Co-reporter:Kevin L. Vickerman and Levi M. Stanley
Organic Letters October 6, 2017 Volume 19(Issue 19) pp:
Publication Date(Web):September 21, 2017
DOI:10.1021/acs.orglett.7b02230
Enantioselective synthesis of polycyclic nitrogen, oxygen, and sulfur heterocycles by rhodium-catalyzed intramolecular alkene hydroacylation is reported. The intramolecular hydroacylation reactions generate 1,4-dihydrocyclopenta[b]indol-3(2H)-ones and 3,4-dihydrocyclopenta[b]indol-1(2H)-one in moderate-to-high yields (65–99%) with good-to-excellent enantioselectivities (84–99% ee). The catalyst system also promotes alkene hydroacylation of 3-vinylfuran-, 3-vinylbenzothiophene-, and 3-vinylthiophene-2-carboxaldehydes to generate the corresponding ketone products in moderate-to-high yields (71–91% yield) with excellent enantioselectivities (97–99% ee).
Co-reporter:Abhishek A. Kadam, Arkady Ellern, and Levi M. Stanley
Organic Letters August 4, 2017 Volume 19(Issue 15) pp:
Publication Date(Web):July 24, 2017
DOI:10.1021/acs.orglett.7b01825
We report enantioselective, palladium-catalyzed conjugate additions of arylboronic acids to β-aryl, β,β-disubstituted enones to generate ketones containing bis-benzylic quaternary stereocenters. A catalyst generated from palladium trifluoroacetate and (S)-4-tert-butyl-2-(2-pyridyl)oxazoline ligand ((S)-t-BuPyOx) promotes conjugate additions of a wide range of arylboronic acids to a variety of β-aryl, β,β-disubstituted enones. Iterative addition of the arylboronic acid to minimize undesired protodeboronation pathways leads to efficient formation of the corresponding ketones containing bis-benzylic quaternary stereocenters in up to 92% yield and up to 93% enantioselectivity.
Co-reporter:James A. Walker Jr., Kevin L. Vickerman, Jenna N. Humke, and Levi M. Stanley
Journal of the American Chemical Society August 2, 2017 Volume 139(Issue 30) pp:10228-10228
Publication Date(Web):July 14, 2017
DOI:10.1021/jacs.7b06191
We report Ni-catalyzed formal carboacylation of o-allylbenzamides with arylboronic acid pinacol esters. The reaction is triggered by oxidative addition of an activated amide C–N bond to a Ni(0) catalyst and proceeds via alkene insertion into a Ni(II)–acyl bond. The exo-selective carboacylation reaction generates 2-benzyl-2,3-dihydro-1H-inden-1-ones in moderate to high yields (46–99%) from a variety of arylboronic acid pinacol esters and substituted o-allylbenzamides. These results show that amides are practical substrates for alkene carboacylation via amide C–N bond activation, and this approach bypasses challenges associated with alkene carboacylation triggered by C–C bond activation.
Co-reporter:Xinle Li, Ryan Van Zeeland, Raghu V. Maligal-Ganesh, Yuchen Pei, Gregory Power, Levi Stanley, and Wenyu Huang
ACS Catalysis 2016 Volume 6(Issue 9) pp:6324
Publication Date(Web):August 9, 2016
DOI:10.1021/acscatal.6b01753
A series of mixed-linker bipyridyl metal–organic framework (MOF)-supported palladium(II) catalysts were used to elucidate the electronic and steric effects of linker substitution on the activity of these catalysts in the context of Suzuki–Miyaura cross-coupling reactions. m-6,6′-Me2bpy-MOF-PdCl2 exhibited 110- and 496-fold enhancements in activity compared to nonfunctionalized m-bpy-MOF-PdCl2 and m-4,4′-Me2bpy-MOF-PdCl2, respectively. This result clearly demonstrates that the stereoelectronic properties of metal-binding linker units are critical to the activity of single-site organometallic catalysts in MOFs and highlights the importance of linker engineering in the design and development of efficient MOF catalysts.Keywords: bipyridyl linker; heterogeneous catalysis; isoreticular metal−organic frameworks; single-site catalyst; structure−activity relationship; Suzuki−Miyaura cross-coupling
Co-reporter:Avipsa Ghosh, James A. Walker Jr., Arkady Ellern, and Levi M. Stanley
ACS Catalysis 2016 Volume 6(Issue 4) pp:2673
Publication Date(Web):March 15, 2016
DOI:10.1021/acscatal.6b00365
