Co-reporter:Rasika S. Phansalkar, Charlotte Simmler, Jonathan Bisson, Shao-Nong Chen, David C. Lankin, James B. McAlpine, Matthias Niemitz, and Guido F. Pauli
Journal of Natural Products March 24, 2017 Volume 80(Issue 3) pp:634-634
Publication Date(Web):January 9, 2017
DOI:10.1021/acs.jnatprod.6b00923
Chemical standardization, along with morphological and DNA analysis ensures the authenticity and advances the integrity evaluation of botanical preparations. Achievement of a more comprehensive, metabolomic standardization requires simultaneous quantitation of multiple marker compounds. Employing quantitative 1H NMR (qHNMR), this study determined the total isoflavone content (TIfCo; 34.5–36.5% w/w) via multimarker standardization and assessed the stability of a 10-year-old isoflavone-enriched red clover extract (RCE). Eleven markers (nine isoflavones, two flavonols) were targeted simultaneously, and outcomes were compared with LC-based standardization. Two advanced quantitative measures in qHNMR were applied to derive quantities from complex and/or overlapping resonances: a quantum mechanical (QM) method (QM-qHNMR) that employs 1H iterative full spin analysis, and a non-QM method that uses linear peak fitting algorithms (PF-qHNMR). A 10 min UHPLC-UV method provided auxiliary orthogonal quantitation. This is the first systematic evaluation of QM and non-QM deconvolution as qHNMR quantitation measures. It demonstrates that QM-qHNMR can account successfully for the complexity of 1H NMR spectra of individual analytes and how QM-qHNMR can be built for mixtures such as botanical extracts. The contents of the main bioactive markers were in good agreement with earlier HPLC-UV results, demonstrating the chemical stability of the RCE. QM-qHNMR advances chemical standardization by its inherent QM accuracy and the use of universal calibrants, avoiding the impractical need for identical reference materials.
Co-reporter:Yang Liu, J. Brent Friesen, Edyta M. Grzelak, Qingfei Fan, Ting Tang, Kemal Durić, Birgit U. Jaki, James B. McAlpine, Scott G. Franzblau, Shao-Nong Chen, Guido F. Pauli
Journal of Chromatography A 2017 Volume 1504(Volume 1504) pp:
Publication Date(Web):30 June 2017
DOI:10.1016/j.chroma.2017.04.055
•TLC method of solvent system selection for countercurrent separation is revisited.•A broader approach to the GUESS method is implemented.•The theory supporting TLC and CCS sweet spot matching is presented.•Application of the GUESS method to natural product separation is demonstrated.TLC-based strategies were proposed in 1979 (Hostettmann et al.) and 2005 (Friesen & Pauli; GUESS method) to minimize the number of partitioning experiments required for countercurrent separation (CCS) solvent system selection. As semi-empirical approaches, both proposed that the K values defining the sweet spot of optimal CCS corresponded to a matching Rf value range from the silica gel TLC plate developed in the organic phase of a biphasic or a corresponding monophasic solvent system. Despite their simplicity, there has been an absence of theoretical support and a deficiency of reported experimental evidence. The present study explores the theory required to develop correlations between Rf and K. All theoretical models surmise that the optimal Rf value range should be centered at 0.5. In order to validate the feasibility of the concept of matching Rf and K values, 43 natural products and six solvent system families were investigated. Out of 62 correlations, 45 resulted in matched Rf and K values. Based on this study, practical guidelines for the TLC-based prediction strategy are provided. These approaches will equip CCS users with an updated understanding of how to apply the TLC-based solvent system selection strategy to accelerate a targeted selection of CCS conditions.
Co-reporter:
Magnetic Resonance in Chemistry 2017 Volume 55(Issue 3) pp:239-244
Publication Date(Web):2017/03/01
DOI:10.1002/mrc.4425
Keywords:Oligopeptides;1H iterative Full Spin Analysis (HiFSA);Ecumicin;Computational Iteration;Structure elucidation
Co-reporter:Joo-Won Nam, Rasika S. Phansalkar, David C. Lankin, James B. McAlpine, Ariene A. Leme-Kraus, Cristina M. P. Vidal, Li-She Gan, Ana Bedran-Russo, Shao-Nong ChenGuido F. Pauli
The Journal of Organic Chemistry 2017 Volume 82(Issue 3) pp:
Publication Date(Web):January 18, 2017
DOI:10.1021/acs.joc.6b02161
The structurally complex oligomeric proanthocyanidins (OPACs) are promising biomimetic agents, capable of strengthening the macromolecular backbone of teeth via intermolecular and intermicrofibrillar cross-linking. This study establishes analytical methods capable of determining the absolute configuration of the catechin-type monomeric units of underivatized OPACs. This preserves the capacity of their biological evaluation, aimed at understanding the inevitably stereospecific interactions between the OPACs and dentin collagen. Guided by dental bioassays (modulus of elasticity, long-term stability), two new trimeric and tetrameric A-type OPACs were discovered as dentin biomodifiers from pine (Pinus massoniana) bark: epicatechin-(2β→O→7,4β→8)-epicatechin-(2β→O→7,4β→8)-catechin (5) and epicatechin-(2β→O→7,4β→8)-epicatechin-(2β→O→7,4β→6)-epicatechin-(2β→O→7,4β→8)-catechin (6), respectively. Combining 1D/2D NMR, HRESIMS, ECD, 1H iterative full spin analysis (HiFSA), and gauge-invariant atomic orbital (GIAO) δ calculations, we demonstrate how 13C NMR chemical shifts (diastereomeric building blocks (A-type dimers)) empower the determination of the absolute configuration of monomeric units in the higher oligomers 5 and 6. Collectively, NMR with ECD reference data elevates the level of structural information achievable for these structurally demanding molecules when degradation analysis is to be avoided. Considering their numerous and deceptively subtle, but 3D impactful, structural variations, this advances the probing of OPAC chemical spaces for species that bind selectively to collagenous and potentially other biologically important biomacromolecules.
Co-reporter:J. Brent Friesen, James B. McAlpine, Shao-Nong Chen, Guido F. Pauli
Journal of Chromatography A 2017 Volume 1520(Volume 1520) pp:
Publication Date(Web):20 October 2017
DOI:10.1016/j.chroma.2017.08.077
•This is a review of the 9th International Countercurrent Chromatography Conference.•Social Events, Workshop, Scientific Program and Awards were summarized.•A total of 100 participants from 16 countries attended CCC 2016.•Includes a brief review of all articles in the CCC 2016 Special Virtual JCA Issue.•The scientific program covers instrument design, basic science and applications.The 9th International Countercurrent Chromatography Conference (CCC 2016) was held at Dominican University near Chicago, IL (USA), from August 1st–3rd, 2016. The biennial CCC 20XX conferences provide an opportunity for countercurrent chromatography and centrifugal partition chromatography (CCC/CPC) manufactures, marketers, theorists, and research scientists to gather together socially, learn from each other, and advance countercurrent separation technology. A synopsis of the conference proceedings as well as a series of short reviews of the special edition articles is included in this document. Many productive discussions and collegial conversation at CCC 2016 attested to the liveliness, connectivity, and productivity of the global countercurrent research community and bodes well for the success of the 10th conference at the University of Braunschweig, Germany on August 1–3, 2018.
Co-reporter:Jonathan Bisson; James B. McAlpine; J. Brent Friesen; Shao-Nong Chen; James Graham
Journal of Medicinal Chemistry 2016 Volume 59(Issue 5) pp:1671-1690
Publication Date(Web):October 27, 2015
DOI:10.1021/acs.jmedchem.5b01009
High-throughput biology has contributed a wealth of data on chemicals, including natural products (NPs). Recently, attention was drawn to certain, predominantly synthetic, compounds that are responsible for disproportionate percentages of hits but are false actives. Spurious bioassay interference led to their designation as pan-assay interference compounds (PAINS). NPs lack comparable scrutiny, which this study aims to rectify. Systematic mining of 80+ years of the phytochemistry and biology literature, using the NAPRALERT database, revealed that only 39 compounds represent the NPs most reported by occurrence, activity, and distinct activity. Over 50% are not explained by phenomena known for synthetic libraries, and all had manifold ascribed bioactivities, designating them as invalid metabolic panaceas (IMPs). Cumulative distributions of ∼200,000 NPs uncovered that NP research follows power-law characteristics typical for behavioral phenomena. Projection into occurrence–bioactivity–effort space produces the hyperbolic black hole of NPs, where IMPs populate the high-effort base.
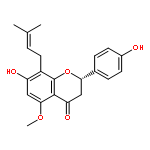
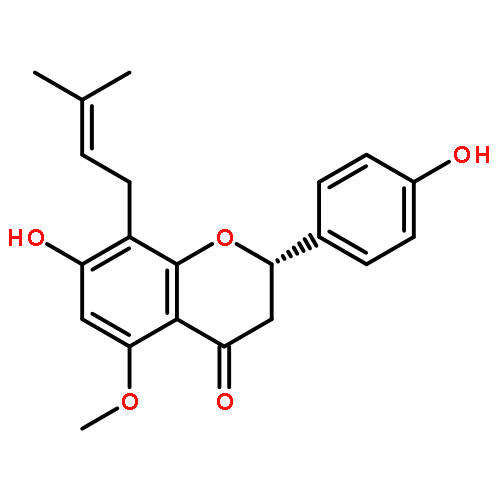
Co-reporter:Ayano Imai; David C. Lankin; Dejan Nikolić; Soyoun Ahn; Richard B. van Breemen; Norman R. Farnsworth; James B. McAlpine; Shao-Nong Chen
Journal of Natural Products 2016 Volume 79(Issue 3) pp:541-554
Publication Date(Web):January 13, 2016
DOI:10.1021/acs.jnatprod.5b00927
Investigating the phytochemical equivalence of the aerial parts of Actaea racemosa (syn. Cimicifuga racemosa) relative to the widely used roots/rhizomes, this study provides a perspective for the potential use of renewable (“green”) plant parts as a source of black cohosh botanical preparations. In addition to the characterization of Nω-methylserotonin as one representative marker of the Actaea alkaloids, nine cycloartane triterpenes were isolated and characterized, including the two new triterpene glycosides (1S,15R)-1,15,25-trihydroxy-3-O-β-d-xylopyranosyl-acta-(16S,23R,24R)-16,23;16,24-binoxoside (1) and 3-O-α-l-arabinopyranosyl-(1S,24R)-1,24,25-trihydroxy-15-oxo-acta-(16R,23R)-16,23-monoxoside (2). Their structures were elucidated by spectroscopic data interpretation. The relative configuration of 1 was deduced by 1H iterative full-spin analysis (HiFSA), making it the first example of the complete analysis of the complex 1H NMR spectrum of a triterpene glycoside. In addition to the new compounds 1 and 2, the aerial plant parts were shown to contain the previously known binoxosides 3, 4, 6, and 7, the monoxoside 8, and the binoxols 5 and 9. Overall, the metabolome of the aerial plant parts consists of a variety of Actaea triterpenes, similar to those found in roots/rhizomes, a tendency toward C-1 and C-7 hydroxylation of the cycloartanol skeleton, a greater abundance of aglycones, and the presence of comparable amounts of Nω-methylserotonin.
Co-reporter:Guido F. Pauli, Matthias Niemitz, Jonathan Bisson, Michael W. Lodewyk, Cristian Soldi, Jared T. Shaw, Dean J. Tantillo, Jordy M. Saya, Klaas Vos, Roel A. Kleinnijenhuis, Henk Hiemstra, Shao-Nong Chen, James B. McAlpine, David C. Lankin, and J. Brent Friesen
The Journal of Organic Chemistry 2016 Volume 81(Issue 3) pp:878-889
Publication Date(Web):January 26, 2016
DOI:10.1021/acs.joc.5b02456
The revision of the structure of the sesquiterpene aquatolide from a bicyclo[2.2.0]hexane to a bicyclo[2.1.1]hexane structure using compelling NMR data, X-ray crystallography, and the recent confirmation via full synthesis exemplify that the achievement of “structural correctness” depends on the completeness of the experimental evidence. Archived FIDs and newly acquired aquatolide spectra demonstrate that archiving and rigorous interpretation of 1D 1H NMR data may enhance the reproducibility of (bio)chemical research and curb the growing trend of structural misassignments. Despite being the most accessible NMR experiment, 1D 1H spectra encode a wealth of information about bonds and molecular geometry that may be fully mined by 1H iterative full spin analysis (HiFSA). Fully characterized 1D 1H spectra are unideterminant for a given structure. The corresponding FIDs may be readily submitted with publications and collected in databases. Proton NMR spectra are indispensable for structural characterization even in conjunction with 2D data. Quantum interaction and linkage tables (QuILTs) are introduced for a more intuitive visualization of 1D J-coupling relationships, NOESY correlations, and heteronuclear experiments. Overall, this study represents a significant contribution to best practices in NMR-based structural analysis and dereplication.
Co-reporter:Guido F. Pauli, Samuel M. Pro, Lucas R. Chadwick, Thomas Burdick, Luke Pro, Warren Friedl, Nick Novak, John Maltby, Feng Qiu, and J. Brent Friesen
Analytical Chemistry 2015 Volume 87(Issue 14) pp:7418
Publication Date(Web):July 8, 2015
DOI:10.1021/acs.analchem.5b01613
Countercurrent separation (CCS) utilizes the differential partitioning behavior of analytes between two immiscible liquid phases. We introduce the first platform (“CherryOne”) capable of real-time monitoring, metering, and control of the dynamic liquid–liquid CCS process. Automated phase monitoring and volumetrics are made possible with an array of sensors, including the new permittivity-based phase metering apparatus (PMA). Volumetric data for each liquid phase are converted into a dynamic real-time display of stationary phase retention (Sf) and eluent partition coefficients (K), which represent critical parameters of CCS reproducibility. When coupled with the elution–extrusion operational mode (EECCC), automated Sf and K determination empowers untargeted and targeted applications ranging from metabolomic analysis to preparative purifications.
Co-reporter:José G. Napolitano; Charlotte Simmler; James B. McAlpine; David C. Lankin; Shao-Nong Chen
Journal of Natural Products 2015 Volume 78(Issue 4) pp:658-665
Publication Date(Web):February 25, 2015
DOI:10.1021/np5008203
This report describes a fragment-based approach to the examination of congeneric organic compounds by NMR spectroscopy. The method combines the classic interpretation of 1D- and 2D-NMR data sets with contemporary computer-assisted NMR analysis. Characteristic NMR profiles of key structural motifs were generated by 1H iterative full spin analysis and then joined together as building blocks to recreate the 1H NMR spectra of increasingly complex molecules. To illustrate the methodology described, a comprehensive analysis of steviol (1), seven steviol glycosides (2–8) and two structurally related isosteviol compounds (9, 10) was carried out. The study also assessed the potential impact of this method on relevant aspects of natural product research including structural verification, chemical dereplication, and mixture analysis.
Co-reporter:Charlotte Simmler, Jeffrey R. Anderson, Laura Gauthier, David C. Lankin, James B. McAlpine, Shao-Nong Chen, and Guido F. Pauli
Journal of Natural Products 2015 Volume 78(Issue 8) pp:2007-2022
Publication Date(Web):August 5, 2015
DOI:10.1021/acs.jnatprod.5b00342
Raw licorice roots represent heterogeneous materials obtained from mainly three Glycyrrhiza species. G. glabra, G. uralensis, and G. inflata exhibit marked metabolite differences in terms of flavanones (Fs), chalcones (Cs), and other phenolic constituents. The principal objective of this work was to develop complementary chemometric models for the metabolite profiling, classification, and quality control of authenticated licorice. A total of 51 commercial and macroscopically verified samples were DNA authenticated. Principal component analysis and canonical discriminant analysis were performed on 1H NMR spectra and area under the curve values obtained from UHPLC-UV chromatograms, respectively. The developed chemometric models enable the identification and classification of Glycyrrhiza species according to their composition in major Fs, Cs, and species specific phenolic compounds. Further key outcomes demonstrated that DNA authentication combined with chemometric analyses enabled the characterization of mixtures, hybrids, and species outliers. This study provides a new foundation for the botanical and chemical authentication, classification, and metabolomic characterization of crude licorice botanicals and derived materials. Collectively, the proposed methods offer a comprehensive approach for the quality control of licorice as one of the most widely used botanical dietary supplements.
Co-reporter:Joo-Won Nam, Rasika S. Phansalkar, David C. Lankin, Jonathan Bisson, James B. McAlpine, Ariene A. Leme, Cristina M. P. Vidal, Benjamin Ramirez, Matthias Niemitz, Ana Bedran-Russo, Shao-Nong Chen, and Guido F. Pauli
The Journal of Organic Chemistry 2015 Volume 80(Issue 15) pp:7495-7507
Publication Date(Web):July 27, 2015
DOI:10.1021/acs.joc.5b01082
The ability of certain oligomeric proanthocyanidins (OPACs) to enhance the biomechanical properties of dentin involves collagen cross-linking of the 1.3–4.5 nm wide space via protein–polyphenol interactions. A systematic interdisciplinary search for the bioactive principles of pine bark has yielded the trimeric PAC, ent-epicatechin-(4β→8)-epicatechin-(2β→O→7,4β→8)-catechin (3), representing the hitherto most potent single chemical entity capable of enhancing dentin stiffness. Building the case from two congeneric PAC dimers, a detailed structural analysis decoded the stereochemistry, spatial arrangement, and chemical properties of three dentin biomodifiers. Quantum-mechanics-driven 1H iterative full spin analysis (QM-HiFSA) of NMR spectra distinguished previously unrecognized details such as higher order J coupling and provided valuable information about 3D structure. Detection and quantification of H/D-exchange effects by QM-HiFSA identified C-8 and C-6 as (re)active sites, explain preferences in biosynthetic linkage, and suggest their involvement in dentin cross-linking activity. Mapping of these molecular properties underscored the significance of high δ precision in both 1H and 13C NMR spectroscopy. Occurring at low- to subppb levels, these newly characterized chemical shift differences in ppb are small but diagnostic measures of dynamic processes inherent to the OPAC pharmacophores and can help augment our understanding of nanometer-scale intermolecular interactions in biomodified dentin macromolecules.
Co-reporter:Wei Gao, Jin-Yong Kim, Shao-Nong Chen, Sang-Hyun Cho, Jongkeun Choi, Birgit U. Jaki, Ying-Yu Jin, David C. Lankin, Ji-Ean Lee, Sun-Young Lee, James B. McAlpine, José G. Napolitano, Scott G. Franzblau, Joo-Won Suh, and Guido F. Pauli
Organic Letters 2014 Volume 16(Issue 23) pp:6044-6047
Publication Date(Web):November 19, 2014
DOI:10.1021/ol5026603
The new tuberculosis (TB) lead ecumicin (1), a cyclic tridecapeptide, was isolated from Nonomuraea sp. MJM5123, following a high-throughput campaign for anti-TB activity. The large molecular weight of 1599 amu detected by LC-HR-MS precluded the initial inference of its molecular formula. The individual building blocks were identified by extensive NMR experiments. The resulting two possible planar structures were distinguished by LC-MS2. Determination of absolute configuration and unambiguous structural confirmation were carried out by X-ray crystallography and Marfey’s analysis.
Co-reporter:Guido F. Pauli ; Shao-Nong Chen ; Charlotte Simmler ; David C. Lankin ; Tanja Gödecke ; Birgit U. Jaki ; J. Brent Friesen ; James B. McAlpine ;José G. Napolitano
Journal of Medicinal Chemistry 2014 Volume 57(Issue 22) pp:9220-9231
Publication Date(Web):October 8, 2014
DOI:10.1021/jm500734a
In any biomedical and chemical context, a truthful description of chemical constitution requires coverage of both structure and purity. This qualification affects all drug molecules, regardless of development stage (early discovery to approved drug) and source (natural product or synthetic). Purity assessment is particularly critical in discovery programs and whenever chemistry is linked with biological and/or therapeutic outcome. Compared with chromatography and elemental analysis, quantitative NMR (qNMR) uses nearly universal detection and provides a versatile and orthogonal means of purity evaluation. Absolute qNMR with flexible calibration captures analytes that frequently escape detection (water, sorbents). Widely accepted structural NMR workflows require minimal or no adjustments to become practical 1H qNMR (qHNMR) procedures with simultaneous qualitative and (absolute) quantitative capability. This study reviews underlying concepts, provides a framework for standard qHNMR purity assays, and shows how adequate accuracy and precision are achieved for the intended use of the material.
