Co-reporter:Daniel Healion, Yu Zhang, Jason D. Biggs, Niranjan Govind, and Shaul Mukamel
The Journal of Physical Chemistry Letters September 6, 2012 Volume 3(Issue 17) pp:
Publication Date(Web):August 8, 2012
DOI:10.1021/jz300981w
We show that broadband X-ray pulses can create wavepackets of valence electrons and holes localized in the vicinity of a selected atom (nitrogen, oxygen, or sulfur in cysteine) by stimulated resonant Raman scattering. The subsequent dynamics reveals highly correlated motions of entangled electron and hole quasiparticles. This information goes beyond the time-dependent total charge density derived from X-ray diffraction.Keywords: concurrence; cysteine; electronic wavepackets; natural orbitals; quasiparticles; stimulated Raman;
Co-reporter:Yu Zhang;Jérémy R. Rouxel;Jochen Autschbach;Niranjan Govind
Chemical Science (2010-Present) 2017 vol. 8(Issue 9) pp:5969-5978
Publication Date(Web):2017/08/21
DOI:10.1039/C7SC01347G
Core-resonant circular dichroism (CD) signals are induced by molecular chirality and vanish for achiral molecules and racemic mixtures. The highly localized nature of core excitations makes them ideal probes of local chirality within molecules. Simulations of the circular dichroism spectra of several molecular families illustrate how these signals vary with the electronic coupling to substitution groups, the distance between the X-ray chromophore and the chiral center, geometry, and chemical structure. Clear insight into the molecular structure is obtained through analysis of the X-ray CD spectra.
Co-reporter:Markus Kowalewski
PNAS 2017 114 (13 ) pp:3278-3280
Publication Date(Web):2017-03-28
DOI:10.1073/pnas.1702160114
Co-reporter:Weijie Hua, Kochise Bennett, Yu Zhang, Yi Luo and Shaul Mukamel
Chemical Science 2016 vol. 7(Issue 9) pp:5922-5933
Publication Date(Web):12 May 2016
DOI:10.1039/C6SC01571A
The multi-configurational self-consistent field method is employed to simulate the two-dimensional all-X-ray double-quantum-coherence (XDQC) spectroscopy, a four-wave mixing signal that provides direct signatures of double core hole (DCH) states. The valence electronic structure is probed by capturing the correlation between the single (SCH) and double core hole states. The state-averaged restricted-active-space self-consistent field (SA-RASSCF) approach is used which can treat the valence, SCH, and DCH states at the same theoretical level, and applies to all types of DCHs (located on one or two atoms, K-edge or L-edge), with both accuracy and efficiency. Orbital relaxation introduced by the core hole(s) and the static electron correlation is properly accounted for. The XDQC process can take place via different intermediate DCH state channels by tuning the pulse frequencies. We simulate the XDQC signals for the three isomers of aminophenol at 8 pulse frequency configurations, covering all DCH pathways involving the N1s and O1s core hole (N1sN1s, O1sO1s and N1sO1s), which reveal different patterns of valence excitations.
Co-reporter:Jérémy R. Rouxel, Markus Kowalewski, and Shaul Mukamel
Journal of Chemical Theory and Computation 2016 Volume 12(Issue 8) pp:3959-3968
Publication Date(Web):June 27, 2016
DOI:10.1021/acs.jctc.6b00279
Stimulated (coherent) and spontaneous (incoherent) nonlinear X-ray signals are expressed using a spatially nonlocal response tensor which directly connects them to the time evolving current j and charge σ densities rather than to electric and magnetic multipoles. The relative contributions of the σA2 and j · A minimal coupling terms, where A is the vector potential, are demonstrated. The two dominate off-resonant and resonant scattering, respectively, and make comparable contributions at near resonant detunings.
Co-reporter:Kochise Bennett, Markus Kowalewski, and Shaul Mukamel
Journal of Chemical Theory and Computation 2016 Volume 12(Issue 2) pp:740-752
Publication Date(Web):December 21, 2015
DOI:10.1021/acs.jctc.5b00824
We present a hierarchy of Fermi golden rules (FGRs) that incorporate strongly coupled electronic/nuclear dynamics in time-resolved photoelectron spectroscopy (TRPES) signals at different levels of theory. Expansion in the joint electronic and nuclear eigenbasis yields the numerically most challenging exact FGR (eFGR). The quasistatic Fermi Golden Rule (qsFGR) neglects nuclear motion during the photoionization process but takes into account electronic coherences as well as populations initially present in the pumped matter as well as those generated internally by coupling between electronic surfaces. The standard semiclassical Fermi Golden Rule (scFGR) neglects the electronic coherences and the nuclear kinetic energy during the ionizing pulse altogether, yielding the classical Condon approximation. The coherence contributions depend on the phase-profile of the ionizing field, allowing coherent control of TRPES signals. The photoelectron spectrum from model systems is simulated using these three levels of theory. The eFGR and the qsFGR show temporal oscillations originating from the electronic or vibrational coherences generated as the nuclear wave packet traverses a conical intersection. These oscillations, which are missed by the scFGR, directly reveal the time-evolving splitting between electronic states of the neutral molecule in the curve-crossing regime.
Co-reporter:Konstantin E. Dorfman;Yu Zhang
PNAS 2016 Volume 113 (Issue 36 ) pp:10001-10006
Publication Date(Web):2016-09-06
DOI:10.1073/pnas.1610729113
We show that X-ray pulses resonant with selected core transitions can manipulate electron transfer (ET) in molecules with
ultrafast and atomic selectivity. We present possible protocols for coherently controlling ET dynamics in donor–bridge–acceptor
(DBA) systems by stimulated X-ray resonant Raman processes involving various transitions between the D, B, and A sites. Simulations
presented for a Ru(II)–Co(III) model complex demonstrate how the shapes, phases and amplitudes of the X-ray pulses can be
optimized to create charge on demand at selected atoms, by opening up otherwise blocked ET pathways.
Co-reporter:Yu Zhang, Shaul Mukamel, Munira Khalil, and Niranjan Govind
Journal of Chemical Theory and Computation 2015 Volume 11(Issue 12) pp:5804-5809
Publication Date(Web):November 9, 2015
DOI:10.1021/acs.jctc.5b00763
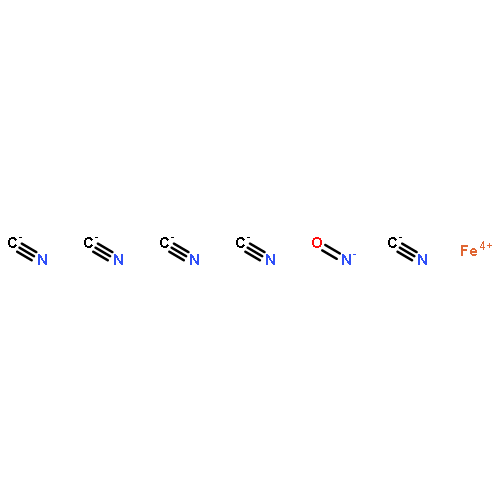
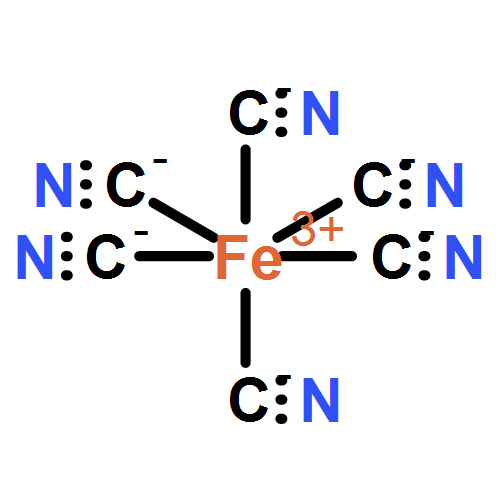
Valence-to-core (VtC) X-ray emission spectroscopy (XES) has emerged as a powerful technique for the structural characterization of complex organometallic compounds in realistic environments. Since the spectrum represents electronic transitions from the ligand molecular orbitals to the core holes of the metal centers, the approach is more chemically sensitive to the metal–ligand bonding character compared with conventional X-ray absorption techniques. In this paper we study how linear-response time-dependent density functional theory (LR-TDDFT) can be harnessed to simulate K-edge VtC X-ray emission spectra reliably. LR-TDDFT allows one to go beyond the single-particle picture that has been extensively used to simulate VtC-XES. We consider seven low- and high-spin model complexes involving chromium, manganese, and iron transition metal centers. Our results are in good agreement with experiment.