We report a strategy that combines alkene hydroacylation and enantioselective α-(hetero)arylation reactions to form a wide variety of nitrogen-containing heterocyclic ketones bearing α-chiral quaternary stereogenic centers. Exo-selective, intramolecular Ni-catalyzed hydroacylations of N-homoallylindole- and N-homoallylpyrrole-2-carboxaldehydes form α-substituted six-membered heterocyclic ketones in up to 95% yield, while N-heterocyclic carbene (NHC) catalyzed hydroacylations of N-allylindole- and N-allylpyrrole-2-carboxaldehydes form α-substituted five-membered heterocyclic ketones in up to 99% yield. The racemic five- and six-membered products of Ni- and NHC-catalyzed hydroacylation reactions are readily transformed into heterocyclic ketones containing an α-chiral quaternary stereogenic center by enantioselective Ni-catalyzed α-arylation and α-heteroarylation reactions. The chiral, nonracemic products formed through a combination of alkene hydroacylation and α-(hetero)arylation reactions are formed in moderate to high yields (44–99%) with excellent enantioselectivities (typically >95% ee). The identity of the precatalyst for Ni-catalyzed α-(hetero)arylation is dictated by the identity of the α-substituted heterocyclic ketone starting material. α-(Hetero)arylations of six-membered heterocyclic ketones occur at 65–85 °C in the presence of a catalyst generated in situ from Ni(COD)2 and (R)-BINAP or (R)-DIFLUORPHOS. α-(Hetero)arylation of five-membered heterocyclic ketones must be conducted at room temperature in the presence of an [((R)-BINAP)Ni(η2-NC-Ph)] precatalyst or a catalyst generated in situ from Ni(COD)2, (R)-DIFLUORPHOS, and benzonitrile.Keywords: alkene hydroacylation; enantioselective catalysis; heterocycles; ketone α-arylation; quaternary stereocenters
Co-reporter:Anthony L. Gerten, Levi M. Stanley
Tetrahedron Letters 2016 Volume 57(Issue 49) pp:5460-5463
Publication Date(Web):7 December 2016
DOI:10.1016/j.tetlet.2016.10.084
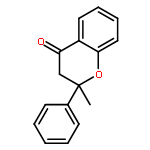
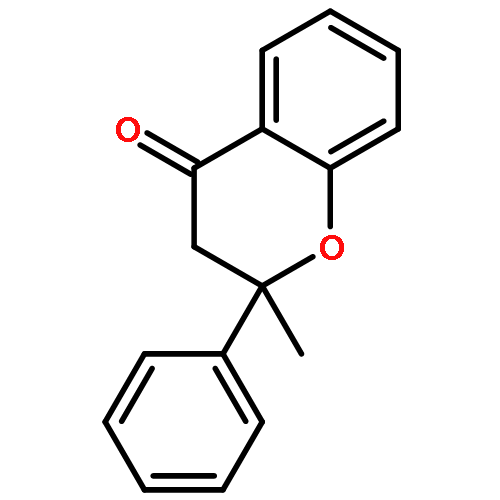
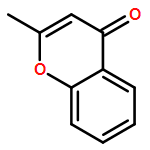
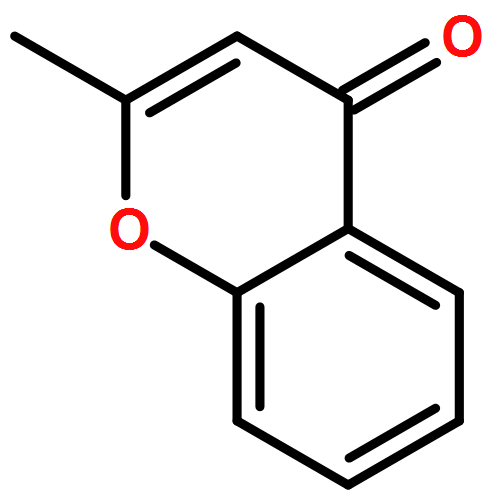
•A catalytic method for synthesis of substituted flavanone derivatives is reported.•The catalyst system minimizes protodeboronation and biaryl formation processes.•The reactions encompass a range of arylboronic acids and 2-alkylchromanones.Palladium-catalyzed conjugate additions of arylboronic acids to 2-alkylchromones are reported. The conjugate additions occur in aqueous media in the presence of a catalyst generated from palladium trifluoroacetate and 1,10-phenanthroline, and the flavanone derivatives containing a fully-substituted carbon center are formed in up to 90% yield.
Co-reporter:Avipsa Ghosh, David T. Bainbridge, and Levi M. Stanley
The Journal of Organic Chemistry 2016 Volume 81(Issue 17) pp:7945-7951
Publication Date(Web):August 5, 2016
DOI:10.1021/acs.joc.6b01730
An enantioselective model synthesis of the 2,3-dihydro-1H-pyrrolo[1,2-a]indole core of the putative structure of yuremamine is reported in 39% overall yield and 96% ee over five steps. The model synthesis leverages enantioselective, rhodium-catalyzed hydroacylation of an N-vinylindole-2-carboxaldehyde as the key step in the installation of the stereochemical triad. An enantioselective synthesis of a densely functionalized dihydropyrroloindolone that maps onto the putative structure of yuremamine is demonstrated in 26% yield and 97% ee over eight steps.
Co-reporter:Ryan Van Zeeland and Levi M. Stanley
ACS Catalysis 2015 Volume 5(Issue 9) pp:5203
Publication Date(Web):August 3, 2015
DOI:10.1021/acscatal.5b01272
Palladium-catalyzed conjugate addition of arylboronic acids to β,β-disubstituted enones in aqueous media is reported. Additions of a wide range of arylboronic acids to β,β-disubstituted enones occur to form ketone products bearing benzylic all-carbon quaternary centers. These reactions are promoted by a simple catalyst prepared from palladium trifluoracetate and 2,2′-bipyridine. The use of aqueous sodium trifluoracetate as the reaction medium significantly enhances reactivity and enables the formation of challenging bis-benzylic and ortho-substituted benzylic all-carbon quaternary centers.Keywords: aqueous media; arylboronic acid; conjugate addition; palladium; quaternary center