Co-reporter:Feng Qiu, James B. McAlpine, David C. Lankin, Ian Burton, Tobias Karakach, Shao-Nong Chen, and Guido F. Pauli
Analytical Chemistry 2014 Volume 86(Issue 8) pp:3964
Publication Date(Web):March 27, 2014
DOI:10.1021/ac500188j
The interpretation of NMR spectroscopic information for structure elucidation involves decoding of complex resonance patterns that contain valuable molecular information (δ and J), which is not readily accessible otherwise. We introduce a new concept of 2D-NMR barcoding that uses clusters of fingerprint signals and their spatial relationships in the δ−δ coordinate space to facilitate the chemical identification of complex mixtures. Similar to widely used general barcoding technology, the structural information of individual compounds is encoded as a specifics pattern of their C,H correlation signals. Software-based recognition of these patterns enables the structural identification of the compounds and their discrimination in mixtures. Using the triterpenes from various Actaea (syn. Cimicifuga) species as a test case, heteronuclear multiple-bond correlation (HMBC) barcodes were generated on the basis of their structural subtypes from a statistical investigation of their δH and δC data in the literature. These reference barcodes allowed in silico identification of known triterpenes in enriched fractions obtained from an extract of A. racemosa (black cohosh). After dereplication, a differential analysis of heteronuclear single-quantum correlation (HSQC) spectra even allowed for the discovery of a new triterpene. The 2D barcoding concept has potential application in a natural product discovery project, allowing for the rapid dereplication of known compounds and as a tool in the search for structural novelty within compound classes with established barcodes.
Co-reporter:René F. Ramos Alvarenga ; J. Brent Friesen ; Dejan Nikolić ; Charlotte Simmler ; José G. Napolitano ; Richard van Breemen ; David C. Lankin ; James B. McAlpine ; Guido F. Pauli ;Shao-Nong Chen
Journal of Natural Products 2014 Volume 77(Issue 12) pp:2595-2604
Publication Date(Web):December 1, 2014
DOI:10.1021/np500376g
This study introduces a flexible and compound targeted approach to Deplete and Enrich Select Ingredients to Generate Normalized Extract Resources, generating DESIGNER extracts, by means of chemical subtraction or augmentation of metabolites. Targeting metabolites based on their liquid–liquid partition coefficients (K values), K targeting uses countercurrent separation methodology to remove single or multiple compounds from a chemically complex mixture, according to the following equation: DESIGNER extract = total extract ± target compound(s). Expanding the scope of the recently reported depletion of extracts by immunoaffinity or solid phase liquid chromatography, the present approach allows a more flexible, single- or multi-targeted removal of constituents from complex extracts such as botanicals. Chemical subtraction enables both chemical and biological characterization, including detection of synergism/antagonism by both the subtracted targets and the remaining metabolite mixture, as well as definition of the residual complexity of all fractions. The feasibility of the DESIGNER concept is shown by K-targeted subtraction of four bioactive prenylated phenols, isoxanthohumol (1), 8-prenylnaringenin (2), 6-prenylnaringenin (3), and xanthohumol (4), from a standardized hops (Humulus lupulus L.) extract using specific solvent systems. Conversely, adding K-targeted isolates allows enrichment of the original extract and hence provides an augmented DESIGNER material. Multiple countercurrent separation steps were used to purify each of the four compounds, and four DESIGNER extracts with varying depletions were prepared. The DESIGNER approach innovates the characterization of chemically complex extracts through integration of enabling technologies such as countercurrent separation, K-by-bioactivity, the residual complexity concepts, as well as quantitative analysis by 1H NMR, LC-MS, and HiFSA-based NMR fingerprinting.
Co-reporter:Yang Liu ; Shao-Nong Chen ; James B. McAlpine ; Larry L. Klein ; J. Brent Friesen ; David C. Lankin
Journal of Natural Products 2014 Volume 77(Issue 3) pp:611-617
Publication Date(Web):January 16, 2014
DOI:10.1021/np400874z
A new strategy for the analysis of natural products uses a combination of quantitative 1H NMR (qHNMR) and adsorbent-free countercurrent separation (CS) methodology to establish a quantification method for ginkgotoxin (4′-O-methylpyridoxine) in Ginkgo biloba preparations. The target analyte was concentrated in a one-step CS process using the ChMWat +2 solvent system (CHCl3–MeOH–H2O, 10:5:5) and subsequently assayed by qHNMR. While commercial G. biloba seeds contained 59 μg of ginkgotoxin per seed, the compound was below the limit of detection (9 ppm) in a typical leaf extract. Due to the enrichment potential and loss-free operation of CS, the combination of CS and qHNMR is a generally suitable approach for threshold assays aimed at quantifying target compounds such as botanical negative markers at the low ppm level. As the proof of principle is demonstrated for relatively small CS capacities (20 mL, 1:40 loading) and modest NMR sensitivity (n = 16, 400 MHz, 5 mm RT probe), the approach can be adapted to quantification at the ppb level. The procedure enables the quantification of a botanical negative marker in the absence of identical reference material, which otherwise is a prerequisite for LC-based assays.
Co-reporter:Guido F. Pauli ; Shao-Nong Chen ; David C. Lankin ; Jonathan Bisson ; Ryan J. Case ; Lucas R. Chadwick ; Tanja Gödecke ; Taichi Inui ; Aleksej Krunic ; Birgit U. Jaki ; James B. McAlpine ; Shunyan Mo ; José G. Napolitano ; Jimmy Orjala ; Juuso Lehtivarjo ; Samuli-Petrus Korhonen ;Matthias Niemitz
Journal of Natural Products 2014 Volume 77(Issue 6) pp:1473-1487
Publication Date(Web):June 4, 2014
DOI:10.1021/np5002384
The present study demonstrates the importance of adequate precision when reporting the δ and J parameters of frequency domain 1H NMR (HNMR) data. Using a variety of structural classes (terpenoids, phenolics, alkaloids) from different taxa (plants, cyanobacteria), this study develops rationales that explain the importance of enhanced precision in NMR spectroscopic analysis and rationalizes the need for reporting Δδ and ΔJ values at the 0.1–1 ppb and 10 mHz level, respectively. Spectral simulations paired with iteration are shown to be essential tools for complete spectral interpretation, adequate precision, and unambiguous HNMR-driven dereplication and metabolomic analysis. The broader applicability of the recommendation relates to the physicochemical properties of hydrogen (1H) and its ubiquity in organic molecules, making HNMR spectra an integral component of structure elucidation and verification. Regardless of origin or molecular weight, the HNMR spectrum of a compound can be very complex and encode a wealth of structural information that is often obscured by limited spectral dispersion and the occurrence of higher order effects. This altogether limits spectral interpretation, confines decoding of the underlying spin parameters, and explains the major challenge associated with the translation of HNMR spectra into tabulated information. On the other hand, the reproducibility of the spectral data set of any (new) chemical entity is essential for its structure elucidation and subsequent dereplication. Handling and documenting HNMR data with adequate precision is critical for establishing unequivocal links between chemical structure, analytical data, metabolomes, and biological activity. Using the full potential of HNMR spectra will facilitate the general reproducibility for future studies of bioactive chemicals, especially of compounds obtained from the diversity of terrestrial and marine organisms.
Co-reporter:José G. Napolitano, Tanja Gödecke, David C. Lankin, Birgit U. Jaki, James B. McAlpine, Shao-Nong Chen, Guido F. Pauli
Journal of Pharmaceutical and Biomedical Analysis 2014 Volume 93() pp:59-67
Publication Date(Web):May 2014
DOI:10.1016/j.jpba.2013.06.017
The development of analytical methods for parallel characterization of multiple phytoconstituents is essential to advance the quality control of herbal products. While chemical standardization is commonly carried out by targeted analysis using gas or liquid chromatography-based methods, more universal approaches based on quantitative 1H NMR (qHNMR) measurements are being used increasingly in the multi-targeted assessment of these complex mixtures. The present study describes the development of a 1D qHNMR-based method for simultaneous identification and quantification of green tea constituents. This approach utilizes computer-assisted 1H iterative Full Spin Analysis (HiFSA) and enables rapid profiling of seven catechins in commercial green tea extracts. The qHNMR results were cross-validated against quantitative profiles obtained with an orthogonal LC–MS/MS method. The relative strengths and weaknesses of both approaches are discussed, with special emphasis on the role of identical reference standards in qualitative and quantitative analyses.
Co-reporter:Charlotte Simmler;Tristesse Jones;Jeffrey R. Anderson;Dejan C. Nikoli&x107;;Richard B. van Breemen;Djaja D. Soejarto;Shao-Nong Chen
Phytochemical Analysis 2014 Volume 25( Issue 4) pp:378-388
Publication Date(Web):
DOI:10.1002/pca.2472
ABSTRACT
Introduction
Major phenolics from licorice roots (Glycyrrhiza sp.) are glycosides of the flavanone liquiritigenin (F) and its 2′-hydroxychalcone isomer, isoliquiritigenin (C). As the F and C contents fluctuate between batches of licorice, both quality control and standardisation of its preparations become complex tasks.
Objective
To characterise the F and C metabolome in extracts from Glycyrrhiza glabra L. and Glycyrrhiza uralensis Fisch. ex DC. by addressing their composition in major F–C pairs and defining the total F:C proportion.
Material and methods
Three types of extracts from DNA-authenticated samples were analysed by a validated UHPLC/UV method to quantify major F and C glycosides. Each extract was characterised by the identity of major F–C pairs and the proportion of Fs among all quantified Fs:Cs.
Results
The F and C compositions and proportions were found to be constant for all extracts from a Glycyrrhiza species. All G. uralensis extracts contained up to 2.5 more Fs than G. glabra extracts. Major F–C pairs were B-ring glycosidated in G. uralensis, and A-/B-ring apiosyl-glucosidated in the G. glabra extracts. The F:C proportion was found to be linked to the glycosidation site: the more B-ring F-C glycosides were present, the higher was the final F:C proportion in the extract. These results enable the chemical differentiation of extracts from G. uralensis and G. glabra, which are characterised by total F:C proportions of 8.37:1.63 and 7.18:2.82, respectively.
Conclusion
Extracts from G. glabra and G. uralensis can be differentiated by their respective F and C compositions and proportions, which are both useful for further standardisation of licorice botanicals. Copyright © 2013 John Wiley & Sons, Ltd.
Co-reporter:Feng Qiu, Geping Cai, Birgit U. Jaki, David C. Lankin, Scott G. Franzblau, and Guido F. Pauli
Journal of Natural Products 2013 Volume 76(Issue 3) pp:413-419
Publication Date(Web):January 28, 2013
DOI:10.1021/np3007809
The present study provides an extension of the previously developed concept of purity–activity relationships (PARs) and enables the quantitative evaluation of the effects of multiple minor components on the bioactivity of residually complex natural products. The anti-tuberculosis active triterpenes from the Alaskan ethnobotanical Oplopanax horridus were selected as a case for the development of the quantitative PAR (QPAR) concept. The residual complexity of the purified triterpenes was initially evaluated by 1D- and 2D-NMR and identified as a combination of structurally related and unrelated impurities. Using a biochemometric approach, the qHNMR purity and anti-TB activity of successive chromatographic fractions of O. horridus triterpenes were correlated by linear regression analysis to generate a mathematical QPAR model. The results demonstrate that impurities, such as widely occurring monoglycerides, can have a profound impact on the observed antimycobacterial activity of triterpene-enriched fractions. The QPAR concept is shown to be capable of providing a quantitative assessment in situations where residually complex constitution contributes toward the biological activity of natural products.
Co-reporter:Chang Hwa Hwang ; Birgit U. Jaki ; Larry L. Klein ; David C. Lankin ; James B. McAlpine ; José G. Napolitano ; Nicole A. Fryling ; Scott G. Franzblau ; Sang Hyun Cho ; Paul E. Stamets ; Yuehong Wang
Journal of Natural Products 2013 Volume 76(Issue 10) pp:1916-1922
Publication Date(Web):October 2, 2013
DOI:10.1021/np400497f
An EtOH extract of the polypore mushroom Fomitopsis officinalis afforded two new naturally occurring chlorinated coumarins, which were identified as the previously synthesized compounds 6-chloro-4-phenyl-2H-chromen-2-one (1) and ethyl 6-chloro-2-oxo-4-phenyl-2H-chromen-3-carboxylate (2). The structures of the two isolates were deduced by ab initio spectroscopic methods and confirmed by chemical synthesis. In addition, an analogue of each was synthesized as 7-chloro-4-phenyl-2H-chromen-2-one (3) and ethyl 7-chloro-2-oxo-4-phenyl-2H-chromen-3-carboxylate (4). All four compounds were characterized physicochemically, and their antimicrobial activity profiles revealed a narrow spectrum of activity with lowest MICs against the Mycobacterium tuberculosis complex.
Co-reporter:Geping Cai, José G. Napolitano, James B. McAlpine, Yuehong Wang, Birgit U. Jaki, Joo-Won Suh, Seung Hwan Yang, In-Ae Lee, Scott G. Franzblau, Guido F. Pauli, and Sanghyun Cho
Journal of Natural Products 2013 Volume 76(Issue 11) pp:2009-2018
Publication Date(Web):November 13, 2013
DOI:10.1021/np400145u
Thirty-five thousand actinomycete extracts were screened for anti-Mycobacterium tuberculosis (M. tb) activity, followed by C18 cartridge fractionation of 37 prioritized extracts. Based on MICs against replicating and nonreplicating M. tb, and IC50 values against Vero cells to generate selectivity indices, seven fractions from seven different strains were selected for further examination. When cultured in G.S.S. media and extracted with ethyl acetate, the Streptomyces hygroscopicus strain ECUM 14046 yielded an extract with promising anti-M. tb activity and a well-defined chromatographic profile. Fractionation by preparative HPLC and subsequent structure elucidation of two active fractions using 1D- and 2D-NMR and MS methods revealed the presence of two cyclohexapeptides, hytramycins V and I, each containing three unusual piperazic acid moieties. The use of 1H iterative full spin analysis (HiFSA) on both hytramycins confirmed that quantum mechanics-simulated spectra match the experimental data, and all JH,H and δH values are consistent with the proposed structures. The absolute configuration of each amino acid moiety was determined by Marfey’s method. The MICs against replicating and, more importantly, nonreplicating M. tb fall into the range of some existing second-line anti-TB drugs, such as streptomycin and capreomycin, respectively. The activities were maintained against M. tb strains that represent the major global clades, as well as H37Rv-isogenic strains that are resistant to individual clinical anti-TB drugs.
Co-reporter:Charlotte Simmler, Atieh Hajirahimkhan, David C. Lankin, Judy L. Bolton, Tristesse Jones, Djaja D. Soejarto, Shao-Nong Chen, and Guido F. Pauli
Journal of Agricultural and Food Chemistry 2013 Volume 61(Issue 9) pp:2146-2157
Publication Date(Web):February 22, 2013
DOI:10.1021/jf304445p
Bioactive components in food plants can undergo dynamic processes that involve multiple chemical species. For example, 2′-hydroxychalcones can readily isomerize into flavanones. Although chemically well documented, this reaction has barely been explored in the context of cell-based assays. The present time-resolved study fills this gap by investigating the isomerization of isoliquiritigenin (a 2′-hydroxychalcone) and liquiritigenin (a flavanone) in two culture media (Dulbecco’s modified eagle medium and Roswell Park Memorial Institute medium) with and without MCF-7 cells, using high-performance liquid chromatography–diode array detector–electrospray ionization/atmospheric pressure chemical ionization–mass spectrometry for analysis. Both compounds were isomerized and epimerized under all investigated biological conditions, leading to mixtures of isoliquiritigenin and R/S-liquiritigenin, with 19.6% R enantiomeric excess. Consequently, all three species can potentially modulate the biological responses. This exemplifies dynamic residual complexity and demonstrates how both nonchiral reactions and enantiomeric discrimination can occur in bioassay media, with or without cells. The findings highlight the importance of controlling in situ chemical reactivity, influenced by biological systems when evaluating the mode of action of bioactives.
Co-reporter:Tanja Gödecke;José G. Napolitano;María F. Rodríguez-Brasco;Shao-Nong Chen;Birgit U. Jaki;David C. Lankin
Phytochemical Analysis 2013 Volume 24( Issue 6) pp:581-597
Publication Date(Web):
DOI:10.1002/pca.2436
ABSTRACT
Introduction
Nuclear magnetic resonance (NMR) spectroscopy is increasingly employed in the quantitative analysis and quality control (QC) of natural products (NP) including botanical dietary supplements (BDS). The establishment of QC protocols based on quantitative 1H NMR (qHNMR) requires method validation.
Objective
Develop and validate a generic qHNMR method. Optimize acquisition and processing parameters, with specific attention to the requirements for the analysis of complex NP samples, including botanicals and purity assessment of NP isolates.
Methods
In order to establish the validated qHNMR method, samples containing two highly pure reference materials were used. The influence of acquisition and processing parameters on the method validation was examined, and general aspects of method validation of qHNMR methods discussed. Subsequently, the method established was applied to the analysis of two NP samples: a purified reference compound and a crude mixture.
Results
The accuracy and precision of qHNMR using internal or external calibration were compared, using a validated method suitable for complex samples. The impact of post-acquisition processing on method validation was examined using three software packages: TopSpin, Mnova and NUTS. The dynamic range of the qHNMR method developed was 5000:1 with a limit of detection (LOD) of better than 10 µm. The limit of quantification (LOQ) depends on the desired level of accuracy and experiment time spent.
Conclusion
This study revealed that acquisition parameters, processing parameters and processing software all contribute to qHNMR method validation. A validated method with a high dynamic range and general workflow for qHNMR analysis of NP is proposed. Copyright © 2013 John Wiley & Sons, Ltd.
Co-reporter:José G. Napolitano, David C. Lankin, Tyler N. Graf, J. Brent Friesen, Shao-Nong Chen, James B. McAlpine, Nicholas H. Oberlies, and Guido F. Pauli
The Journal of Organic Chemistry 2013 Volume 78(Issue 7) pp:2827-2839
Publication Date(Web):March 5, 2013
DOI:10.1021/jo302720h