Co-reporter:Hideo Ando ; Benjamin P. Fingerhut ; Konstantin E. Dorfman ; Jason D. Biggs
Journal of the American Chemical Society 2014 Volume 136(Issue 42) pp:14801-14810
Publication Date(Web):September 19, 2014
DOI:10.1021/ja5063955
Cyclobutane thymine dimer, one of the major lesions in DNA formed by exposure to UV sunlight, is repaired in a photoreactivation process, which is essential to maintain life. The molecular mechanism of the central step, i.e., intradimer C—C bond splitting, still remains an open question. In a simulation study, we demonstrate how the time evolution of characteristic marker bands (C═O and C═C/C—C stretch vibrations) of cyclobutane thymine dimer and thymine dinucleotide radical anion, thymidylyl(3′→5′)thymidine, can be directly probed with femtosecond stimulated Raman spectroscopy (FSRS). We construct a DFT(M05-2X) potential energy surface with two minor barriers for the intradimer C5—C5′ splitting and a main barrier for the C6—C6′ splitting, and identify the appearance of two C5═C6 stretch vibrations due to the C6—C6′ splitting as a spectroscopic signature of the underlying bond splitting mechanism. The sequential mechanism shows only absorptive features in the simulated FSRS signals, whereas the fast concerted mechanism shows characteristic dispersive line shapes.
Co-reporter:Benjamin P. Fingerhut, Konstantin E. Dorfman, and Shaul Mukamel
Journal of Chemical Theory and Computation 2014 Volume 10(Issue 3) pp:1172-1188
Publication Date(Web):January 22, 2014
DOI:10.1021/ct401012u
Nonadiabatic electron and nuclear dynamics of photoexcited molecules involving conical intersections is of fundamental importance in many reactions such as the self-protection mechanism of DNA and RNA bases against UV irradiation. Nonlinear vibrational spectroscopy can provide an ultrafast sensitive probe for these processes. We employ a simulation protocol that combines nonadiabatic on-the-fly molecular dynamics with a mode-tracking algorithm for the simulation of femtosecond stimulated Raman spectroscopy (SRS) signals of the high frequency C–H- and N–H-stretch vibrations of the photoexcited RNA base uracil. The simulations rely on a microscopically derived expression that takes into account the path integral of the excited state evolution and the pulse shapes. Analysis of the joint time/frequency resolution of the technique reveals a matter chirp contribution that limits the inherent temporal resolution. Characteristic signatures of relaxation dynamics mediated in the vicinity of conical intersection are predicted. The C–H and N–H spectator modes provide high sensitivity to their local environment and act as local probes with submolecular and high temporal resolution.
Co-reporter:Yu Zhang, Jason D. Biggs, Weijie Hua, Konstantin E. Dorfman and Shaul Mukamel
Physical Chemistry Chemical Physics 2014 vol. 16(Issue 44) pp:24323-24331
Publication Date(Web):19 Sep 2014
DOI:10.1039/C4CP03361B
We investigate computationally the valence electronic excitations of the amino acid glycine prepared by a sudden nitrogen core ionization induced by an attosecond X-ray pump pulse. The created superposition of cationic excited states is probed by two-dimensional transient X-ray absorption and by three dimensional attosecond stimulated X-ray Raman signals. The latter, generated by applying a second broadband X-ray pulse combined with a narrowband pulse tuned to the carbon K-edge, reveal the complex coupling between valence and core-excited manifolds of the cation.
Co-reporter:Dr. Artur Nenov;Dr. Silvio a Beccara;Dr. Ivan Rivalta; Giulio Cerullo; Shaul Mukamel; Marco Garavelli
ChemPhysChem 2014 Volume 15( Issue 15) pp:3282-3290
Publication Date(Web):
DOI:10.1002/cphc.201402374
Abstract
The ability of nonlinear electronic spectroscopy to track folding/unfolding processes of proteins in solution by monitoring aromatic interactions is investigated by first-principles simulations of two-dimensional (2D) electronic spectra of a model peptide. A dominant reaction pathway approach is employed to determine the unfolding pathway of a tetrapeptide, which connects the initial folded configuration with stacked aromatic side chains and the final unfolded state with distant noninteracting aromatic residues. The π-stacking and excitonic coupling effects are included through ab initio simulations based on multiconfigurational methods within a hybrid quantum mechanics/molecular mechanics scheme. It is shown that linear absorption spectroscopy in the ultraviolet (UV) region is unable to resolve the unstacking dynamics characterized by the three-step process: T-shapedtwisted offset stackingunstacking. Conversely, pump–probe spectroscopy can be used to resolve aromatic interactions by probing in the visible region, the excited-state absorptions (ESAs) that involve charge-transfer states. 2D UV spectroscopy offers the highest sensitivity to the unfolding process, by providing the disentanglement of ESA signals belonging to different aromatic chromophores and high correlation between the conformational dynamics and the quartic splitting.
Co-reporter:Ivan Rivalta, Artur Nenov, Oliver Weingart, Giulio Cerullo, Marco Garavelli, and Shaul Mukamel
The Journal of Physical Chemistry B 2014 Volume 118(Issue 28) pp:8396-8405
Publication Date(Web):May 2, 2014
DOI:10.1021/jp502538m
Time-resolved two-dimensional (2D) electronic spectra (ES) tracking the evolution of the excited state manifolds of the retinal chromophore have been simulated along the photoisomerization pathway in bovine rhodopsin, using a state-of-the-art hybrid QM/MM approach based on multiconfigurational methods. Simulations of broadband 2D spectra provide a useful picture of the overall detectable 2D signals from the near-infrared (NIR) to the near-ultraviolet (UV). Evolution of the stimulated emission (SE) and excited state absorption (ESA) 2D signals indicates that the S1 → SN (with N ≥ 2) ESAs feature a substantial blue-shift only after bond inversion and partial rotation along the cis → trans isomerization angle, while the SE rapidly red-shifts during the photoinduced skeletal relaxation of the polyene chain. Different combinations of pulse frequencies are proposed in order to follow the evolution of specific ESA signals. These include a two-color 2DVis/NIR setup especially suited for tracking the evolution of the S1 → S2 transitions that can be used to discriminate between different photochemical mechanisms of retinal photoisomerization as a function of the environment. The reported results are consistent with the available time-resolved pump–probe experimental data, and may be used for the design of more elaborate transient 2D electronic spectroscopy techniques.
