variation in adsorption equilibrium constants for C3–C6 alkanes at temperatures relevant for monomolecular cracking; the size-independent activation energy for CC bond activation in C3–C6 alkanes indicates that the marked increase in monomolecular cracking turnover rates observed with alkane chain size reflects a concurrent increase in activation entropies. Thermodynamic treatments for non-ideal systems rigorously describe confinement effects within zeolite channels and show that pre-exponential factors depend on solvation effects of the zeolite-host environment through variations in the thermodynamic activity of the zeolitic proton. Observed differences in rates and selectivities of monomolecular alkane activation with zeolite structure, after normalization to intrazeolitic concentrations, reflect differences in intrinsic rate constants.
Co-reporter:Xuebing Li, Enrique Iglesia
Journal of Catalysis (1 April 2008) Volume 255(Issue 1) pp:134-137
Publication Date(Web):1 April 2008
DOI:10.1016/j.jcat.2008.01.021
An equilibrated mixture of pentene isomers was produced by dehydroisomerization of n-pentane on catalysts consisting of Pt clusters within [Fe]ZSM-5 channels. These catalysts showed high isomerization rates, excellent stability even without added H2, and isopentene selectivities above 60%. Metal sites on Pt clusters dehydrogenate n-alkanes and n-alkenes formed undergo skeletal rearrangements with high selectivity on weak acid sites prevalent in Na-[Fe]ZSM-5 after reduction of exchanged Pt cations. Zeolite channels inhibit side reactions and sintering processes that cause rapid deactivation on unprotected Pt clusters.
Co-reporter:Janine Lichtenberger, Doohwan Lee and Enrique Iglesia
Physical Chemistry Chemical Physics 2007 - vol. 9(Issue 35) pp:NaN4906-4906
Publication Date(Web):2007/07/27
DOI:10.1039/B707465D
Supported Pd clusters catalyze methanol oxidation to methyl formate with high turnover rates and >90% selectivity at near ambient temperatures (313 K). Metal clusters are much more reactive than PdO clusters and rates are inhibited by the reactant O2. These data suggest that ensembles of Pd metal atoms on surfaces nearly saturated with chemisorbed oxygen are required for kinetically-relevant C–H bond activation in chemisorbed methoxide intermediates. Pd metal surfaces become more reactive with increasing metal particle size. The higher coordination of surface atoms on larger clusters leads to more weakly-bound chemisorbed species and to a larger number of Pd metal ensembles available during steady-state catalysis. Chemisorbed oxygen removes H-atoms formed in C–H bond activation steps and inhibits methoxide decomposition and CO2 formation, two functions essential for the high turnover rates and methyl formate selectivities reported here.
Co-reporter:Tatiana Luts, Enrique Iglesia and Alexander Katz
Journal of Materials Chemistry A 2011 - vol. 21(Issue 4) pp:NaN990-990
Publication Date(Web):2010/11/24
DOI:10.1039/C0JM02826F
The design of new materials for gaseous NOx (NO and NO2) removal at ambient temperature using organic active sites is reported. The materials consist of unfunctionalized silica and silica modified by immobilized aminoxyls and function via sequential processes consisting of (i) NO oxidation to NO2 and (ii) NO2 storage. NOx removal by physical mixtures of immobilized PTIO (2-phenyl-4,4,5,5-tetramethyl-imidazoline-3-oxide-1-oxyl) sites on silica as the NO oxidant and hydrated silica as the NO2 trap occurs with significant degradation of the PTIO oxidant via undesired side reactions with NO2 when NO2 adsorption sites are fewer than required for its complete removal along the packed bed. The use of packed beds with sufficient NO2 adsorption sites requires a large excess of unfunctionalized silica, because of its low surface density of geminal silanols, which are shown to be the relevant sites for NO2 storage on silica at ambient temperature based on density functional theory calculations. This PTIO degradation is circumvented by the design of NOx traps consisting of immobilized PTIO on silica as the NO oxidant and immobilized TEMPO (2,2,6,6-tetramethylpiperidine-1-oxyl) on silica as an adsorbent with a high density of strong NO2 binding sites. Packed beds consisting of a 2:1 molar mixture of PTIO and TEMPO sites consume NOx as predicted by stoichiometry without detectable PTIO degradation and also without a contribution from geminal silanols as NO2 storage sites. This result requires that PTIO and TEMPO sites on silica render geminal silanols as inactive towards NO2 storage presumably because of the titration of these silanols by immobilized aminoxyls.
Co-reporter:Akio Ishikawa and Enrique Iglesia
Chemical Communications 2007(Issue 28) pp:NaN2993-2993
Publication Date(Web):2007/05/11
DOI:10.1039/B702693E
Mixtures of Pt clusters dispersed on γ-Al2O3 and additional γ-Al2O3 led to much higher DME combustion turnover rates than on the individual components or on Pt clusters supported on non-acidic oxides.
Co-reporter:Xuebing Li and Enrique Iglesia
Chemical Communications 2008(Issue 5) pp:NaN596-596
Publication Date(Web):2007/11/27
DOI:10.1039/B715543C
Pt clusters within [Fe]ZSM-5 channels provide active and stable sites for the selective catalytic dehydrogenation of n-alkanes to n-alkenes. Cs and Na cations titrate acid sites and inhibit skeletal isomerization and cracking side reactions.
Co-reporter:Manuel Ojeda and Enrique Iglesia
Chemical Communications 2009(Issue 3) pp:NaN354-354
Publication Date(Web):2008/12/03
DOI:10.1039/B813589D
Au/TiO2catalysts form hydroperoxy species from H2O–O2 mixtures at near-ambient temperatures; these species can be used in the selective epoxidation of propene to propylene oxide.
Co-reporter:Rajamani Gounder and Enrique Iglesia
Chemical Communications 2013 - vol. 49(Issue 34) pp:NaN3509-3509