This study demonstrates how regio- and diastereo-isomers with near-identical NMR spectra can be distinguished and unambiguously assigned using quantum mechanical driven 1H iterative Full Spin Analysis (HiFSA). The method is illustrated with four natural products, the flavonolignans silybin A, silybin B, isosilybin A, and isosilybin B, which exhibit extremely similar coupling patterns and chemical shift differences well below the commonly reported level of accuracy of 0.01 ppm. The HiFSA approach generated highly reproducible 1H NMR fingerprints that enable distinction of all four isomers at 1H frequencies from 300 to 900 MHz. Furthermore, it is demonstrated that the underlying numeric 1H NMR profiles, combined with iterative computational analysis, allow parallel quantification of all four isomers, even in difficult to characterize reference materials and mixtures. The results shed new light on the historical challenges to the qualitative and quantitative analysis of these therapeutically relevant flavonolignans and open new opportunities to explore hidden diversity in the chemical space of organic molecules.
Co-reporter:José G. Napolitano, David C. Lankin, James B. McAlpine, Matthias Niemitz, Samuli-Petrus Korhonen, Shao-Nong Chen, and Guido F. Pauli
The Journal of Organic Chemistry 2013 Volume 78(Issue 19) pp:9963-9968
Publication Date(Web):September 6, 2013
DOI:10.1021/jo4011624
The characteristic signals observed in NMR spectra encode essential information on the structure of small molecules. However, extracting all of this information from complex signal patterns is not trivial. This report demonstrates how computer-aided spectral analysis enables the complete interpretation of 1D 1H NMR data. The effectiveness of this approach is illustrated with a set of organic molecules, for which replicas of their 1H NMR spectra were generated. The potential impact of this methodology on organic chemistry research is discussed.
Co-reporter:Guido F. Pauli ; Shao-Nong Chen ; J. Brent Friesen ; James B. McAlpine ;Birgit U. Jaki
Journal of Natural Products 2012 Volume 75(Issue 6) pp:1243-1255
Publication Date(Web):May 23, 2012
DOI:10.1021/np300066q
Based on a meta-analysis of data mined from almost 2000 publications on bioactive natural products (NPs) from >80 000 pages of 13 different journals published in 1998–1999, 2004–2005, and 2009–2010, the aim of this systematic review is to provide both a survey of the status quo and a perspective for analytical methodology used for isolation and purity assessment of bioactive NPs. The study provides numerical measures of the common means of sourcing NPs, the chromatographic methodology employed for NP purification, and the role of spectroscopy and purity assessment in NP characterization. A link is proposed between the observed use of various analytical methodologies, the challenges posed by the complexity of metabolomes, and the inescapable residual complexity of purified NPs and their biological assessment. The data provide inspiration for the development of innovative methods for NP analysis as a means of advancing the role of naturally occurring compounds as a viable source of biologically active agents with relevance for human health and global benefit.
Co-reporter:Feng Qiu, J. Brent Friesen, James B. McAlpine, Guido F. Pauli
Journal of Chromatography A 2012 Volume 1242() pp:26-34
Publication Date(Web):15 June 2012
DOI:10.1016/j.chroma.2012.03.081
Terpene lactones such as bilobalide, ginkgolides A, B, C, and J are major bioactive compounds of Ginkgo biloba L. Purification of these compounds is tedious due to their similar chemical properties. For the purpose of developing an effective and efficient method for both analytical and preparative separation of terpene lactones in G. biloba, an innovative orthogonality-enhanced high-speed countercurrent chromatography (HSCCC) method was established. Taking advantage of quantitative 1H NMR (qHNMR) methodology, partition coefficients (K) of individual terpene lactones were calculated directly from crude G. biloba leaf extract, using their H-12 signals as distinguishing feature. The partitioning experiment assisted the design of a two dimensional (2D) HSCCC procedure using a pair of orthogonal HSCCC solvent systems (SSs), ChMWat +4 and HEMSoWat +3/0.05%. It was surprising that the resolution of ginkgolides A and B was improved by 25% in the HEMWat +3 SS modified with 0.5% DMSO. Consequently, all five terpene lactones could be well separated with qHNMR purity > 95% from G. biloba leaf extract. The separation was further evaluated by offline qHNMR analysis of HSCCC fractions associated with Gaussian curve fitting. The results showed less than 2% error in HSCCC retention predicted from the partitioning experiment. This compelling consistency demonstrates that qHNMR-derived K determination (“K-by-NMR”) can be used to predict CCC fractionation and target purification of analytes from complex mixtures. Furthermore, Gaussian curve fitting enabled an accurate prediction of less than 2% impurity in the CCC fraction, which demonstrates its potential as a powerful tool to study the presence of minor constituents, especially when they are beyond the detection limit of conventional spectroscopic detectors.Highlights► Novel qHNMR-based approach to countercurrent partition coefficients (K values). ► Enabled targeted countercurrent separation (CS) of Ginkgo biloba terpene lactones. ► Established orthogonal solvent systems (SSs) for separation of Ginkgo lactones. ► Developed DMSO-modified SSs as a new means of resolution enhancement in CS. ► Evaluated ID and residual complexity of eluents by qHNMR with Gaussian fitting.
Co-reporter:José G. Napolitano, Tanja Gödecke, María F. Rodríguez-Brasco, Birgit U. Jaki, Shao-Nong Chen, David C. Lankin, and Guido F. Pauli
Journal of Natural Products 2012 Volume 75(Issue 2) pp:238-248
Publication Date(Web):February 14, 2012
DOI:10.1021/np200949v
Botanical dietary supplements and herbal remedies are widely used for health promotion and disease prevention. Due to the high chemical complexity of these natural products, it is essential to develop new analytical strategies to guarantee their quality and consistency. In particular, the precise characterization of multiple botanical markers remains a challenge. This study demonstrates how a combination of computer-aided spectral analysis and 1D quantitative 1H NMR spectroscopy (qHNMR) generates the analytical foundation for innovative means of simultaneously identifying and quantifying botanical markers in complex mixtures. First, comprehensive 1H NMR profiles (fingerprints) of selected botanical markers were generated via 1H iterative full spin analysis (HiFSA) with PERCH. Next, the 1H fingerprints were used to assign specific 1H resonances in the NMR spectra of reference materials, enriched fractions, and crude extracts of Ginkgo biloba leaves. These 1H fingerprints were then used to verify the assignments by 2D NMR. Subsequently, a complete purity and composition assessment by means of 1D qHNMR was conducted. As its major strengths, this tandem approach enables the simultaneous quantification of multiple constituents without the need for identical reference materials, the semiquantitative determination of particular subclasses of components, and the detection of impurities and adulterants.
Co-reporter:Feng Qiu ; Ayano Imai ; James B. McAlpine ; David C. Lankin ; Ian Burton ; Tobias Karakach ; Norman R. Farnsworth ; Shao-Nong Chen
Journal of Natural Products 2012 Volume 75(Issue 3) pp:432-443
Publication Date(Web):February 9, 2012
DOI:10.1021/np200878s
The genus Actaea (including Cimicifuga) has been the source of ∼200 cycloartane triterpenes. While they are major bioactive constituents of complementary and alternative medicines, their structural similarity is a major dereplication problem. Moreover, their trivial names seldom indicate the actual structure. This project develops two new tools for Actaea triterpenes that enable rapid dereplication of more than 170 known triterpenes and facilitates elucidation of new compounds. A predictive computational model based on classification binary trees (CBTs) allows in silico determination of the aglycone type. This tool utilizes the Me 1H NMR chemical shifts and has potential to be applicable to other natural products. Actaea triterpene dereplication is supported by a new systematic naming scheme. A combination of CBTs, 1H NMR deconvolution, characteristic 1H NMR signals, and quantitative 1H NMR (qHNMR) led to the unambiguous identification of minor constituents in residually complex triterpene samples. Utilizing a 1.7 mm cryo-microprobe at 700 MHz, qHNMR enabled characterization of residual complexity at the 10–20 μg level in a 1–5 mg sample. The identification of five co-occurring minor constituents, belonging to four different triterpene skeleton types, in a repeatedly purified natural product emphasizes the critical need for the evaluation of residual complexity of reference materials, especially when used for biological assessment.
Co-reporter:Guido F. Pauli, Tanja Gödecke, Birgit U. Jaki, and David C. Lankin
Journal of Natural Products 2012 Volume 75(Issue 4) pp:834-851
Publication Date(Web):April 6, 2012
DOI:10.1021/np200993k
Covering the literature from mid-2004 until the end of 2011, this review continues a previous literature overview on quantitative 1H NMR (qHNMR) methodology and its applications in the analysis of natural products. Among the foremost advantages of qHNMR is its accurate function with external calibration, the lack of any requirement for identical reference materials, a high precision and accuracy when properly validated, and an ability to quantitate multiple analytes simultaneously. As a result of the inclusion of over 170 new references, this updated review summarizes a wealth of detailed experiential evidence and newly developed methodology that supports qHNMR as a valuable and unbiased analytical tool for natural product and other areas of research.
Co-reporter:José G. Napolitano;David C. Lankin;Shao-Nong Chen
Magnetic Resonance in Chemistry 2012 Volume 50( Issue 8) pp:569-575
Publication Date(Web):
DOI:10.1002/mrc.3829
The complete and unambiguous 1H NMR assignments of ten marker constituents of Ginkgo biloba are described. The comprehensive 1H NMR profiles (fingerprints) of ginkgolide A, ginkgolide B, ginkgolide C, ginkgolide J, bilobalide, quercetin, kaempferol, isorhamnetin, isoquercetin, and rutin in DMSO-d6 were obtained through the examination of 1D 1H NMR and 2D 1H,1H-COSY data, in combination with 1H iterative full spin analysis (HiFSA). The computational analysis of discrete spin systems allowed a detailed characterization of all the 1H NMR signals in terms of chemical shifts (δH) and spin-spin coupling constants (JHH), regardless of signal overlap and higher order coupling effects. The capability of the HiFSA-generated 1H fingerprints to reproduce experimental 1H NMR spectra at different field strengths was also evaluated. As a result of this analysis, a revised set of 1H NMR parameters for all ten phytoconstituents was assembled. Furthermore, precise 1H NMR assignments of the sugar moieties of isoquercetin and rutin are reported for the first time. Copyright © 2012 John Wiley & Sons, Ltd.
Co-reporter:Gloria M. Molina-Salinas, Verónica M. Rivas-Galindo, Salvador Said-Fernández, David C. Lankin, Marcelo A. Muñoz, Pedro Joseph-Nathan, Guido F. Pauli, and Noemí Waksman
Journal of Natural Products 2011 Volume 74(Issue 9) pp:1842-1850
Publication Date(Web):August 22, 2011
DOI:10.1021/np2000667
Bioactivity-guided fractionation of the methanolic root bark extract of Leucophyllum frutescens led to the identification of leubethanol (1), a new serrulatane-type diterpene with activity against both multi-drug-resistant and drug-sensitive strains of virulent Mycobacterium tuberculosis. Leubethanol (1) was identified by 1D/2D NMR data, as a serrulatane closely related to erogorgiane (2), and exhibited anti-TB activity with minimum inhibitory concentrations in the range 6.25–12.50 μg/mL. Stereochemical evidence for 1 was gleaned from 1D and 2D NOE experiments, from 1H NMR full spin analysis, and by comparison of the experimental vibrational circular dichroism (VCD) spectrum to density functional theory calculated VCD spectra of two diastereomers.
Co-reporter:Guido F. Pauli, J. Brent Friesen, Tanja Gödecke, Norman R. Farnsworth and Bernhard Glodny
Journal of Natural Products 2010 Volume 73(Issue 3) pp:338-345
Publication Date(Web):January 28, 2010
DOI:10.1021/np9007415
Previously, the presence of a wide variety of chemically diverse steroids has been identified in both flora and fauna. Despite the relatively small differences in chemical structures and large differences in physiological function of steroids, new discoveries indicate that plants and animals are more closely related than previously thought. In this regard, the present study gathers supporting evidence for shared phylogenetic roots of structurally similar steroids produced by these two eukaryotic taxa. Definitive proof for the presence of progesterone (3) in a vascular plant, Juglans regia, is provided. Additional evidence is gleaned from the characterization of five new plant steroids from Adonis aleppica: three 3-O-sulfated pregnenolones (6a/b, 7), a sulfated H-5β cardenolide, strophanthidin-3-O-sulfate (8), and spirophanthigenin (10), a novel C-18 oxygenated spirocyclic derivative of strophanthidin. The ab initio isolation and structure elucidation (NMR, MS) of these genuine minor plant steroids offers information on preparative metabolomic profiling at the ppm level and provides striking evidence for the conserved structural space of pregnanes and its congeners across the phylogenetic tree.
Co-reporter:Taichi Inui, Yuehong Wang, Dejan Nikolic, David C. Smith, Scott G. Franzblau and Guido F. Pauli
Journal of Natural Products 2010 Volume 73(Issue 4) pp:563-567
Publication Date(Web):March 10, 2010
DOI:10.1021/np900674d
From the anti-TB active fractions of the inner stem bark of Oplopanax horridus, two new heterocyclic nerolidol derivatives, 3,10-epoxy-3,7,11-trimethyldodeca-1,6-dien-11-ol, named neroplomacrol (1), and rel-(3S,6R,7S,10R)-7,10-epoxy-3,7,11-trimethyldodec-1-ene-3,6,11-triol, named neroplofurol (2), were isolated together with oplopandiol (3), falcarindiol (4), and sesamin (5). Extensive spectroscopic analysis revealed that 1 possesses a novel 3,10-oxanonacyclic ring system. Computer-iterated full spin system analysis led to the generation of 1H NMR fingerprints that will facilitate future dereplication of analogues by providing characteristic spin−spin coupling patterns. The full spin analysis of 5 revealed asymmetric coupling patterns among the chemically equivalent spins, thus confirming the magnetic asymmetry of 5. It was further demonstrated that 1H NMR fingerprints and MS data enable dereplication of isolates at a submilligram levels including their relative configuration. Countercurrent concentration of the anti-TB activity of the ethnobotanical O. horridus versus the Mycobacterium tuberculosis Erdman strain led to polyynes 3 and 4 as main anti-TB active principles. Their synergistic behavior is linked to a complex fraction containing the new nerolidiol sesquiterpenes, 1 and 2, as phytochemical marker compounds.
Co-reporter:Tanja Gödecke, David C. Lankin, Dejan Nikolic, Shao-Nong Chen, Richard B. van Breemen, Norman R. Farnsworth and Guido F. Pauli
Journal of Natural Products 2009 Volume 72(Issue 3) pp:433-437
Publication Date(Web):February 16, 2009
DOI:10.1021/np8006952
As an extension of work on the recently discovered nitrogenous metabolites from Cimicifuga/Actaea species, three new guanidine alkaloids have been isolated and characterized from C. racemosa (syn. A. racemosa) roots. Of these, cyclo-cimipronidine (1) and cimipronidine methyl ester (2) are congeners of cimipronidine (3), whereas dopargine (5) is a derivative of dopamine. By employing NMR- and MS-guided chemodiversity profiling of a polar serotonergic (5-HT7) fraction, the guanidine alkaloids were initially detected in a clinical extract of black cohosh and were isolated along with a congener of salsolinol 4, 5, and 3-hydroxytyrosol 3-O-glucoside (7). The structures of 1, 2, and 5 were confirmed by 1D and 2D NMR spectroscopy as well as LC-MS and HRMS spectroscopy. A plausible biosynthetic relationship may be inferred between the homoproline-analogue cimipronidines and the dopamine-derived Cimicifuga alkaloids. These strongly basic and frequently zwitterionic nitrogenous metabolites contribute considerable chemical diversity to the polar serotonergic fraction of black cohosh.
Co-reporter:J.B. Friesen, G.F. Pauli
Journal of Chromatography A 2009 Volume 1216(Issue 19) pp:4237-4244
Publication Date(Web):8 May 2009
DOI:10.1016/j.chroma.2009.01.048
Counter-current separation (CS) technology is currently faced with the challenge of being fit for the purpose of omics analysis, which involves highly complex samples and digitized research environments. Resembling a network of binary decisions, CS requires standardization of operation parameters in order to be efficient. While recent CS engineering solutions uniformly involve centrifugal force designs to overcome the limitation of the earth's 1 × g force, factors of instrument design, operation, and graphical representation of the outcome are equally important targets for standardization. For example, chromatograms that emphasize the unique K-based nature of CS, such as reciprocal symmetry (ReS) plots, foster the fundamental understanding of CS operation. Because significant differences exist in underlying mechanism (e.g., stationary phase volume), outcome (e.g., construction of chromatograms), and scale (e.g., factors affecting overall method sensitivity) of solid–liquid vs. liquid–liquid chromatography technologies, standardization will enable the systematic exploration of the differential properties of the two LC technologies, and will be key to making CS fit for the digital omics age.
Co-reporter:Tanja Gödecke;Dejan Nikolic;David C. Lankin;Shao-Nong Chen;Sharla L. Powell;Birgit Dietz;Judy L. Bolton;Richard B. van Breemen;Norman R. Farnsworth ;Guido F. Pauli
Phytochemical Analysis 2009 Volume 20( Issue 2) pp:120-133
Publication Date(Web):
DOI:10.1002/pca.1106
Abstract
Introduction
Earlier studies reported serotonergic activity for cimicifugic acids (CA) isolated from Cimicifuga racemosa. The discovery of strongly basic alkaloids, cimipronidines, from the active extract partition and evaluation of previously employed work-up procedures has led to the hypothesis of strong acid/base association in the extract.
Objective
Re-isolation of the CAs was desired to permit further detailed studies. Based on the acid/base association hypothesis, a new separation scheme of the active partition was required, which separates acids from associated bases.
Methodology
A new 5-HT7 bioassay guided work-up procedure was developed that concentrates activity into one partition. The latter was subjected to a new two-step centrifugal partitioning chromatography (CPC) method, which applies pH zone refinement gradient (pHZR CPC) to dissociate the acid/base complexes. The resulting CA fraction was subjected to a second CPC step. Fractions and compounds were monitored by 1H NMR using a structure-based spin-pattern analysis facilitating dereplication of the known acids. Bioassay results were obtained for the pHZR CPC fractions and for purified CAs.
Results
A new CA was characterised. While none of the pure CAs was active, the serotonergic activity was concentrated in a single pHZR CPC fraction, which was subsequently shown to contain low levels of the potent 5-HT7 ligand, Nω-methylserotonin.
Conclusion
This study shows that CAs are not responsible for serotonergic activity in black cohosh. New phytochemical methodology (pHZR CPC) and a sensitive dereplication method (LC-MS) led to the identification of Nω-methylserotonin as serotonergic active principle. Copyright © 2009 John Wiley & Sons, Ltd.