Co-reporter:Artur Nenov, Ivan Rivalta, Giulio Cerullo, Shaul Mukamel, and Marco Garavelli
The Journal of Physical Chemistry Letters 2014 Volume 5(Issue 4) pp:767-771
Publication Date(Web):February 7, 2014
DOI:10.1021/jz5002314
Two-dimensional (2D) optical spectroscopy techniques based on ultrashort laser pulses have been recently extended to the optical domain in the ultraviolet (UV) spectral region. UV-active aromatic side chains can thus be used as local highly specific markers for tracking dynamics and structural rearrangements of proteins. Here we demonstrate that 2D electronic spectra of a model proteic system, a tetrapeptide with two aromatic side chains, contain enough structural information to distinguish between two different configurations with distant and vicinal side chains. For accurate simulations of the 2DUV spectra in solution, we combine a quantum mechanics/molecular mechanics approach based on wave function methods, accounting for interchromophores coupling and environmental effects, with nonlinear response theory. The proposed methodology reveals effects, such as charge transfer between vicinal aromatic residues that remain concealed in conventional exciton Hamiltonian approaches. Possible experimental setups are discussed, including multicolor experiments and signal manipulation techniques for limiting undesired background contributions and enhancing 2DUV signatures of specific electronic couplings.Keywords: aromatic amino acid side chains; multiconfigurational methods; protein structure; SOS//QM/MM method; ultrafast nonlinear spectroscopy;
Co-reporter:Yu Zhang, Jason D. Biggs, Niranjan Govind, and Shaul Mukamel
The Journal of Physical Chemistry Letters 2014 Volume 5(Issue 21) pp:3656-3661
Publication Date(Web):October 9, 2014
DOI:10.1021/jz501966h
Long-range electron transfer (ET) is a crucial step in many energy conversion processes and biological redox reactions in living organisms. We show that newly developed X-ray pulses can directly probe the evolving oxidation states and the electronic structure around selected atoms with detail not available through conventional time-resolved infrared or optical techniques. This is demonstrated in a simulation study of the stimulated X-ray Raman (SXRS) signals in Re-modified azurin, which serves as a benchmark system for photoinduced ET in proteins. Nonlinear SXRS signals offer a direct novel window into the long-range ET mechanism.Keywords: azurin; cupredoxin; electron hopping; TDDFT; transient X-ray absorption spectroscopy;
Co-reporter:Jun Jiang, Zaizhi Lai, Jin Wang, and Shaul Mukamel
The Journal of Physical Chemistry Letters 2014 Volume 5(Issue 8) pp:1341-1346
Publication Date(Web):March 19, 2014
DOI:10.1021/jz5002264
The function of protein relies on their folding to assume the proper structure. Probing the structural variations during the folding process is crucial for understanding the underlying mechanism. We present a combined quantum mechanics/molecular dynamics simulation study that demonstrates how coherent resonant nonlinear ultraviolet spectra can be used to follow the fast folding dynamics of a mini-protein, Trp-cage. Two dimensional ultraviolet signals of the backbone transitions carry rich information of both local (secondary) and global (tertiary) structures. The complexity of signals decreases as the conformational entropy decreases in the course of the folding process. We show that the approximate entropy of the signals provides a quantitative marker of protein folding status, accessible by both theoretical calculations and experiments.Keywords: approximate entropy; conformational entropy; protein folding; quantum mechanics/molecular dynamics; two-dimensional ultraviolet spectroscopy;
Co-reporter:Weijie Hua, Jason D. Biggs, Yu Zhang, Daniel Healion, Hao Ren, and Shaul Mukamel
Journal of Chemical Theory and Computation 2013 Volume 9(Issue 12) pp:5479-5489
Publication Date(Web):October 21, 2013
DOI:10.1021/ct400767g
Attosecond Stimulated X-ray Raman Spectroscopy (SXRS) is a promising technique for investigating molecular electronic structure and photochemical processes with high spatial and temporal resolution. We present a theoretical study of SXRS from multiple core excitation sites of the same element. Two issues are addressed: interference between pathways contributing the signals from different sites and how nuclear vibrations influence the signals. Taking furan as a model system, which contains two types of carbons, Cα and Cβ, we performed time-dependent density functional theory calculations and computed the SXRS signals with two pulses tuned at the carbon K-edge. Our simulations demonstrate that the SXRS signal from the Cα and Cβ sites are nonadditive, owing to the significant mixed contributions (Cα 1s excitations by the pump pulse followed by Cβ 1s excitations by the probe, or vice verse). Harmonic vibrations linearly coupled to the electronic transitions are incorporated using the cumulant expansion. The nuclei act as a bath for electronic transitions which accelerate the decay of the time-domain signal. The frequency-domain spectrum is modified by a small red shift, and high-resolution fine-structure features are introduced.
Co-reporter:Hao Ren, Zaizhi Lai, Jason D. Biggs, Jin Wang and Shaul Mukamel
Physical Chemistry Chemical Physics 2013 vol. 15(Issue 44) pp:19457-19464
Publication Date(Web):20 Sep 2013
DOI:10.1039/C3CP51347E
We report a combined molecular dynamics (MD) and ab initio simulation study of the ultrafast broadband ultraviolet (UV) stimulated resonance Raman (SRR) spectra of the Trp-cage mini protein. Characteristic two dimensional (2D) SRR features of various folding states are identified. Structural fluctuations erode the cross peaks and the correlation between diagonal peaks is a good indicator of the α-helix formation.
Co-reporter:Konstantin E. Dorfman, Benjamin P. Fingerhut and Shaul Mukamel
Physical Chemistry Chemical Physics 2013 vol. 15(Issue 29) pp:12348-12359
Publication Date(Web):29 May 2013
DOI:10.1039/C3CP51117K
Vibrational motions in electronically excited states can be observed either by time and frequency resolved infrared absorption or by off resonant stimulated Raman techniques. Multipoint correlation function expressions are derived for both signals. Three representations which suggest different simulation protocols for the signals are developed. These are based on the forward and the backward propagation of the wavefunction, sum over state expansion using an effective vibrational Hamiltonian or a semiclassical treatment of a bath. We show that the effective temporal (Δt) and spectral (Δω) resolution of the techniques is not controlled solely by experimental knobs but also depends on the system dynamics being probed. The Fourier uncertainty ΔωΔt > 1 is never violated.
Co-reporter:Hao Ren, Benjamin P. Fingerhut, and Shaul Mukamel
The Journal of Physical Chemistry A 2013 Volume 117(Issue 29) pp:6096-6104
Publication Date(Web):March 21, 2013
DOI:10.1021/jp400044t
The excited state isomerization of thioflavin T (ThT) is responsible for the quenching of its fluorescence in a nonrestricted environment. The fluorescence quantum yield increases substantially upon binding to amyloid fibers. Simulations reveal that the variation of the twisting angle between benzothiazole and benzene groups (ϕ1) is responsible for the subpicosecond fluorescence quenching. The evolution of the twisting process can be directly probed by photoelectron emission with energies ε ≥ 1.0 eV before the molecule reaches the ϕ1-twisted configuration (∼300 fs).
Co-reporter:Jun Jiang, Kory J. Golchert, Carolyn N. Kingsley, William D. Brubaker, Rachel W. Martin, and Shaul Mukamel
The Journal of Physical Chemistry B 2013 Volume 117(Issue 46) pp:14294-14301
Publication Date(Web):November 12, 2013
DOI:10.1021/jp408000k
The formation of amyloid fibrils is associated with many serious diseases as well as diverse biological functions. Despite the importance of these aggregates, predicting the aggregation propensity of a particular sequence is a major challenge. We report a joint 2D nuclear magnetic resonance (NMR) and ultraviolet (2DUV) study of fibrillization in the wild-type and two aggregation-prone mutants of the eye lens protein γS-crystallin. Simulations show that the complexity of 2DUV signals as measured by their “approximate entropy” is a good indicator for the conformational entropy and in turn is strongly correlated with its aggregation propensity. These findings are in agreement with high-resolution NMR experiments and are corroborated for amyloid fibrils. The 2DUV technique is complementary to high-resolution structural methods and has the potential to make the evaluation of the aggregation propensity for protein variant propensity of protein structure more accessible to both theory and experiment. The approximate entropy of experimental 2DUV signals can be used for fast screening, enabling identification of variants with high fibrillization propensity for the much more time-consuming NMR structural studies, potentially expediting the characterization of protein variants associated with cataract and other protein aggregation diseases.
Co-reporter:Benjamin P. Fingerhut, Konstantin E. Dorfman, and Shaul Mukamel
The Journal of Physical Chemistry Letters 2013 Volume 4(Issue 11) pp:1933-1942
Publication Date(Web):May 20, 2013
DOI:10.1021/jz400776r
Resolving the excited-state dynamics of DNA and RNA nucleobases has attracted considerable attention. UV irradiation of the isolated nucleobases leads to the population of an electronic excited state, which is quenched by internal conversion mediated by conical intersections on an ultrafast time scale. We present nonadiabatic on-the-fly molecular dynamics simulations of the UV pump–IR probe signal of the pyrimidine nucleobase uracil using a novel semiclassical protocol that takes into account the path integral over the excited-state vibrational dynamics and properly describes the joint temporal and spectral resolution of the technique. Simulations of vibrational motions of carbonyl fingerprint modes in the electronically excited states reveal clear signatures of different relaxation pathways on a time scale of hundreds of femtoseconds, which arise from an ultrafast branching in the excited state. We show that the inherent temporal and spectral resolution of the technique is not purely instrumental but also depends on the vibrational fluctuation time scale.Keywords: DNA; excited states; nonadiabatic on-the-fly dynamics; time-resolved IR spectroscopy;
Co-reporter:Jason D. Biggs;Daniel Healion;Yu Zhang
PNAS 2013 Volume 110 (Issue 39 ) pp:15597-15601
Publication Date(Web):2013-09-24
DOI:10.1073/pnas.1308604110
Understanding the excitation energy transfer mechanism in multiporphyrin arrays is key for designing artificial light-harvesting
devices and other molecular electronics applications. Simulations of the stimulated X-ray Raman spectroscopy signals of a
Zn/Ni porphyrin heterodimer induced by attosecond X-ray pulses show that these signals can directly reveal electron–hole pair
motions. These dynamics are visualized by a natural orbital decomposition of the valence electron wavepackets.