Co-reporter:Xiang-Wei Du and Levi M. Stanley
Organic Letters 2015 Volume 17(Issue 13) pp:3276-3279
Publication Date(Web):June 22, 2015
DOI:10.1021/acs.orglett.5b01447
Tandem reactions involving Rh-catalyzed intermolecular hydroacylations of alkynes with salicylaldehydes followed by intramolecular oxo-Michael additions are described for the diastereoselective synthesis of 2,3-disubstituted chroman-4-ones. The tandem hydroacylation/oxo-Michael additions occur to form 2,3-disubstituted chroman-4-ones in high yields from a range of 1,2-disubstituted acetylenes and substituted salicylaldehyes. The resulting 2,3-disubstituted chroman-4-ones are readily fluorinated to form trans-3-fluoro-2,3-disubstituted chroman-4-ones in high yields with excellent diastereoselectivity.
Co-reporter:Kirsten F. Johnson, Adam C. Schmidt, and Levi M. Stanley
Organic Letters 2015 Volume 17(Issue 19) pp:4654-4657
Publication Date(Web):September 23, 2015
DOI:10.1021/acs.orglett.5b02559
The development of a rhodium catalyst for endo- and enantioselective hydroacylation of ortho-allylbenzaldehydes is reported. A catalyst generated in situ from [Rh(COD)Cl]2, (R)-DTBM-SEGPHOS, and NaBARF promotes the desired hydroacylation reactions and minimizes the formation of byproducts from competitive alkene isomerization and ene/dehydration pathways. These rhodium-catalyzed processes generate the 3,4-dihydronaphthalen-1(2H)-one products in moderate-to-high yields (49–91%) with excellent enantioselectivities (96–99% ee).
Co-reporter:Xiang-Wei Du, Avipsa Ghosh, and Levi M. Stanley
Organic Letters 2014 Volume 16(Issue 15) pp:4036-4039
Publication Date(Web):July 14, 2014
DOI:10.1021/ol501869s
Catalytic, enantioselective hydroacylations of N-allylindole-2-carboxaldehydes and N-allylpyrrole-2-carboxaldehydes are reported. In contrast to many alkene hydroacylations that form six-membered rings, these annulative processes occur in the absence of ancillary functionality to stabilize the acylrhodium(III) hydride intermediate. The intramolecular hydroacylation reactions generate 7,8-dihydropyrido[1,2-a]indol-9(6H)ones and 6,7-dihydroindolizin-8(5H)-ones in moderate to high yields with excellent enantioselectivities.
Co-reporter:Avipsa Ghosh and Levi M. Stanley
Chemical Communications 2014 vol. 50(Issue 21) pp:2765-2768
Publication Date(Web):24 Jan 2014
DOI:10.1039/C4CC00210E
We report catalytic, enantioselective intramolecular hydroacylation of N-vinylindole-2-carboxaldehydes. These hydroacylation reactions occur in the presence of a readily accessible rhodium catalyst and form chiral, non-racemic 2,3-dihydro-1H-pyrrolo[1,2-a]indol-1-ones in high yields with excellent enantioselectivities.
Co-reporter:Kirsten F. Johnson, Ryan Van Zeeland, and Levi M. Stanley
Organic Letters 2013 Volume 15(Issue 11) pp:2798-2801
Publication Date(Web):May 28, 2013
DOI:10.1021/ol4011344
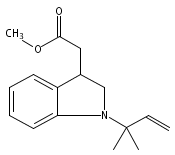
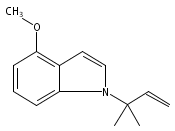
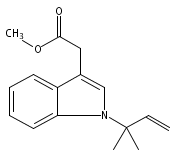
Palladium-catalyzed N-tert-prenylations of indoles, tricarbonylchromium-activated indoles, and indolines that occur in high yields (up to 94%) with high tert-prenyl-to-n-prenyl selectivity (up to 12:1) are reported.
Co-reporter:Anthony L. Gerten, Michael C. Slade, Kelsie M. Pugh and Levi M. Stanley
Organic & Biomolecular Chemistry 2013 vol. 11(Issue 45) pp:7834-7837
Publication Date(Web):09 Oct 2013
DOI:10.1039/C3OB41815D
Catalytic, enantioselective 1,3-dipolar cycloadditions of nitrile imines with methyleneindolinones are reported. The spiro[pyrazolin-3,3′-oxindole] products are formed in good yields (up to 98%) and high enantioselectivity (up to 99% ee).