Co-reporter:Guido F. Pauli, Samuel M. Pro and J. Brent Friesen
Journal of Natural Products 2008 Volume 71(Issue 8) pp:1489-1508
Publication Date(Web):July 31, 2008
DOI:10.1021/np800144q
An assessment of the technology and method development in countercurrent chromatography (CCC) and centrifugal partition chromatography (CPC), collectively referred to as countercurrent separation (CS), is provided. More than six decades of CS theory and applications are critically reviewed and developed into a practical guide to CS for natural products research. The necessary theoretical foundation is given for better use of CS in the separation of biological molecules of any size, small to large, and from any matrix, simple to complex. The three operational fundamentals of CS—instrumentation, biphasic solvent systems, and theory—are covered in a prismatic fashion. The goal of this review is to provide the necessary background and references for an up-to-date perspective of CS and to point out its potential for the natural products scientist for applications in natural products chemistry, metabolome, and proteome research involving organisms from terrestrial and marine sources.
Co-reporter:Andreas Schinkovitz, Samuel M. Pro, Matthew Main, Shao-Nong Chen, Birgit U. Jaki, David C. Lankin and Guido F. Pauli
Journal of Natural Products 2008 Volume 71(Issue 9) pp:1604-1611
Publication Date(Web):September 10, 2008
DOI:10.1021/np800137n
Monomeric phthalides such as Z-ligustilide (1) and Z-butylidenephthalide (2) are major constituents of medicinal plants of the Apiaceae family. While 1 has been associated with a variety of observed biological effects, it is also known for its instability and rapid chemical degradation. For the purpose of isolating pure 1 and 2, a gentle and rapid two-step countercurrent isolation procedure was developed. From a supercritical CO2 fluid extract of Angelica sinensis roots, the phthalides were isolated with high GC-MS purities of 99.4% for 1 and 98.9% for 2 and consistently lower qHNMR purities of 98.1% and 96.4%, respectively. Taking advantage of molarity-based qHNMR methodology, a time-resolved study of the dynamic changes and residual complexity of pure 1 was conducted. GC-MS and (qH)NMR analysis of artificially degraded 1 provided evidence for the phthalide degradation pathways and optimized storing conditions. Parallel qHNMR analysis led to the recognition of variations in time- and process-dependent sample purity and has impact on the overall assessment of time-dependent changes in complex natural products systems. The study underscores the importance of independent quantitative monitoring as a prerequisite for the biological evaluation of labile natural products such as monomeric phthalides.
Co-reporter:Birgit U. Jaki, Scott G. Franzblau, Lucas R. Chadwick, David C. Lankin, Fangqiu Zhang, Yuehong Wang and Guido F. Pauli
Journal of Natural Products 2008 Volume 71(Issue 10) pp:1742-1748
Publication Date(Web):September 18, 2008
DOI:10.1021/np800329j
The present study explores the variability of biological responses from the perspective of sample purity and introduces the concept of purity−activity relationships (PARs) in natural product research. The abundant plant triterpene ursolic acid (1) was selected as an exemplary natural product due to the overwhelming number yet inconsistent nature of its approximate 120 reported biological activities, which include anti-TB potential. Nine different samples of ursolic acid with purity certifications were obtained, and their purity was independently assessed by means of quantitative 1H NMR (qHNMR). Biological evaluation consisted of determining MICs against two strains of virulent Mycobacterium tuberculosis and IC50 values in Vero cells. Ab initio structure elucidation provided unequivocal structural confirmation and included an extensive 1H NMR spin system analysis, determination of nearly all J couplings and the complete NOE pattern, and led to the revision of earlier reports. As a net result, a sigmoid PAR profile of 1 was obtained, demonstrating the inverse correlation of purity and anti-TB bioactivity. The results imply that synergistic effects of 1 and its varying impurities are the likely cause of previously reported antimycobacterial potential. Generating PARs is a powerful extension of the routinely performed quantitative correlation of structure and activity ([Q]SAR). Advanced by the use of primary analytical methods such as qHNMR, PARs enable the elucidation of cases like 1 when increasing purity voids biological activity. This underlines the potential of PARs as a tool in drug discovery and synergy research and accentuates the need to routinely combine biological testing with purity assessment.
Co-reporter:J. Brent Friesen and Guido F. Pauli
Journal of Agricultural and Food Chemistry 2008 Volume 56(Issue 1) pp:19-28
Publication Date(Web):December 11, 2007
DOI:10.1021/jf072415a
A standard test mix consisting of 21 commercially available natural products of agricultural significance, termed the GUESSmix, was employed to measure the countercurrent chromatography performance characteristics of a very popular quaternary solvent system family made up of hexane−ethyl acetate−methanol–water (HEMWat). The polarity range of the GUESSmix combined with the elution−extrusion countercurrent chromatography (EECCC) technique and the newly developed reciprocal symmetry (ReS) and reciprocal shifted symmetry (ReSS) plots allow liquid–liquid distribution ratios ( K D) to be plotted for every compound eluted on a scale of zero to infinity. It was demonstrated that 16 of the 21 GUESSmix compounds are found in the optimal range of resolution (0.25 < K D < 16) of at least one HEMWat solvent system. The HEMWat solvent systems represented by the ratios 4:6:5:5, 4:6:4:6, and 3:7:4:6 possess the most densely populated optimal ranges of resolution for this standard mix. ReS plots have been shown to reveal the symmetrical reversibility of the EECCC method in reference to K D = 1. This study lays the groundwork for evaluation and comparison of solvent system families proposed in the literature, as well as the creation of new solvent system families with desired performance characteristics.
Co-reporter:J. Brent Friesen, Guido F. Pauli
Journal of Chromatography A 2007 Volume 1151(1–2) pp:51-59
Publication Date(Web):1 June 2007
DOI:10.1016/j.chroma.2007.01.126
Application of a mixture of 21 commercially available natural products, termed the GUESSmix, established a standard test that allows for a systematic analysis and comparison of the properties of biphasic solvent systems in counter-current/partition chromatography. Because the GUESSmix is comprised of compounds with varying polarities, functional groups, and structural features, it proves to be a rational method for mapping the optimal resolution polarity range of a particular solvent system. The mapping of optimal resolution polarity ranges of solvent systems provided for the description of the overall optimal resolution polarity range of a solvent system family, comprised of the same solvents in different proportions. By comparing the GUESSmix performance in the individual members of a solvent system family, the solvent system that best functions as the representative of, or portal to, the solvent system families was determined. The GUESSmix also afforded a method to compare the overall optimal resolution polarity ranges of solvent system families. Based on performance of GUESSmix chromatograms, the counter-current chromatography (CCC) properties of a two ternary literature solvent systems, ethyl acetate/n-butanol/water (EBuWat) and t-butylmethylether/acetonitrile/water (terAcWat), were explored in order to contrast and compare their CCC potential. A quaternary solvent system, hexane/t-butylmethylether/acetonitrile/water (HterAcWat), was also formulated and studied. The results indicated that the GUESSmix is fit for the purpose of developing and evaluating CCC solvent system families with desired performance characteristics.
Co-reporter:Ryan J. Case, Yuehong Wang, Scott G. Franzblau, D. Doel Soejarto, Lohi Matainaho, Pius Piskaut, Guido F. Pauli
Journal of Chromatography A 2007 Volume 1151(1–2) pp:169-174
Publication Date(Web):1 June 2007
DOI:10.1016/j.chroma.2007.01.022
Through several steps of in vitro based bioassay-guided fractionation, three highly potent anti-mycobacterial constituents (1–2 μg/mL minimum inhibitory concentrations) were isolated from Dracaena angustifolia Roxb. (Dracaenaceae). Isolation was expedited due to the application of new techniques in counter-current chromatography (CCC). The first of these applications, gradient-array CCC, enables the expansion of the high-resolution window (sweet spot) by applying successive CCC separations in different solvent systems to a crude extract. Further fractionation was also performed using the recently designed 1 L fast-centrifugal partition chromatography (FCPC) rotor with the solvent system hexane:methyl-tert butylether:acetonitrile (10:1:10). Results indicated that gradient-array CCC and high-capacity FCPC can facilitate drug discovery efforts from complex natural products, increase reproducibility of separation schemes, and provide more rigid dereplication of previously isolated bioactive compounds.
Co-reporter:Taichi Inui, Yuehong Wang, Shixin Deng, David C. Smith, Scott G. Franzblau, Guido F. Pauli
Journal of Chromatography A 2007 Volume 1151(1–2) pp:211-215
Publication Date(Web):1 June 2007
DOI:10.1016/j.chroma.2007.01.127
The crude extract of an Alaskan ethnobotanical plant, Oplopanax horridus, was subjected to counter-current chromatography (CCC), and the selected active regions were evaluated for their synergistic effects with an in vitro model of anti-tubercular efficacy. CCC as a support-free high-resolution separation method was employed to preclude potential irreversible absorption to a solid stationary phase. The microplate Alamar blue assay and the isobole method were used to measure the biological activity and eliminate dose–response dependent errors, respectively. Using the combination of CCC, bioassay and isobole method, significant synergistic effects were observed. Among the entire polarity range, fractions with distribution constant between 0.44 and 0.81 showed the most synergistic enhancement with an increase in potency by 108% for the recombined fractions.
Co-reporter:Matthias Niemitz;Robert Kleps;Reino Laatikainen;Shao-Nong Chen;Alan P. Kozikowski
Magnetic Resonance in Chemistry 2007 Volume 45(Issue 10) pp:878-882
Publication Date(Web):29 AUG 2007
DOI:10.1002/mrc.2061
Complete analysis of the 1H NMR spectrum of huperzine A, 1-amino-13-ethylidene-11-methyl-6-aza-tricyclo[7.3.1.02, 7]trideca-2(7),3,10-trien-5-one, a Lycopodium alkaloid and anti-Alzheimer drug lead containing an ABCD(E)(MN)(OP)X3Y3–type system of 15 nonexchangeable proton spins, is reported for the first time, and earlier assignments are corrected. The complete 1H parameter set of 11 chemical shifts clarifies the diastereotopism of both methylene groups, and provides a total of 38 observed H,H-couplings including 31 long-range (4–6J) connectivities. The NMR data is consistent with the comparatively rigid alicyclic backbone predicted by molecular mechanics calculations, and forms the basis for 1H NMR fingerprint analysis for the purpose of dereplication, purity analysis, and elucidation of structural analogs. Copyright © 2007 John Wiley & Sons, Ltd.
Co-reporter:Shixin Deng;Shao-Nong Chen;Jian Lu;Zaijie Jim Wang;Dejan Nikolic;Richard B. van Breemen;Bernard D. Santarsiero;Andrew Mesecar;Harry H.S. Fong;Norman R. Farnsworth
Phytochemical Analysis 2006 Volume 17(Issue 6) pp:398-405
Publication Date(Web):7 AUG 2006
DOI:10.1002/pca.937
The methanol extract of Angelica sinensis (Oliv.) Diels roots (Dang Gui) has been shown to exhibit competitive binding to the GABAa receptor, suggesting the presence of GABAergic ligands. Chromatographic fractionation of the methanol extract led to the isolation of two GABAergic dimeric phthalides 1 and 2. Gelispirolide (1) was elucidated as a new phthalide dimer composed of a Z-ligustilide and a Z-butylidenephthalide unit on the basis of spectroscopic approaches including one- and two-dimensional NMR, HRESIMS and HRESIMS-MS. Compound 2 was identified as the known dimeric phthalide, riligustilide, by comparison of its spectroscopic data with literature values. Its dimeric linkage and stereochemistry were ascertained by a single crystal X-ray diffraction experiment. Both dimers 1 and 2 were found to be active in an in vitro GABAa receptor-binding assay with IC50 values of 29 and 24 µm, respectively. Copyright © 2006 John Wiley & Sons, Ltd.
Co-reporter:Birgit Jaki;Scott Franzblau
Phytochemical Analysis 2004 Volume 15(Issue 4) pp:213-219
Publication Date(Web):28 JUN 2004
DOI:10.1002/pca.760
State-of-the-art structure elucidation and dereplication of natural products is incomplete without the determination of enantiomeric purity, especially when compounds are to be biologically evaluated. An NMR procedure is presented in order to distinguish and determine enantiomers in natural product samples. The method is also of value in the structure elucidation process by providing information, which is otherwise of a non-routine nature. Using enantiomeric 1-acetoxychavicol acetates and carvones as model compounds, this study presents a chiral NMR procedure that allows distinction and determination of chiral antipodes of natural products in a routine set-up. Copyright © 2004 John Wiley & Sons, Ltd.
Co-reporter:Yang Liu, Jahir Garzon, J. Brent Friesen, Yu Zhang, James B. McAlpine, David C. Lankin, Shao-Nong Chen, Guido F. Pauli
Fitoterapia (July 2016) Volume 112() pp:30-37
Publication Date(Web):1 July 2016
DOI:10.1016/j.fitote.2016.04.019
NAtural Deep Eutectic Solvents (NADES) are chemically simple but physiologically important plant constituents that exhibit unique solubilizing properties of other metabolites, including bioactive constituents. The high polarity of NADES introduces a challenge in the ability of conventional solid-support based chromatography to recover potential bioactive metabolites. This complicates the systematic explanation of the NADES' functions in botanical extracts. The present study utilizes countercurrent separation (CCS) methodology to overcome the recovery challenge. To demonstrate its feasibility, Glucose-Choline chloride-Water (GCWat, 2:5:5, mole/mole) served as a model NADES, and four widely used marker flavonoids with different polarities (rutin, quercetin, kaempferol, and daidzein) were chosen as model target analytes. In order to prepare GCWat with high consistency, a water drying study was performed. The unique capabilities of the recently introduced CherryOne system, offering volumetric phase metering, were used to monitor the CCS operations. The collected fractions were analyzed using UHPLC and NMR/quantitative NMR. CCS was able to recover the analytes from the NADES matrix with quantitative recoveries of 95.7%, 94.6%, 97.0%, and 96.7% for rutin, quercetin, kaempferol, and daidzein respectively. The CCS strategy enables recovery of target metabolites from NADES-containing crude extracts as well as from other chemical mixtures, and moreover offers a means of using NADES as environmentally friendly extraction solvents.Download high-res image (154KB)Download full-size image
Co-reporter:Charlotte Simmler, José G Napolitano, James B McAlpine, Shao-Nong Chen, Guido F Pauli
Current Opinion in Biotechnology (February 2014) Volume 25() pp:51-59
Publication Date(Web):1 February 2014
DOI:10.1016/j.copbio.2013.08.004
•qNMR is a powerful analytical tool for structure and purity determination.•qNMR is suitable for the quality control of complex natural samples.•qNMR and MS-hyphenated chromatography are complementary, orthogonal techniques.•qNMR has a strong track record for metabolomic studies of biological systems.•NMR-based metabolomics is a diagnostic tool for human diseases.Nuclear Magnetic Resonance (NMR) is a universal and quantitative analytical technique. Being a unique structural tool, NMR also competes with metrological techniques for purity determination and reference material analysis. In pharmaceutical research, applications of quantitative NMR (qNMR) cover mostly the identification and quantification of drug and biological metabolites. Offering an unbiased view of the sample composition, and the possibility to simultaneously quantify multiple compounds, qNMR has become the method of choice for metabolomic studies and quality control of complex natural samples such as foods, plants or herbal remedies, and biofluids. In this regard, NMR-based metabolomic studies, dedicated to both the characterization of herbal remedies and clinical diagnosis, have increased considerably.Download high-res image (306KB)Download full-size image
Co-reporter:Guido F. Pauli
European Journal of Integrative Medicine (December 2014) Volume 6(Issue 6) pp:
Publication Date(Web):1 December 2014
DOI:10.1016/j.eujim.2014.09.022
Co-reporter:Bethany G. Elkington, Kongmany Sydara, Andrew Newsome, Chang Hwa Hwang, David C. Lankin, Charlotte Simmler, José G. Napolitano, Richard Ree, James G. Graham, Charlotte Gyllenhaal, Somsanith Bouamanivong, Onevilay Souliya, Guido F. Pauli, Scott G. Franzblau, Djaja Djendoel Soejarto
Journal of Ethnopharmacology (3 February 2014) Volume 151(Issue 2) pp:903-911
Publication Date(Web):3 February 2014
DOI:10.1016/j.jep.2013.11.057
Ethnopharmacological relevanceThere is widespread use of traditional herbal remedies in the Lao PDR (Laos). It is common practice to treat many diseases with local plants. This research project documented and analysed some of these traditional remedies used to treat symptoms of tuberculosis (TB).Materials and methodsThis research was executed by interviewing healers about plants used traditionally to treat the symptoms of TB. Samples of some of the plants were collected, and extracts of 77 species were submitted to various in vitro assays in order to determine the amount of growth inhibition of virulent Mycobacterium tuberculosis H37Rv (Mtb), as opposed to other microbes and mammalian Vero cells.ResultsInterviews took place with 58 contemporary healers in 5 different provinces about plants currently used, giving a list of 341 plants. Bioassay-guided fractionation was performed on Marsypopetalum modestum (Pierre) B. Xue and R.M.K. Saunders (Annonaceae), leading to the isolation of dipyrithione, an anti-mycobacterial compound isolated for the first time from the genus Marsypopetalum through this research.ConclusionsThis research has helped to increase awareness of Laos’ rich diversity of medicinal plants and will hopefully provide incentive to preserve the undeveloped forested areas that remain, which still hold a wealth of medical information for future discoveries.Download high-res image (195KB)Download full-size image
Co-reporter:J. Brent Friesen; James B. McAlpine; Shao-Nong Chen
Journal of Natural Products () pp:
Publication Date(Web):
DOI:10.1021/np501065h
This work assesses the current instrumentation, method development, and applications in countercurrent chromatography (CCC) and centrifugal partition chromatography (CPC), collectively referred to as countercurrent separation (CCS). The article provides a critical review of the CCS literature from 2007 since our last review (J. Nat. Prod. 2008, 71, 1489–1508), with a special emphasis on the applications of CCS in natural products research. The current state of CCS is reviewed in regard to three continuing topics (instrumentation, solvent system development, theory) and three new topics (optimization of parameters, workflow, bioactivity applications). The goals of this review are to deliver the necessary background with references for an up-to-date perspective of CCS, to point out its potential for the natural product scientist, and thereby to induce new applications in natural product chemistry, metabolome, and drug discovery research involving organisms from terrestrial and marine sources.