Co-reporter:Benjamin P. Fingerhut and Shaul Mukamel
The Journal of Physical Chemistry Letters 2012 Volume 3(Issue 13) pp:1798-1805
Publication Date(Web):June 18, 2012
DOI:10.1021/jz3006282
At the heart of photosynthesis is excitation energy transfer toward and charge separation within highly conserved reaction centers (RCs). The function principles of RCs in purple bacteria offer a blueprint for an optoelectronic device, which efficiently utilizes the near-IR region of the solar spectrum. We present theoretical modeling of the nonlinear optical response of the bacterial RC B. viridis incorporating electron and energy transfer on equal footing. The splitting of special pair excitons P is the origin of distinct cross peaks, which allow monitoring of the kinetics of charge separation. The xxyy – xyxy signal, obtained from sequences of orthogonal polarized laser pulses, highlights the kinetics of the secondary, subpicosecond electron transfer from the accessory bacteriochlorophyll BClL to the bacteriopheophytine BPL. The increased selectivity is explained by the relative orientation of exciton transitions. The technique can resolve complex kinetics in congested signals of photosynthetic complexes that are otherwise hardly accessible.Keywords: coherent 2D nonlinear optical spectroscopy; electron and energy transfer; photosynthesis; reaction center; ultrafast spectroscopy;
Co-reporter:Jun Jiang and Shaul Mukamel
Physical Chemistry Chemical Physics 2011 vol. 13(Issue 6) pp:2394-2400
Publication Date(Web):06 Dec 2010
DOI:10.1039/C0CP02047H
We report a first principles study of two dimensional electronic spectroscopy of aromatic side chain transitions in the 32-residue β-amyloid (Aβ9–40) fibrils in the near ultraviolet (250–300 nm). An efficient exciton Hamiltonian with electrostatic fluctuations (EHEF) algorithm is used to compute the electronic excitations in the presence of environmental fluctuations. The through-space inter- and intra-molecular interactions are calculated with high level quantum mechanics (QM) approaches, and interfaced with molecular mechanics (MM) simulations. Distinct two dimensional near ultraviolet (2DNUV) spectroscopic signatures are identified for different aromatic transitions, and the couplings between them. 2DNUV signals associated with the transition couplings are shown to be very sensitive to the change of residue-residue interactions induced by residue mutations. Our simulations suggest that 2DNUV spectra could provide a useful local probe for the structure and kinetics of fibrils.
Co-reporter:A. R. Lam, J. Jiang, and S. Mukamel
Biochemistry 2011 Volume 50(Issue 45) pp:
Publication Date(Web):September 30, 2011
DOI:10.1021/bi201317c
Understanding the aggregation mechanism of amyloid fibrils and characterizing their structures are important steps in the investigation of several neurodegenerative disorders associated with the misfolding of proteins. We report a simulation study of coherent two-dimensional chiral signals of three NMR structures of Aβ protein fibrils associated with Alzheimer’s Disease, two models for Aβ(8–40) peptide wild-type (WT) and one for the Iowa (D23N) Aβ(15–40) mutant. Both far-ultraviolet (FUV) signals (λ = 190–250 nm), which originate from the backbone nπ* and ππ* transitions, and near-ultraviolet (NUV) signals (λ ≥ 250 nm) associated with aromatic side chains (Phe and Tyr) show distinct cross-peak patterns that can serve as novel signatures for the secondary structure.
Co-reporter:Hao Ren, Jun Jiang, and Shaul Mukamel
The Journal of Physical Chemistry B 2011 Volume 115(Issue 47) pp:13955-13962
Publication Date(Web):October 18, 2011
DOI:10.1021/jp207849u
We present a combined quantum mechanics and molecular mechanics study of the deep ultraviolet ππ* resonance Raman spectra of β-sheet amyloid fibrils Aβ34–42 and Aβ1–40. Effects of conformational fluctuations are described using a Ramachandran angle map, thus avoiding repeated ab initio calculations. Experimentally observed effects of hydrogen–deuterium exchange are reproduced. We propose that the AmIII band redshift upon deuteration is caused by the loss of coupling between Cα–H bending and N–D bending modes, rather than by peptide bond hydration.
Co-reporter:Jun Jiang and Shaul Mukamel
The Journal of Physical Chemistry B 2011 Volume 115(Issue 19) pp:6321-6328
Publication Date(Web):April 25, 2011
DOI:10.1021/jp201164u
Keeping track of the aggregation kinetics of amyloid fibrils is essential for understanding their formation mechanism and eventually developing treatments for misfolded protein-related diseases. A simulation study of a series of Aβ9–40 amyloid fibrils with different size shows that novel two-dimensional near-ultraviolet (2DNUV) spectra contain characteristic signatures of interactions between peptides. Chiral 2DNUV signals show a larger degree of exciton delocalization compared to their nonchiral counterparts. Intensities of specific peaks provide a direct measure of the number of peptides in a fibril. These signals could be used to monitor the fibril growth kinetics, one peptide at a time.
Co-reporter:Yuki Nagata
Journal of the American Chemical Society 2010 Volume 132(Issue 18) pp:6434-6442
Publication Date(Web):April 15, 2010
DOI:10.1021/ja100508n
The sum-frequency generation (SFG) spectrum from the water/[1,2-dimyristoyl-sn-glycero-3-phosphatidylcholine] (DMPC) interface in the OH stretching mode region of water is simulated and shows three spectral peaks which are assigned to different environments. The weak 3590 cm−1 peak is attributed to a few water molecules coupled to the glycerol backbone of DMCP. The 3470 cm−1 feature comes from the top water layer adjacent to the hydrophilic headgroup of DMPC. The 3290 cm−1 peak arises from the near-bulk water nonadjacent to DMPC. The stretching mode corresponding to the 3290 cm−1 peak is strongly coupled to the neighboring water molecules. In contrast, the 3470 cm−1 mode is decoupled from the surrounding water molecules, and the orientation of water is governed by DMPC. This decoupling explains the slow relaxation dynamics of water measured in the time-resolved SFG experiment. Despite the similarity of the SFG spectra, the peak origins of water/lipid and water/vapor interfaces are different.
Co-reporter:Darius Abramavicius ; Jun Jiang ; Benjamin M. Bulheller ; Jonathan D. Hirst
Journal of the American Chemical Society 2010 Volume 132(Issue 22) pp:7769-7775
Publication Date(Web):May 18, 2010
DOI:10.1021/ja101968g
Amide n−π* and π−π* excitations around 200 nm are prominent spectroscopic signatures of the protein backbone, which are routinely used in ultraviolet (UV) circular dichroism for structure characterization. Recently developed ultrafast laser sources may be used to extend these studies to two dimensions. We apply a new algorithm for modeling protein electronic transitions to simulate two-dimensional UV photon echo signals in this regime and to identify signatures of protein backbone secondary (and tertiary) structure. Simulated signals for a set of globular and fibrillar proteins and their specific regions reveal characteristic patterns of helical and sheet secondary structures. We investigate how these patterns vary and converge with the size of the structural motif. Specific chiral polarization configurations of the UV pulses are found to be sensitive to aspects of the protein structure. This information significantly augments that available from linear circular dichroism.
Co-reporter:Benoit Palmieri, Darius Abramavicius and Shaul Mukamel
Physical Chemistry Chemical Physics 2010 vol. 12(Issue 1) pp:108-114
Publication Date(Web):10 Nov 2009
DOI:10.1039/B916723D
Recently developed approaches to simulate environment-induced fluctuation effects in two-dimensional (2D) spectroscopy of excitons are compared for the Fenna-Matthews-Olson light-harvesting complex. Fast fluctuations induce population redistribution between exciton energy-levels and raise homogeneous line widths of various peaks in 2D spectra. These effects are easily accounted for in sum-over-eigenstates (SOS) approach and the quasi-particle (QP) scattering approach through relaxation and dephasing rate constants. Slow fluctuations cause correlations of energies at various delay times in 2D photon-echo spectra. These may be calculated either by doing cumulant expansion in SOS techniques or by statistical averaging over static disorder in SOS and QP approaches. We compare the 2D photon-echo signal simulated using two levels of cumulant expansion approaches and two statistical averaging approaches for the same system. These levels differ by the treatment of energy-level correlations at different delay times and give rise to different cross-peak shapes: the cross-peaks retain their original diagonally elongated shapes when correlations are included, while they are more spherically broadened when correlations are neglected. Statistical averaging over disorder give very similar results but requires much higher computational effort. The peak redistribution timescales are very similar for all levels of theory. The spectral signatures at these different levels of theory are compared and simulation cost is estimated. Approaches which do require statistical averaging over disorder are orders of magnitude slower.