Publication Date(Web):2013/03/04
DOI:10.1039/C3CC40731D
The ability of molecular sieves to control the access and egress of certain reactants and products and to preferentially contain certain transition states while excluding others based on size were captured as shape selectivity concepts early in the history of zeolite catalysis. The marked consequences for reactivity and selectivity, specifically in acid catalysis, have since inspired and sustained many discoveries of novel silicate frameworks and driven the engineering of hierarchical structures and void size to influence catalysis. The catalytic diversity of microporous voids is explored and extended here in the context of their solvating environments, wherein voids act as hosts and stabilize guests, whether reactive intermediates or transition states, by van der Waals forces. We use specific examples from acid catalysis, including activation of C–C and C–H bonds in alkanes, alkylation and hydrogenation of alkenes, carbonylation of dimethyl ether, and elimination and homologation reactions of alkanols and ethers, which involve transition states and adsorbed precursors of varying size and composition. Mechanistic interpretations of measured turnover rates enable us to assign precise chemical origins to kinetic and thermodynamic constants in rate equations and, in turn, to identify specific steps and intermediates that determine the free energy differences responsible for chemical reactivity and selectivity. These free energy differences reflect the stabilization of transition states and their relevant precursors via electrostatic interactions that depend on acid strength and van der Waals interactions that depend on confinement within voids. Their respective contributions to activation free energies are examined by Born–Haber thermochemical cycles by considering plausible transition states and the relevant precursors. These examples show that zeolite voids solvate transition states and precursors differently, and markedly so for guest moieties of different size and chemical composition, thus enabling voids of a given size and shape to provide the “right fit” for a given elementary step, defined as that which minimizes Gibbs free energies of activation. Tighter confinement is preferred at low temperatures because enthalpic gains prevail over concomitant entropic losses, while looser fits are favored at high temperatures because entropy gains offset losses in enthalpic stabilization. Confinement and solvation by van der Waals forces are not directly involved in the making or breaking of strong chemical bonds; yet, they confer remarkable diversity to zeolites, in spite of their structural rigidity and their common aluminosilicate composition. A single zeolite can itself contain a range of local void environments, each with distinct reactivity and selectivity; as a result, varying the distribution of protons among these locations within a given framework or modifying a given location by partial occlusion of the void space can extend the range of catalytic opportunities for zeolites. Taken together with theoretical tools that accurately describe van der Waals interactions between zeolite voids and confined guests and with synthetic protocols that place protons or space-filling moieties at specific locations, these concepts promise to broaden the significant impact and catalytic diversity already shown by microporous solids.
Co-reporter:Josef Macht and Enrique Iglesia
Physical Chemistry Chemical Physics 2008 - vol. 10(Issue 35) pp:NaN5343-5343
Publication Date(Web):2008/07/09
DOI:10.1039/B805251D
Redox and acid–base properties of dispersed oxide nanostructures change markedly as their local structure and electronic properties vary with domain size. These changes give rise to catalytic behavior, site structures, and reaction chemistries often unavailable on bulk crystalline oxides. Turnover rates for redox and acid catalysis vary as oxide domains evolve from isolated monomers to two-dimensional oligomers, and ultimately into clusters with bulk-like properties. These reactivity changes reflect the ability of oxide domains to accept or redistribute electron density in kinetically-relevant reduction steps, in the formation of temporary acid sites via reductive processes, and in the stabilization of cationic transition states. Reduction steps are favored by low-lying empty orbitals prevalent in larger clusters, which also favor electron delocalization, stable anions, and strong Brønsted acidity. Isomerization of xylenes and alkanes, elimination reactions of alkanols, and oxidation of alkanes to alkenes on V, Mo, Nb, and W oxide domains are used here to demonstrate the remarkable catalytic diversity made available by changes in domain size. The reactive and disordered nature of small catalytic domains introduces significant challenges in their synthesis and their structural and mechanistic characterization, which require in situprobes and detailed kinetic analysis. The local structure and electronic properties of these materials must be probed during catalysis and their catalytic function be related to specific kinetically-relevant steps. Structural uniformity can be imposed on oxide clusters by the use of polyoxometalate clusters with thermodynamically stable and well-defined size and connectivity. These clusters provide the compositional diversity and the structural fidelity required to develop composition-function relations from synergistic use of experiments and theory. In these clusters, the valence and electronegativity of the central atom affects the acid strength of the polyoxometalate clusters and the rate constants for acid catalyzed elementary steps via the specific stabilization of cationic transition states in isomerization and elimination reactions.
.jpg)


