![Bicyclo[7.2.0]undec-3-en-5-ol, 4,8,11,11-tetramethyl-, (3Z)-](http://img.cochemist.com/ccimg/913200/913176-41-7.png)
![Bicyclo[7.2.0]undec-3-en-5-ol, 4,8,11,11-tetramethyl-, (3Z)-](http://img.cochemist.com/ccimg/913200/913176-41-7_b.png)



![5H-Imidazo[2,1-b][1,3]oxazine, 6,7-dihydro-2-nitro-6-[[4-(trifluoromethoxy)phenyl]methoxy]-, (6S)-](http://img.cochemist.com/ccimg/187300/187235-37-6.png)
![5H-Imidazo[2,1-b][1,3]oxazine, 6,7-dihydro-2-nitro-6-[[4-(trifluoromethoxy)phenyl]methoxy]-, (6S)-](http://img.cochemist.com/ccimg/187300/187235-37-6_b.png)
![[1(2H),2'-Bi-3H-indole]-3,3'-dione, 2,2-dichloro-](http://img.cochemist.com/ccimg/186200/186136-15-2.png)
![[1(2H),2'-Bi-3H-indole]-3,3'-dione, 2,2-dichloro-](http://img.cochemist.com/ccimg/186200/186136-15-2_b.png)