Co-reporter:Avipsa Ghosh and Levi M. Stanley
Chemical Communications 2014 - vol. 50(Issue 21) pp:NaN2768-2768
Publication Date(Web):2014/01/24
DOI:10.1039/C4CC00210E
We report catalytic, enantioselective intramolecular hydroacylation of N-vinylindole-2-carboxaldehydes. These hydroacylation reactions occur in the presence of a readily accessible rhodium catalyst and form chiral, non-racemic 2,3-dihydro-1H-pyrrolo[1,2-a]indol-1-ones in high yields with excellent enantioselectivities.
Co-reporter:Anthony L. Gerten and Levi M. Stanley
Inorganic Chemistry Frontiers 2016 - vol. 3(Issue 3) pp:NaN343-343
Publication Date(Web):2016/01/04
DOI:10.1039/C5QO00346F
Catalytic, enantioselective [3 + 2] cycloadditions of azomethine ylides derived from alanine imino esters with 3-nitroindoles are reported. The dearomative cycloaddition reactions occur in the presence of a catalyst generated in situ from Cu(OTf)2 and (R)-Difluorphos to form exo′-pyrroloindoline cycloadducts and establish four contiguous stereogenic centers, two of which are fully substituted. The exo′-pyrroloindoline products are formed in moderate-to-good yields (39–85%) with high diastereoselectivities (up to 98:1:1 dr) and enantioselectivities (up to 96% ee).
Co-reporter:Avipsa Ghosh, Kirsten F. Johnson, Kevin L. Vickerman, James A. Walker and Levi M. Stanley
Inorganic Chemistry Frontiers 2016 - vol. 3(Issue 5) pp:NaN644-644
Publication Date(Web):2016/02/11
DOI:10.1039/C6QO00023A
This highlight discusses developments in transition metal-catalysed alkene and alkyne hydroacylation reactions over the past three years. The discussion summarizes the development of new catalyst systems for alkene and alkyne hydroacylation and applications to the synthesis of important ketone building blocks. The highlight captures transition metal-catalysed alkene and alkyne hydroacylation at a time of impressive growth when cobalt, nickel, ruthenium, and iridium catalysts are emerging as complements or replacements for traditional rhodium catalysts.
Co-reporter:Anthony L. Gerten, Michael C. Slade, Kelsie M. Pugh and Levi M. Stanley
Organic & Biomolecular Chemistry 2013 - vol. 11(Issue 45) pp:NaN7837-7837
Publication Date(Web):2013/10/09
DOI:10.1039/C3OB41815D
Catalytic, enantioselective 1,3-dipolar cycloadditions of nitrile imines with methyleneindolinones are reported. The spiro[pyrazolin-3,3′-oxindole] products are formed in good yields (up to 98%) and high enantioselectivity (up to 99% ee).
Co-reporter:James A. Walker and Levi M. Stanley
Organic & Biomolecular Chemistry 2016 - vol. 14(Issue 42) pp:NaN9984-9984
Publication Date(Web):2016/09/28
DOI:10.1039/C6OB01956K
We report catalytic, intramolecular hydroacylations of N-allylimidazole-2-carboxaldehydes and N-allylbenzimidazole-2-carboxaldehydes. These exo-selective hydroacylations occur in the presence of a N-heterocyclic carbene catalyst to generate 5,6-dihydro-7H-pyrrolo[1,2-α]imidazol-7-ones and 1,2-dihydro-3H-benzo[d]pyrrolo[1,2-α]imidazol-2-ones in high yields (66–99%). In addition, hydroacylations of N-allylimidazole-2-carboxaldehydes in the presence of a chiral, non-racemic NHC catalyst occur, forming 5,6-dihydro-7H-pyrrolo[1,2-α]imidazol-7-ones in moderate-to-high yields (39–98%) with modest enantioselectivities (56–79% ee).
.jpg)
![[2,2'-Bipyridine]-5,5'-dicarboxylic acid, 6,6'-dimethyl-, 5,5'-dimethyl ester](/data/chemimg/3721400/2050905-18-3.png)
![[2,2'-Bipyridine]-5,5'-dicarboxylic acid, 6,6'-dimethyl-, 5,5'-dimethyl ester](/data/chemimg/3721400/2050905-18-3_b.png)
![[2,2'-Bipyridine]-5,5'-dicarboxylic acid, 4,4'-dimethyl-](/data/chemimg/3721400/2027535-09-5.png)
![[2,2'-Bipyridine]-5,5'-dicarboxylic acid, 4,4'-dimethyl-](/data/chemimg/3721400/2027535-09-5_b.png)
![[2,2'-Bipyridine]-5,5'-dicarboxylic acid, 4,4'-dimethyl-, 5,5'-dimethyl ester](/data/chemimg/3721400/2027535-08-4.png)
![[2,2'-Bipyridine]-5,5'-dicarboxylic acid, 4,4'-dimethyl-, 5,5'-dimethyl ester](/data/chemimg/3721400/2027535-08-4_b.png)