Co-reporter:Dr. Jun Jiang ;Dr. Shaul Mukamel
Angewandte Chemie International Edition 2010 Volume 49( Issue 50) pp:9666-9669
Publication Date(Web):
DOI:10.1002/anie.201005093
Co-reporter:Dr. Jun Jiang ;Dr. Shaul Mukamel
Angewandte Chemie 2010 Volume 122( Issue 50) pp:9860-9863
Publication Date(Web):
DOI:10.1002/ange.201005093
Co-reporter:Saar Rahav
PNAS 2010 Volume 107 (Issue 11 ) pp:4825-4829
Publication Date(Web):2010-03-16
DOI:10.1073/pnas.0910120107
Coherent anti-Stokes Raman spectroscopy (CARS) uses vibrational resonances to study nuclear wavepacket motions and is widely
used in cell imaging and other applications. The resonances usually lie on top of a parametric component that involves no
change in the molecular state and creates an undesirable background which reduces the sensitivity of the technique. Here,
by examining the process from the perspective of the molecule, rather than the field, we are able to separate the two components
and recast each resonance as a modulus square of a transition amplitude which contains an interference between two Stokes
pathways, each involving a different pair of field modes. We further propose that dissipative signals obtained by measuring
the total absorption of all field modes in a convenient collinear pulse geometry can eliminate the parametric component and
retain the purely resonant contributions. Specific vibrational resonances may then be readily detected using pulse shapers
through derivatives with respect to pulse parameters.
Co-reporter:Jun Jiang, Darius Abramavicius, Cyril Falvo, Benjamin M. Bulheller, Jonathan D. Hirst, and Shaul Mukamel
The Journal of Physical Chemistry B 2010 Volume 114(Issue 37) pp:12150-12156
Publication Date(Web):August 26, 2010
DOI:10.1021/jp1046968
Revealing the structure and aggregation mechanism of amyloid fibrils is essential for the treatment of over 20 diseases related to protein misfolding. Coherent two-dimensional (2D) infrared spectroscopy is a novel tool that provides a wealth of new insight into the structure and dynamics of biomolecular systems. Recently developed ultrafast laser sources are extending multidimensional spectroscopy into the ultraviolet (UV) region, and this opens up new opportunities for probing fibrils. In a simulation study, we show that 2DUV spectra of the backbone of a 32-residue β-amyloid (Aβ9−40) fibril associated with Alzheimer’s disease and two intermediate prefibrillar structures carry characteristic signatures of fibril size and geometry that could be used to monitor its formation kinetics. The dependence of these signals on the fibril size and geometry is explored. We demonstrate that the dominant features of the β-amyloid fibril spectra are determined by intramolecular interactions within a single Aβ9−40, and intermolecular interactions at the “external interface” have clear signatures in the fine details of these signals.
Co-reporter:Jun Jiang, Darius Abramavicius, Benjamin M. Bulheller, Jonathan D. Hirst and Shaul Mukamel
The Journal of Physical Chemistry B 2010 Volume 114(Issue 24) pp:8270-8277
Publication Date(Web):May 26, 2010
DOI:10.1021/jp101980a
A generalized approach combining quantum mechanics (QM) and molecular mechanics (MM) calculations is developed to simulate the n → π* and π → π* backbone transitions of proteins in aqueous solution. These transitions, which occur in the ultraviolet (UV) at 180−220 nm, provide a sensitive probe for secondary structures. The excitation Hamiltonian is constructed using high-level electronic structure calculations of N-methylacetamide (NMA). Its electrostatic fluctuations are modeled using a new algorithm, EHEF, which combines a molecular dynamics (MD) trajectory obtained with a MM forcefield and electronic structures of sampled MD snapshots calculated by QM. The lineshapes and excitation splittings induced by the electrostatic environment in the experimental UV linear absorption (LA) and circular dichroism (CD) spectra of several proteins in aqueous solution are reproduced by our calculations. The distinct CD features of α-helix and β-sheet protein structures are observed in the simulations and can be assigned to different backbone geometries. The fine structure of the UV spectra is accurately characterized and enables us to identify signatures of secondary structures.
Co-reporter:Wei Zhuang;Angel E. Garcia;Zhenyu Li;Nikolaos G. Sgourakis
PNAS 2010 Volume 107 (Issue 36 ) pp:15687-15692
Publication Date(Web):2010-09-07
DOI:10.1073/pnas.1002131107
Elucidating the structural features of the Aβ monomer, the peptide constituent of amyloid fibrils found in Alzheimer’s disease,
can enable a direct characterization of aggregation pathways. Recent studies support the view that the ensemble of Aβ42 monomers
is a mixture of diverse ordered and disordered conformational species, which can be classified according to the formation
of a characteristic β-hairpin conformation in a certain region. Despite the disparity in the structural features of these
species, commonly used spectroscopic techniques such as NMR may not directly trace the conformational dynamics in the ensemble
due to the limited time resolution and the lack of well-resolved spectral features for different comformers. Here we use molecular
dynamics simulations combined with simulations of two-dimensional IR (2DIR) spectra to investigate the structure of these
species, their interchange kinetics, and their spectral features. We show that while the discrimination efficiency of the
ordinary, nonchiral 2DIR signal is limited due to its intrinsic dependence on common order parameters that are dominated by
the generally unstructured part of the sequence, signals with carefully designed chirality-sensitive pulse configurations
have the high resolution required for differentiating the various monomer structures. Our combined simulation studies indicate
the power of the chirality-induced (CI) 2DIR technique in investigating early events in Aβ42 aggregation and open the possibility
for their use as a novel experimental tool.
Co-reporter:Shaul Mukamel, Darius Abramavicius, Lijun Yang, Wei Zhuang, Igor V. Schweigert and Dmitri V. Voronine
Accounts of Chemical Research 2009 Volume 42(Issue 4) pp:553
Publication Date(Web):March 26, 2009
DOI:10.1021/ar800258z
Over the past 15 years, researchers have extended the multidimensional techniques which originated with NMR in the 1970s to infrared and visible coherent spectroscopy. These advances have dramatically enhanced the temporal resolution from the microsecond to the femtosecond regime. NMR spectroscopists have developed principles for the design of pulse sequences that enhance selected spectral features and reveal desired dynamical events. Extending these principles to the optical regime offers numerous opportunities for narrowing the line shapes in specific directions, unraveling weak cross-peaks from otherwise congested spectra, and controlling the interferences between quantum pathways. We can achieve these enhancements by shaping the spectral and temporal profiles of the pulses. Pulse polarization shaping may lead to unique probes of time-dependent chirality. In this Account, we compare two types of signals. The first, the photon echo, is generated in the direction −k1 + k2 + k3, and the second, double quantum coherence, is detected at +k1 + k2 − k3. Here k1, k2, and k3 are the wave vectors of the three incoming pulses in chronological order. We illustrate the novel information extracted from these signals by simulations of three physical systems. In the first system, spectra of GaAs semiconductor quantum wells provide a direct look at many-body electron correlation effects. We directly observe specific projections of the many-electron wave function, which we can use to test the quality of various levels of computational techniques for electronic structure. Secondly, the spectra of photosynthetic aggregates reveal couplings between chromophores, quantum coherence signatures of chromophore entanglement, and energy-transfer pathways. Using some fundamental symmetries of pulse polarization configurations of nonlinear signals, we can construct superpositions of signals designed to better distinguish among various coherent and incoherent exciton transport pathways and amplify subtle variations among different species of the Fenna−Matthews−Olson (FMO) antenna complex. Both of the first two applications require femtosecond pulses of light in the visible range. The third application demonstrates how resonant core spectroscopy may be used to generate core excitations that are highly localized at selected atoms. Such signals can monitor the motions of valence electron wavepackets in real space with atomic spatial resolution. These future X-ray applications will require attosecond bright X-ray sources, which are currently being developed in several labs. Common principles underlie these techniques in coherent spectroscopy for spins, valence electrons, and core electronic excitations, spanning frequencies from radiowaves to hard X-rays.