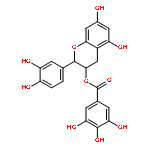
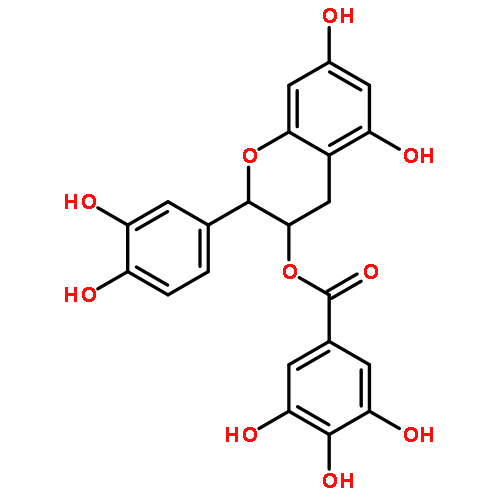




![12-Oxabicyclo[9.1.0]dodeca-3,7-diene, 1,5,5,8-tetramethyl-](http://img.cochemist.com/ccimg/90900/90820-79-4.png)
![12-Oxabicyclo[9.1.0]dodeca-3,7-diene, 1,5,5,8-tetramethyl-](http://img.cochemist.com/ccimg/90900/90820-79-4_b.png)