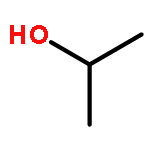
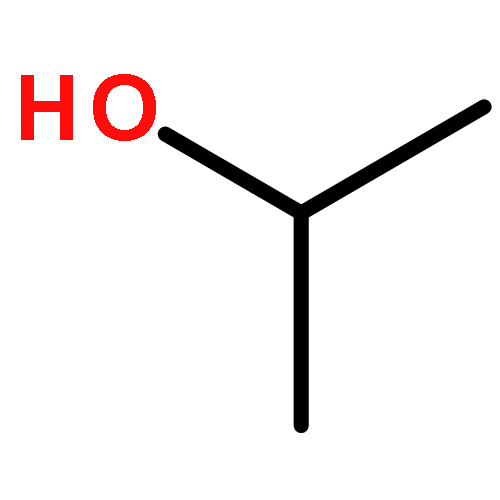



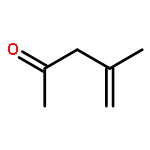

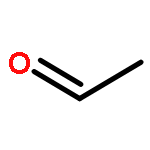
































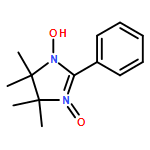
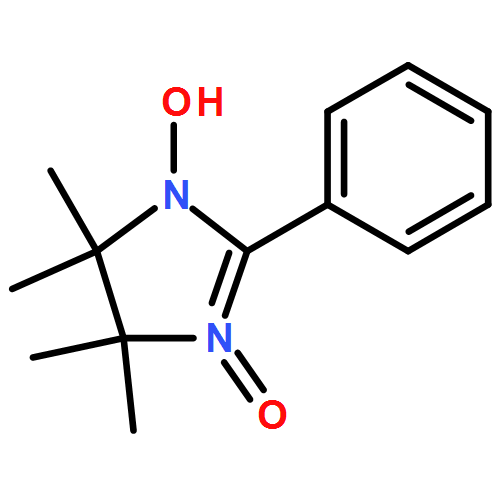

















![Tungstate(3-),tetracosa-m-oxododecaoxo[m12-[phosphato(3-)-kO:kO:kO:kO':kO':kO':kO'':kO'':kO'':kO''':kO''':kO''']]dodeca-,hydrogen (1:3)](http://img.cochemist.com/ccimg/1400/1343-93-7.png)
![Tungstate(3-),tetracosa-m-oxododecaoxo[m12-[phosphato(3-)-kO:kO:kO:kO':kO':kO':kO'':kO'':kO'':kO''':kO''':kO''']]dodeca-,hydrogen (1:3)](http://img.cochemist.com/ccimg/1400/1343-93-7_b.png)