Co-reporter:Darius Abramavicius and Shaul Mukamel
The Journal of Physical Chemistry B 2009 Volume 113(Issue 17) pp:6097-6108
Publication Date(Web):April 7, 2009
DOI:10.1021/jp811339p
Electronic excitations and the optical properties of the photosynthetic complex PSI are analyzed using an effective exciton model developed by Vaitekonis et al. [Photosynth. Res. 2005, 86, 185]. States of the reaction center, the linker states, the highly delocalized antenna states and the red states are identified and assigned in absorption and circular dichroism spectra by taking into account the spectral distribution of density of exciton states, exciton delocalization length, and participation ratio in the reaction center. Signatures of exciton cooperative dynamics in nonchiral and chirality-induced two-dimensional (2D) photon-echo signals are identified. Nonchiral signals show resonances associated with the red, the reaction center, and the bulk antenna states as well as transport between them. Spectrally overlapping contributions of the linker and the delocalized antenna states are clearly resolved in the chirality-induced signals. Strong correlations are observed between the delocalized antenna states, the linker states, and the RC states. The active space of the complex covering the RC, the linker, and the delocalized antenna states is common to PSI complexes in bacteria and plants.
Co-reporter:Wei Zhuang Dr.;Tomoyuki Hayashi Dr.
Angewandte Chemie 2009 Volume 121( Issue 21) pp:3804-3838
Publication Date(Web):
DOI:10.1002/ange.200802644
Co-reporter:Wei Zhuang Dr.;Tomoyuki Hayashi Dr.
Angewandte Chemie International Edition 2009 Volume 48( Issue 21) pp:3750-3781
Publication Date(Web):
DOI:10.1002/anie.200802644
Co-reporter:Sven Welack, Jeremy B. Maddox, Massimiliano Esposito, Upendra Harbola and Shaul Mukamel
Nano Letters 2008 Volume 8(Issue 4) pp:1137-1141
Publication Date(Web):March 11, 2008
DOI:10.1021/nl073292b
Electron counting of a single porphyrin molecule between two electrodes shows a crossover from sub- to super-Poissonian statistics as the bias voltage is scanned. This is attributed to the simultaneous activation of states with electron transfer rates spanning several orders of magnitude. Time-series analysis of consecutive single-electron transfer events reveals fast and slow transport channels, which are not resolved by the average current alone.
Co-reporter:Tomoyuki Hayashi, Shaul Mukamel
Journal of Molecular Liquids 2008 Volume 141(Issue 3) pp:149-154
Publication Date(Web):30 June 2008
DOI:10.1016/j.molliq.2008.02.013
An effective exciton Hamiltonian for all amide bands is used to calculate the absorption and photon echo spectra of a 17 residue helical peptide (YKKKH17). The cross peak bandshapes are sensitive to the inter-band couplings. Fluctuations of the local amide frequencies of the all amide fundamental and their overtone and combination states are calculated using the multipole electric field induced by environment employing the electrostatic DFT map of N-methyl acetamide. Couplings between neighboring peptide units are obtained using the anharmonic vibrational Hamiltonian of glycine dipeptide (GLDP) at the BPW91/6-31G(d,p) level. Electrostatic couplings between non-neighboring units are calculated by a fourth rank transition multipole coupling (TMC) expansion including 1/R3 (dipole–dipole), 1/R4 (quadrupole–dipole), and 1/R5 (quadrupole–quadrupole and octapole–dipole) interactions.
Co-reporter:Zhenyu Li, Haibo Yu, Wei Zhuang, Shaul Mukamel
Chemical Physics Letters 2008 Volume 452(1–3) pp:78-83
Publication Date(Web):4 February 2008
DOI:10.1016/j.cplett.2007.12.022
Molecular dynamics (MD) simulations are performed for N-methylacetamide (NMA) in water at 300 K with different force fields. Compared to the three all-atom force fields (CHARMM22, AMBER03, and OPLS-AA), the united-atom force field (GROMOS96) predicts a broader distribution of the peptide OCNH dehedral angle. A map constructed by fitting the nπ∗nπ∗ and ππ∗ππ∗ transition energies as quadratic functions of the NMA geometric variables is used to simulate the excitation energy fluctuations. GROMOS96 predicts blue shifted nπ∗nπ∗ and ππ∗ππ∗ energies and stronger fluctuations compared to the other three force fields, which indicates that different force fields may predict different spectral lineshapes for proteins.Different force fields are compared in the context of electronic spectrum simulation.
Co-reporter:Darius Abramavicius;Dmitri V. Voronine
PNAS 2008 Volume 105 (Issue 25 ) pp:8525-8530
Publication Date(Web):2008-06-24
DOI:10.1073/pnas.0802926105
A simulation study demonstrates how the nonlinear optical response of the Fenna–Matthews–Olson photosynthetic light-harvesting
complex may be explored by a sequence of laser pulses specifically designed to probe the correlated dynamics of double excitations.
Cross peaks in the 2D correlation plots of the spectra reveal projections of the double-exciton wavefunctions onto a basis
of direct products of single excitons. An alternative physical interpretation of these signals in terms of quasiparticle scattering
is developed.
Co-reporter:Cyril Falvo, Tomoyuki Hayashi, Wei Zhuang and Shaul Mukamel
The Journal of Physical Chemistry B 2008 Volume 112(Issue 39) pp:12479-12490
Publication Date(Web):September 9, 2008
DOI:10.1021/jp801493y
The two-dimensional infrared photon echo spectrum of Antamanide (-1Val-2Pro-3Pro-4Ala-5Phe-6Phe-7Pro-8Pro-9Phe-10Pro-) in chloroform is calculated using an explicit solvent molecular dynamics (MD) simulation combined with a density functional theory (DFT) map for the effective vibrational Hamiltonian. Evidence for a strong intramolecular hydrogen bonding network is found. Comparison with experimental absorption allows the identification of the dominant conformation. Multidimensional spectroscopy reveals intramolecular couplings and gives information on its dynamics. A two-color amide-I and amide-A crosspeak is predicted and analyzed in terms of local structure.
Co-reporter:František Šanda and Shaul Mukamel
The Journal of Physical Chemistry B 2008 Volume 112(Issue 45) pp:14212-14220
Publication Date(Web):October 16, 2008
DOI:10.1021/jp801457c
Signatures of chemical exchange and spectral diffusion in 2D photon-echo line shapes of molecular aggregates are studied using model calculations for a dimer whose Hamiltonian parameters are stochastically modulated. Cross peaks induced by chemical exchange and by exciton transport have different dynamics and distinguish two models which have the same absorption spectrum (a two-state jump bath modulation model of a dimer and a four-state jump bath model of a single chromophore). Slow Gaussian−Markovian spectral diffusion of a symmetric dimer induces new peaks which are damped as the dipole moment is equilibrated. These effects require an explicit treatment of the bath and may not be described by lower-level theories such as the Redfield equations, which eliminate the bath.
Co-reporter:Daniel M. Healion, Igor V. Schweigert and Shaul Mukamel
The Journal of Physical Chemistry A 2008 Volume 112(Issue 45) pp:11449-11461
Publication Date(Web):October 17, 2008
DOI:10.1021/jp803824a
Two-dimensional X-ray correlation spectroscopy (2DXCS) signals of the isolated DNA bases and Watson−Crick base pairs which contain multiple absorbing nitrogen atoms are calculated. Core−hole excited states are calculated using density functional theory with the B3LYP functional and 6-311G** basis set. Sum over states calculations of the signals reveal changes in cross-peak intensities between hydrogen-bonded and stacked base pairs. Nucleobase analogues are proposed for investigating base-stacking and hydrogen-bonding interactions.
Co-reporter:J.B. Maddox, U. Harbola, G.C. Bazan, S. Mukamel
Chemical Physics Letters 2007 Volume 450(1–3) pp:144-150
Publication Date(Web):14 December 2007
DOI:10.1016/j.cplett.2007.10.078
The conductance and current-induced fluorescence of a single distyryl-benzene and a single distyryl-paracyclophane molecule in a tunneling junction is simulated. Our approach is formulated in terms of the electronic states of the neutral and charged molecular bridge and is applied to calculate the effective tunneling rates. We find that an orbital picture is adequate for describing the conductance of distyryl-benzene; however, a many-electron picture is needed for the paracyclophane linked complex. A strategy for maximizing electroluminescence by controlling the voltage drop across the junction through chemical modification of the molecular bridge is suggested.Electronic structure calculations of neutral, anionic, and cationic conjugated molecules are used to simulate conductance and current-induced fluorescence spectra.