![Kaur-16-en-18-oic acid,13-[[2-O-(6-deoxy-a-L-mannopyranosyl)-b-D-glucopyranosyl]oxy]-, b-D-glucopyranosyl ester, (4a)-](http://img.cochemist.com/ccimg/64500/64432-06-0.png)
![Kaur-16-en-18-oic acid,13-[[2-O-(6-deoxy-a-L-mannopyranosyl)-b-D-glucopyranosyl]oxy]-, b-D-glucopyranosyl ester, (4a)-](http://img.cochemist.com/ccimg/64500/64432-06-0_b.png)


![(E)-3-(4-hydroxyphenyl)-1-[2-hydroxy-4-[(2S,3R,4S,5S,6R)-3,4,5-trihydroxy-6-(hydroxymethyl)tetrahydropyran-2-yl]oxy-phenyl]prop-2-en-1-one](http://img.cochemist.com/ccimg/59200/59122-93-9.png)
![(E)-3-(4-hydroxyphenyl)-1-[2-hydroxy-4-[(2S,3R,4S,5S,6R)-3,4,5-trihydroxy-6-(hydroxymethyl)tetrahydropyran-2-yl]oxy-phenyl]prop-2-en-1-one](http://img.cochemist.com/ccimg/59200/59122-93-9_b.png)
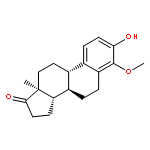
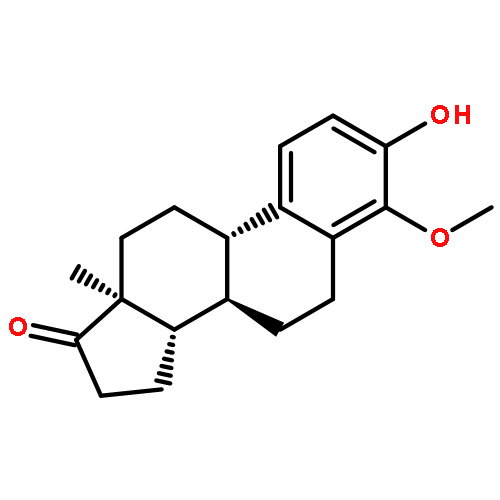





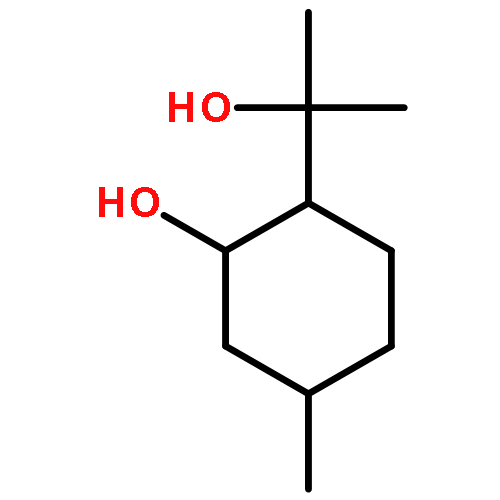
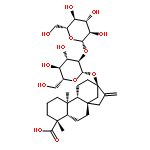
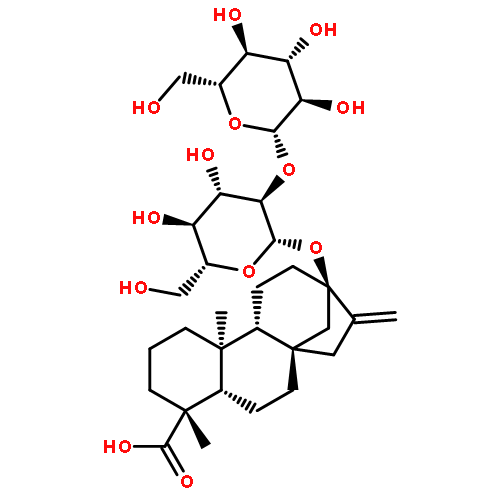