![[2,2'-Bipyridine]-5,5'-dicarboxylic acid, 6,6'-dimethyl-](/data/chemimg/3721200/1190209-37-0.png)
![[2,2'-Bipyridine]-5,5'-dicarboxylic acid, 6,6'-dimethyl-](/data/chemimg/3721200/1190209-37-0_b.png)










![7,8-dihydro-8-methyl-Pyrido[1,2-a]indol-9(6H)-one](http://img.cochemist.com/ccimg/673000/672903-41-2.png)
![7,8-dihydro-8-methyl-Pyrido[1,2-a]indol-9(6H)-one](http://img.cochemist.com/ccimg/673000/672903-41-2_b.png)








![2,3-dihydro-2-methyl-1H-Pyrrolo[1,2-a]indol-1-one](http://img.cochemist.com/ccimg/524700/524689-88-1.png)
![2,3-dihydro-2-methyl-1H-Pyrrolo[1,2-a]indol-1-one](http://img.cochemist.com/ccimg/524700/524689-88-1_b.png)




![1H-Pyrrole-2-carboxaldehyde, 1-[(2E)-3-phenyl-2-propenyl]-](http://img.cochemist.com/ccimg/389700/389621-00-5.png)
![1H-Pyrrole-2-carboxaldehyde, 1-[(2E)-3-phenyl-2-propenyl]-](http://img.cochemist.com/ccimg/389700/389621-00-5_b.png)
![BENZYL OCTAHYDRO-4H-PYRROLO[3,2-B]PYRIDINE-4-CARBOXYLATE](http://img.cochemist.com/ccimg/300800/300706-95-0.png)
![BENZYL OCTAHYDRO-4H-PYRROLO[3,2-B]PYRIDINE-4-CARBOXYLATE](http://img.cochemist.com/ccimg/300800/300706-95-0_b.png)