Co-reporter:Wei Zhuang;Darius Abramavicius;Dimitrii V. Voronine;
Proceedings of the National Academy of Sciences 2007 104(36) pp:14233-14236
Publication Date(Web):August 3, 2007
DOI:10.1073/pnas.0700392104
We propose to use infrared coherent two-dimensional correlation spectroscopy (2DCS) to characterize the fibril structure of
Ab42, the dominant composition of Ab deposit, which is crucial for investigating its toxicity and aggregation mechanism. By
optimizing the pulse polarization configurations with a genetic algorithm combined with sensitivity analysis, we obtained
signals with well resolved cross-peak features attributed to the couplings within and between different structural motifs.
These signals may provide new constraints for refining of the currently available NMR structure. Two-dimensional correlation
spectroscopy also can differentiate the turn structure of Ab42 and other Ab derivatives.
Co-reporter:Darius Abramavicius;Wei Zhuang
PNAS 2006 Volume 103 (Issue 50 ) pp:18934-18938
Publication Date(Web):2006-12-12
DOI:10.1073/pnas.0606912103
A simulation study shows that early protein folding events may be investigated by using a recently developed family of nonlinear
infrared techniques that combine the high temporal and spatial resolution of multidimensional spectroscopy with the chirality-specific
sensitivity of amide vibrations to structure. We demonstrate how the structural sensitivity of cross-peaks in two-dimensional
correlation plots of chiral signals of an α helix and a β hairpin may be used to clearly resolve structural and dynamical
details undetectable by one-dimensional techniques (e.g. circular dichroism) and identify structures indistinguishable by
NMR.
Co-reporter:Shaul Mukamel;Wei Zhuang
PNAS 2005 102 (39 ) pp:13717-13718
Publication Date(Web):2005-09-27
DOI:10.1073/pnas.0506874102
Co-reporter:Wei Zhuang;Darius Abramavicius
PNAS 2005 102 (21 ) pp:7443-7448
Publication Date(Web):2005-05-24
DOI:10.1073/pnas.0408781102
The response of proteins to sequences of femtosecond infrared pulses provides a multidimensional view into their equilibrium
distribution of structures and snapshot pictures of fast-triggered dynamical events. Analyzing these experiments requires
advanced computational tools for assigning regions in the resulting multi-dimensional correlation plots to specific secondary-structure
elements and their couplings. A differential sensitivity analysis technique based on a perturbation of the local (real space)
Hamiltonian is developed to achieve that goal. Application to the amide I region of a small globular protein reveals regions
associated with the α-helix, β-sheet, and their coupling. Comparison of signals generated in different directions shows that
the double-quantum-coherence signal has a higher sensitivity to the couplings compared with the single-quantum-coherence (photon
echo) technique.
Co-reporter:Luke Campbell, Satoshi Tanaka, Shaul Mukamel
Chemical Physics 2004 Volume 299(2–3) pp:225-231
Publication Date(Web):19 April 2004
DOI:10.1016/j.chemphys.2003.08.032
Co-reporter:Tomoyuki Hayashi
Israel Journal of Chemistry 2004 Volume 44(Issue 1‐3) pp:185-191
Publication Date(Web):8 MAR 2010
DOI:10.1560/M6NA-F16J-NVKN-LLM9
The structures of all stable tautomers of guanine·cytosine and adenine·thymine Watson-Crick base pairs produced by single and double proton transfers from the most stable structures are optimized using density functional theory (DFT) at the B3LYP/6–31G(d,p) level. The zwitterionic tautomer of guanine·cyosine is stable in a high-dielectric medium (water), but not in the gas phase. Normal mode calculations show that the infrared peak positions and intensities of the carbonyl, N-H, and O-H stretching modes are sensitive to the tautomer geometries.
Co-reporter:Andrew Moran
PNAS 2004 Volume 101 (Issue 2 ) pp:506-510
Publication Date(Web):2004-01-13
DOI:10.1073/pnas.2533089100
Electrostatic (through-space) and covalent (through-bond) contributions to couplings involving the CO and C—N vibrational stretching modes of the amide group in the α-helix and the parallel and antiparallel β-sheet structures
of alanine polypeptides are analyzed. Coupling constants computed at the density functional theory level are compared with
the transition dipole coupling model and the complete electrostatic interaction between transition densities. We find that
the transition densities of CO modes are localized, and the electrostatic mechanism then holds. In contrast, the C—N mode transition densities are delocalized,
and covalent contributions to the coupling are significant.
Co-reporter:U. Harbola, S. Mukamel
Physics Reports (September 2008) Volume 465(Issue 5) pp:191-222
Publication Date(Web):1 September 2008
DOI:10.1016/j.physrep.2008.05.003
Nonequilibrium Green’s functions provide a powerful tool for computing the dynamical response and particle exchange statistics of coupled quantum systems. We formulate the theory in terms of the density matrix in Liouville space and introduce superoperator algebra that greatly simplifies the derivation and the physical interpretation of all quantities. Expressions for various observables are derived directly in real time in terms of superoperator nonequilibrium Green’s functions (SNGF), rather than the artificial time-loop required in Schwinger’s Hilbert-space formulation. Applications for computing interaction energies, charge densities, average currents, current induced fluorescence, electroluminescence and current fluctuation (electron counting) statistics are discussed.
Co-reporter:Kochise Bennett, Shaul Mukamel
Chemical Physics (20 December 2016) Volume 481() pp:
Publication Date(Web):20 December 2016
DOI:10.1016/j.chemphys.2016.07.001
In the semiclassical theory of multidimensional spectroscopy, which describes a classical field coupled to quantum matter, n-th order signals are calculated as a convolution of n spectral field envelopes and an n-th order matter response function which enforces the time-ordering of interactions. In quantum field spectroscopy, the electromagnetic field is in a nonclassical state and the product of spectral field envelopes is replaced by a correlation function of the associated field operators. In this paper, we introduce a complementary representation in which the roles of field and matter are interchanged and signals are given by an ordinary, correlation function of matter convoluted with a time-ordered response function of the field. This suggests an inverse spectroscopy which uses matter to probe the state of the field.
Co-reporter:Shaul Mukamel
Chemical Physics (20 December 2016) Volume 481() pp:3-4
Publication Date(Web):20 December 2016
DOI:10.1016/j.chemphys.2016.11.009
Co-reporter:Dmitri V. Voronine, Darius Abramavicius, Shaul Mukamel
Biophysical Journal (15 November 2008) Volume 95(Issue 10) pp:
Publication Date(Web):15 November 2008
DOI:10.1529/biophysj.108.134387
Two-dimensional electronic chirality-induced signals of excitons in the photosynthetic Fenna-Matthews-Olson complex from two species of green sulfur bacteria (Chlorobium tepidum and Prosthecochloris aestuarii) are compared. The spectra are predicted to provide sensitive probes of local protein environment of the constituent bacteriochlorophyll a chromophores and reflect electronic structure variations (site energies and couplings) of the two complexes. Pulse polarization configurations are designed that can separate the coherent and incoherent exciton dynamics contributions to the two-dimensional spectra.
Co-reporter:Darius Abramavicius, Dmitri V. Voronine, Shaul Mukamel
Biophysical Journal (1 May 2008) Volume 94(Issue 9) pp:
Publication Date(Web):1 May 2008
DOI:10.1529/biophysj.107.123455
Spectroscopic studies of light harvesting and the subsequent energy conversion in photosynthesis can track quantum dynamics happening on the microscopic level. The Fenna-Matthews-Olson complex of the photosynthetic green sulfur bacteria Chlorobium tepidum is a prototype efficient light-harvesting antenna: it stores the captured photon energy in the form of excitons (collective excitations), which are subsequently converted to chemical energy with almost 100% efficiency. These excitons show an elaborate relaxation pattern involving coherent and incoherent pathways. We make use of the complex chirality and fundamental symmetries of multidimensional optical signals to design new sequences of ultrashort laser pulses that can distinguish between coherent quantum oscillations and incoherent energy dissipation during the exciton relaxation. The cooperative dynamical features, which reflect the coherent nature of excitations, are amplified. The extent of quantum oscillations and their timescales in photosynthesis can be readily extracted from the designed signals, showing that cooperativity is maintained during energy transport in the Fenna-Matthews-Olson complex. The proposed pulse sequences may also be applied to reveal information on the robustness of quantum states in the presence of fluctuating environments in other nanoscopic complexes and devices.