![1H-Indole,3-[2-[[(1,1-dimethylethyl)dimethylsilyl]oxy]ethyl]-1-(phenylsulfonyl)-](http://img.cochemist.com/ccimg/141000/140920-82-7.png)
![1H-Indole,3-[2-[[(1,1-dimethylethyl)dimethylsilyl]oxy]ethyl]-1-(phenylsulfonyl)-](http://img.cochemist.com/ccimg/141000/140920-82-7_b.png)



![Benzene, 1-[1-(bromomethyl)ethenyl]-4-fluoro-](http://img.cochemist.com/ccimg/133000/132927-05-0.png)
![Benzene, 1-[1-(bromomethyl)ethenyl]-4-fluoro-](http://img.cochemist.com/ccimg/133000/132927-05-0_b.png)
![2H-Imidazol-2-ylidene,1,3-dihydro-1,3-bis(tricyclo[3.3.1.13,7]dec-1-yl)-](http://img.cochemist.com/ccimg/131100/131042-77-8.png)
![2H-Imidazol-2-ylidene,1,3-dihydro-1,3-bis(tricyclo[3.3.1.13,7]dec-1-yl)-](http://img.cochemist.com/ccimg/131100/131042-77-8_b.png)




![1-Allyl-1H-benzo[d]imidazole-2-carbaldehyde](http://img.cochemist.com/ccimg/118500/118482-14-7.png)
![1-Allyl-1H-benzo[d]imidazole-2-carbaldehyde](http://img.cochemist.com/ccimg/118500/118482-14-7_b.png)




![Tert-butyl-[2-(1h-indol-3-yl)ethoxy]-dimethylsilane](http://img.cochemist.com/ccimg/101100/101079-48-5.png)
![Tert-butyl-[2-(1h-indol-3-yl)ethoxy]-dimethylsilane](http://img.cochemist.com/ccimg/101100/101079-48-5_b.png)


![1H-INDOLE, 5-BROMO-1-[(4-METHYLPHENYL)SULFONYL]-](http://img.cochemist.com/ccimg/96600/96546-77-9.png)
![1H-INDOLE, 5-BROMO-1-[(4-METHYLPHENYL)SULFONYL]-](http://img.cochemist.com/ccimg/96600/96546-77-9_b.png)
![(6-Formylbenzo[d][1,3]dioxol-5-yl)boronic acid](http://img.cochemist.com/ccimg/94900/94838-88-7.png)
![(6-Formylbenzo[d][1,3]dioxol-5-yl)boronic acid](http://img.cochemist.com/ccimg/94900/94838-88-7_b.png)



















































![5,6-Dihydro[1,1'-biphenyl]-3(4H)-one](http://img.cochemist.com/ccimg/10400/10345-87-6.png)
![5,6-Dihydro[1,1'-biphenyl]-3(4H)-one](http://img.cochemist.com/ccimg/10400/10345-87-6_b.png)



























![Benzene, 1-[(1E)-3-bromo-1-propen-1-yl]-4-chloro-](/data/chemimg/1874500/60691-90-9.png)
![Benzene, 1-[(1E)-3-bromo-1-propen-1-yl]-4-chloro-](/data/chemimg/1874500/60691-90-9_b.png)
![Benzene, 1-[(1E)-3-bromo-1-propen-1-yl]-4-methoxy-](/data/chemimg/926100/16034-99-4.png)
![Benzene, 1-[(1E)-3-bromo-1-propen-1-yl]-4-methoxy-](/data/chemimg/926100/16034-99-4_b.png)


















































































































































































































![2H-Indol-2-one, 3-[(4-bromophenyl)methylene]-1,3-dihydro-, (3E)-](http://img.cochemist.com/ccimg/90900/90828-11-8.png)
![2H-Indol-2-one, 3-[(4-bromophenyl)methylene]-1,3-dihydro-, (3E)-](http://img.cochemist.com/ccimg/90900/90828-11-8_b.png)




![2H-INDOL-2-ONE, 1,3-DIHYDRO-3-[(3-METHOXYPHENYL)METHYLENE]-, (3E)-](http://img.cochemist.com/ccimg/50900/50882-27-4.png)
![2H-INDOL-2-ONE, 1,3-DIHYDRO-3-[(3-METHOXYPHENYL)METHYLENE]-, (3E)-](http://img.cochemist.com/ccimg/50900/50882-27-4_b.png)


![Benzaldehyde, 2-phenylhydrazone, [C(E)]-](/data/chemimg/3730200/41097-65-8.png)
![Benzaldehyde, 2-phenylhydrazone, [C(E)]-](/data/chemimg/3730200/41097-65-8_b.png)


![2H-Indol-2-one, 3-[(4-chlorophenyl)methylene]-1,3-dihydro-, (3E)-](http://img.cochemist.com/ccimg/29600/29551-52-8.png)
![2H-Indol-2-one, 3-[(4-chlorophenyl)methylene]-1,3-dihydro-, (3E)-](http://img.cochemist.com/ccimg/29600/29551-52-8_b.png)
![2H-Indol-2-one, 1,3-dihydro-3-[(4-methoxyphenyl)methylene]-, (3E)-](http://img.cochemist.com/ccimg/29600/29551-48-2.png)
![2H-Indol-2-one, 1,3-dihydro-3-[(4-methoxyphenyl)methylene]-, (3E)-](http://img.cochemist.com/ccimg/29600/29551-48-2_b.png)
![1H-Isoindole-1,3(2H)-dione,2-[2-(1H-indol-3-yl)ethyl]-](http://img.cochemist.com/ccimg/15800/15741-71-6.png)
![1H-Isoindole-1,3(2H)-dione,2-[2-(1H-indol-3-yl)ethyl]-](http://img.cochemist.com/ccimg/15800/15741-71-6_b.png)