Co-reporter:Jun Jiang and Shaul Mukamel
Physical Chemistry Chemical Physics 2011 - vol. 13(Issue 6) pp:NaN2400-2400
Publication Date(Web):2010/12/06
DOI:10.1039/C0CP02047H
We report a first principles study of two dimensional electronic spectroscopy of aromatic side chain transitions in the 32-residue β-amyloid (Aβ9–40) fibrils in the near ultraviolet (250–300 nm). An efficient exciton Hamiltonian with electrostatic fluctuations (EHEF) algorithm is used to compute the electronic excitations in the presence of environmental fluctuations. The through-space inter- and intra-molecular interactions are calculated with high level quantum mechanics (QM) approaches, and interfaced with molecular mechanics (MM) simulations. Distinct two dimensional near ultraviolet (2DNUV) spectroscopic signatures are identified for different aromatic transitions, and the couplings between them. 2DNUV signals associated with the transition couplings are shown to be very sensitive to the change of residue-residue interactions induced by residue mutations. Our simulations suggest that 2DNUV spectra could provide a useful local probe for the structure and kinetics of fibrils.
Co-reporter:Hao Ren, Zaizhi Lai, Jason D. Biggs, Jin Wang and Shaul Mukamel
Physical Chemistry Chemical Physics 2013 - vol. 15(Issue 44) pp:NaN19464-19464
Publication Date(Web):2013/09/20
DOI:10.1039/C3CP51347E
We report a combined molecular dynamics (MD) and ab initio simulation study of the ultrafast broadband ultraviolet (UV) stimulated resonance Raman (SRR) spectra of the Trp-cage mini protein. Characteristic two dimensional (2D) SRR features of various folding states are identified. Structural fluctuations erode the cross peaks and the correlation between diagonal peaks is a good indicator of the α-helix formation.
Co-reporter:Konstantin E. Dorfman, Benjamin P. Fingerhut and Shaul Mukamel
Physical Chemistry Chemical Physics 2013 - vol. 15(Issue 29) pp:NaN12359-12359
Publication Date(Web):2013/05/29
DOI:10.1039/C3CP51117K
Vibrational motions in electronically excited states can be observed either by time and frequency resolved infrared absorption or by off resonant stimulated Raman techniques. Multipoint correlation function expressions are derived for both signals. Three representations which suggest different simulation protocols for the signals are developed. These are based on the forward and the backward propagation of the wavefunction, sum over state expansion using an effective vibrational Hamiltonian or a semiclassical treatment of a bath. We show that the effective temporal (Δt) and spectral (Δω) resolution of the techniques is not controlled solely by experimental knobs but also depends on the system dynamics being probed. The Fourier uncertainty ΔωΔt > 1 is never violated.
Co-reporter:Yu Zhang, Jason D. Biggs, Weijie Hua, Konstantin E. Dorfman and Shaul Mukamel
Physical Chemistry Chemical Physics 2014 - vol. 16(Issue 44) pp:NaN24331-24331
Publication Date(Web):2014/09/19
DOI:10.1039/C4CP03361B
We investigate computationally the valence electronic excitations of the amino acid glycine prepared by a sudden nitrogen core ionization induced by an attosecond X-ray pump pulse. The created superposition of cationic excited states is probed by two-dimensional transient X-ray absorption and by three dimensional attosecond stimulated X-ray Raman signals. The latter, generated by applying a second broadband X-ray pulse combined with a narrowband pulse tuned to the carbon K-edge, reveal the complex coupling between valence and core-excited manifolds of the cation.
Co-reporter:Weijie Hua, Kochise Bennett, Yu Zhang, Yi Luo and Shaul Mukamel
Chemical Science (2010-Present) 2016 - vol. 7(Issue 9) pp:NaN5933-5933
Publication Date(Web):2016/05/12
DOI:10.1039/C6SC01571A
The multi-configurational self-consistent field method is employed to simulate the two-dimensional all-X-ray double-quantum-coherence (XDQC) spectroscopy, a four-wave mixing signal that provides direct signatures of double core hole (DCH) states. The valence electronic structure is probed by capturing the correlation between the single (SCH) and double core hole states. The state-averaged restricted-active-space self-consistent field (SA-RASSCF) approach is used which can treat the valence, SCH, and DCH states at the same theoretical level, and applies to all types of DCHs (located on one or two atoms, K-edge or L-edge), with both accuracy and efficiency. Orbital relaxation introduced by the core hole(s) and the static electron correlation is properly accounted for. The XDQC process can take place via different intermediate DCH state channels by tuning the pulse frequencies. We simulate the XDQC signals for the three isomers of aminophenol at 8 pulse frequency configurations, covering all DCH pathways involving the N1s and O1s core hole (N1sN1s, O1sO1s and N1sO1s), which reveal different patterns of valence excitations.
Co-reporter:Markus Kowalewski; Kochise Bennett
The Journal of Physical Chemistry Letters () pp:
Publication Date(Web):
DOI:10.1021/acs.jpclett.6b00864
Molecular potential energy surfaces can be actively manipulated by light. This is usually done by strong classical laser light but was recently demonstrated for the quantum field in an optical cavity. The photonic vacuum state of a localized cavity mode can be strongly mixed with the molecular degrees of freedom to create hybrid field-matter states known as polaritons. We simulate the avoided crossing of sodium iodide in a cavity by incorporating the quantized cavity field into the nuclear wave packet dynamics calculation. The quantized field is represented on a numerical grid in quadrature space, thus avoiding the limitations set by the rotating wave approximation (RWA) when the field is expanded in Fock space. This approach allows the investigation of cavity couplings in the vicinity of naturally occurring avoided crossings and conical intersections, which is too expensive in the fock space expansion when the RWA does not apply. Numerical results show how the branching ratio between the covalent and ionic dissociation channels can be strongly manipulated by the optical cavity.
Co-reporter:Benoit Palmieri, Darius Abramavicius and Shaul Mukamel
Physical Chemistry Chemical Physics 2010 - vol. 12(Issue 1) pp:NaN114-114
Publication Date(Web):2009/11/10
DOI:10.1039/B916723D
Recently developed approaches to simulate environment-induced fluctuation effects in two-dimensional (2D) spectroscopy of excitons are compared for the Fenna-Matthews-Olson light-harvesting complex. Fast fluctuations induce population redistribution between exciton energy-levels and raise homogeneous line widths of various peaks in 2D spectra. These effects are easily accounted for in sum-over-eigenstates (SOS) approach and the quasi-particle (QP) scattering approach through relaxation and dephasing rate constants. Slow fluctuations cause correlations of energies at various delay times in 2D photon-echo spectra. These may be calculated either by doing cumulant expansion in SOS techniques or by statistical averaging over static disorder in SOS and QP approaches. We compare the 2D photon-echo signal simulated using two levels of cumulant expansion approaches and two statistical averaging approaches for the same system. These levels differ by the treatment of energy-level correlations at different delay times and give rise to different cross-peak shapes: the cross-peaks retain their original diagonally elongated shapes when correlations are included, while they are more spherically broadened when correlations are neglected. Statistical averaging over disorder give very similar results but requires much higher computational effort. The peak redistribution timescales are very similar for all levels of theory. The spectral signatures at these different levels of theory are compared and simulation cost is estimated. Approaches which do require statistical averaging over disorder are orders of magnitude slower.
.jpg)




















![3,5,9-Trioxa-4-phosphaheptacos-18-en-1-aminium,4-hydroxy-N,N,N-trimethyl-10-oxo-7-[[(9Z)-1-oxo-9-octadecen-1-yl]oxy]-, innersalt, 4-oxide, (7R,18Z)-](http://img.cochemist.com/ccimg/4300/4235-95-4.png)
![3,5,9-Trioxa-4-phosphaheptacos-18-en-1-aminium,4-hydroxy-N,N,N-trimethyl-10-oxo-7-[[(9Z)-1-oxo-9-octadecen-1-yl]oxy]-, innersalt, 4-oxide, (7R,18Z)-](http://img.cochemist.com/ccimg/4300/4235-95-4_b.png)
![(6R,9AR)-OCTAHYDRO-2H-PYRIDO[1,2-A]PYRAZIN-6-YLMETHANOL](http://img.cochemist.com/ccimg/13700/13699-48-4.png)
![(6R,9AR)-OCTAHYDRO-2H-PYRIDO[1,2-A]PYRAZIN-6-YLMETHANOL](http://img.cochemist.com/ccimg/13700/13699-48-4_b.png)