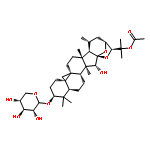
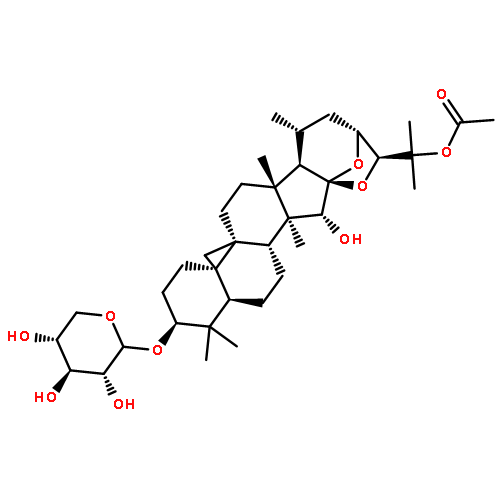





![1H-Cycloprop[e]azulene,decahydro-1,1,7-trimethyl-4-methylene-, (1aR,4aS,7R,7aR,7bS)-](http://img.cochemist.com/ccimg/25300/25246-27-9.png)
![1H-Cycloprop[e]azulene,decahydro-1,1,7-trimethyl-4-methylene-, (1aR,4aS,7R,7aR,7bS)-](http://img.cochemist.com/ccimg/25300/25246-27-9_b.png)
![(2R,2'R,3S,3'S,4S)-2,2'-Bis(3,4-dihydroxyphenyl)-[4,8'-bichroman]-3,3',5,5',7,7'-hexaol](http://img.cochemist.com/ccimg/23600/23567-23-9.png)
![(2R,2'R,3S,3'S,4S)-2,2'-Bis(3,4-dihydroxyphenyl)-[4,8'-bichroman]-3,3',5,5',7,7'-hexaol](http://img.cochemist.com/ccimg/23600/23567-23-9_b.png)





![[4,8'-Bi-2H-1-benzopyran]-3,3',5,5',7,7'-hexol,2'-(3,5-dihydroxy-4-methoxyphenyl)-3,3',4,4'-tetrahydro-2-(4-hydroxyphenyl)-,(2R,2'R,3R,3'R,4R)-](http://img.cochemist.com/ccimg/18300/18206-61-6.png)
![[4,8'-Bi-2H-1-benzopyran]-3,3',5,5',7,7'-hexol,2'-(3,5-dihydroxy-4-methoxyphenyl)-3,3',4,4'-tetrahydro-2-(4-hydroxyphenyl)-,(2R,2'R,3R,3'R,4R)-](http://img.cochemist.com/ccimg/18300/18206-61-6_b.png)
![8,14-Methano-2H,14H-1-benzopyrano[7,8-d][1,3]benzodioxocin-3,5,11,13,15-pentol,2,8-bis(3,4-dihydroxyphenyl)-3,4-dihydro-](http://img.cochemist.com/ccimg/12800/12798-56-0.png)
![8,14-Methano-2H,14H-1-benzopyrano[7,8-d][1,3]benzodioxocin-3,5,11,13,15-pentol,2,8-bis(3,4-dihydroxyphenyl)-3,4-dihydro-](http://img.cochemist.com/ccimg/12800/12798-56-0_b.png)
![[(1s,4ar,5s,7as)-5-hydroxy-1-[(2s,3r,4s,5s,6r)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxy-1,4a,5,7a-tetrahydrocyclopenta[c]pyran-7-yl]methyl 4-hydroxybenzoate](http://img.cochemist.com/ccimg/11100/11027-63-7.png)
![[(1s,4ar,5s,7as)-5-hydroxy-1-[(2s,3r,4s,5s,6r)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxy-1,4a,5,7a-tetrahydrocyclopenta[c]pyran-7-yl]methyl 4-hydroxybenzoate](http://img.cochemist.com/ccimg/11100/11027-63-7_b.png)
![[(2R,3S)-5,7-dihydroxy-2-(3,4,5-trihydroxyphenyl)-3,4-dihydro-2H-chromen-3-yl] 3,4,5-trihydroxybenzoate](http://img.cochemist.com/ccimg/5200/5127-64-0.png)
![[(2R,3S)-5,7-dihydroxy-2-(3,4,5-trihydroxyphenyl)-3,4-dihydro-2H-chromen-3-yl] 3,4,5-trihydroxybenzoate](http://img.cochemist.com/ccimg/5200/5127-64-0_b.png)


![4H-1,3-Dioxino[4,5-c]pyridine-5-methanol,2,2,8-trimethyl-](http://img.cochemist.com/ccimg/1200/1136-52-3.png)
![4H-1,3-Dioxino[4,5-c]pyridine-5-methanol,2,2,8-trimethyl-](http://img.cochemist.com/ccimg/1200/1136-52-3_b.png)
![1H-Indol-5-ol,3-[2-(methylamino)ethyl]-](http://img.cochemist.com/ccimg/1200/1134-01-6.png)
![1H-Indol-5-ol,3-[2-(methylamino)ethyl]-](http://img.cochemist.com/ccimg/1200/1134-01-6_b.png)
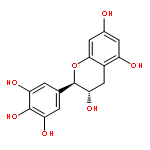
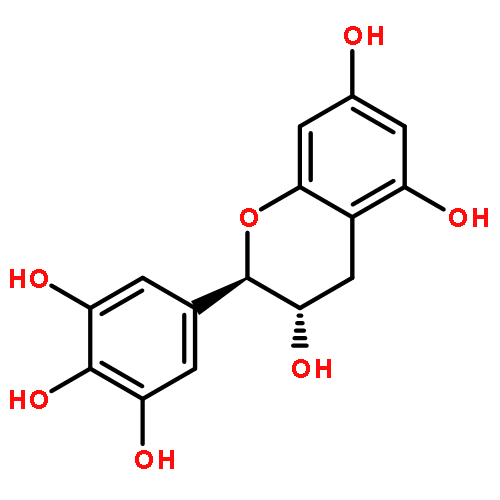




![(1aR,4R,4aS,7R,7aS,7bS)-1,1,4,7-tetramethyldecahydro-1H-cyclopropa[e]azulen-4-ol](http://img.cochemist.com/ccimg/600/577-27-5.png)
![(1aR,4R,4aS,7R,7aS,7bS)-1,1,4,7-tetramethyldecahydro-1H-cyclopropa[e]azulen-4-ol](http://img.cochemist.com/ccimg/600/577-27-5_b.png)
![1-[(4-METHOXYPHENYL)METHYL]-5-PYRIDIN-3-YLPYRROLIDIN-2-ONE](http://img.cochemist.com/ccimg/600/552-02-3.png)
![1-[(4-METHOXYPHENYL)METHYL]-5-PYRIDIN-3-YLPYRROLIDIN-2-ONE](http://img.cochemist.com/ccimg/600/552-02-3_b.png)

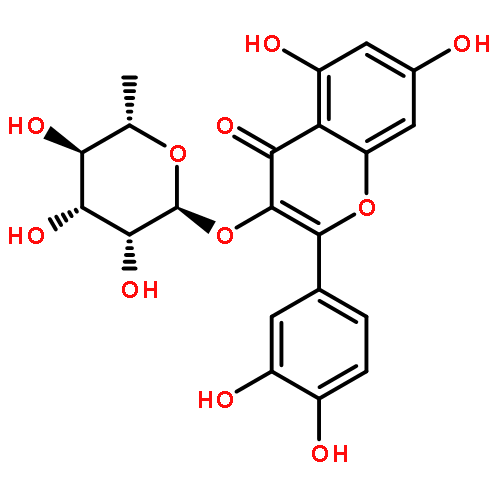


![1H-Cycloprop[e]azulen-4-ol,decahydro-1,1,4,7-tetramethyl-, (1aR,4R,4aR,7R,7aS,7bS)-](http://img.cochemist.com/ccimg/500/489-41-8.png)
![1H-Cycloprop[e]azulen-4-ol,decahydro-1,1,4,7-tetramethyl-, (1aR,4R,4aR,7R,7aS,7bS)-](http://img.cochemist.com/ccimg/500/489-41-8_b.png)
![1H-Cycloprop[e]azulene,decahydro-1,1,7-trimethyl-4-methylene-, (1aR,4aR,7R,7aR,7bS)-](http://img.cochemist.com/ccimg/500/489-39-4.png)
![1H-Cycloprop[e]azulene,decahydro-1,1,7-trimethyl-4-methylene-, (1aR,4aR,7R,7aR,7bS)-](http://img.cochemist.com/ccimg/500/489-39-4_b.png)
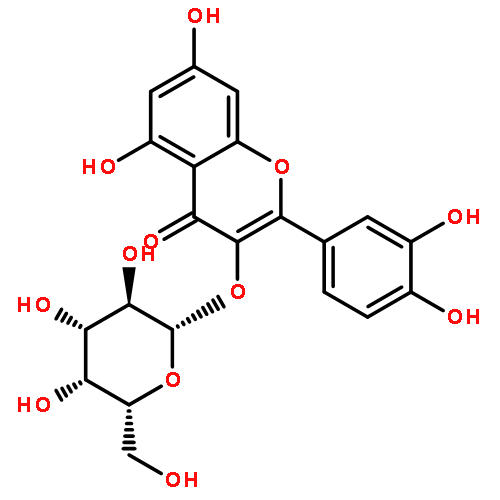
![2-(3,4-dihydroxyphenyl)-5,7-dihydroxy-3-[(2R,3S,4R,5S,6S)-3,4,5-trihydroxy-6-(hydroxymethyl)tetrahydropyran-2-yl]oxy-chromen-4-one](http://img.cochemist.com/ccimg/500/482-35-9.png)
![2-(3,4-dihydroxyphenyl)-5,7-dihydroxy-3-[(2R,3S,4R,5S,6S)-3,4,5-trihydroxy-6-(hydroxymethyl)tetrahydropyran-2-yl]oxy-chromen-4-one](http://img.cochemist.com/ccimg/500/482-35-9_b.png)
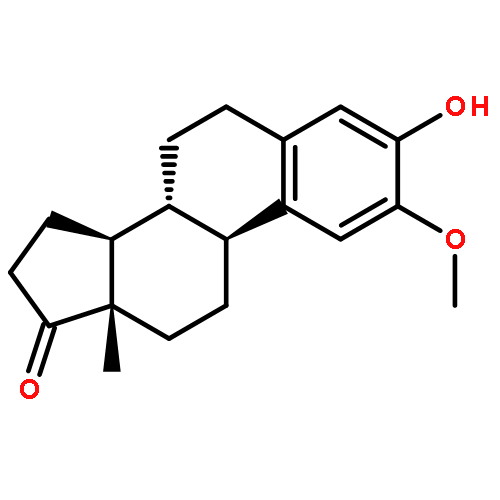
![10H-3,10a-Epidithiopyrazino[1,2-a]indole-1,4-dione,2,3,5a,6-tetrahydro-6-hydroxy-3-(hydroxymethyl)-2-methyl-, (3R,5aS,6S,10aR)-](http://img.cochemist.com/ccimg/100/67-99-2.png)
![10H-3,10a-Epidithiopyrazino[1,2-a]indole-1,4-dione,2,3,5a,6-tetrahydro-6-hydroxy-3-(hydroxymethyl)-2-methyl-, (3R,5aS,6S,10aR)-](http://img.cochemist.com/ccimg/100/67-99-2_b.png)






![PROPANEDIOIC ACID, [(4-HYDROXYPHENYL)METHYL]-](http://img.cochemist.com/ccimg/90900/90844-16-9.png)
![PROPANEDIOIC ACID, [(4-HYDROXYPHENYL)METHYL]-](http://img.cochemist.com/ccimg/90900/90844-16-9_b.png)






![[4,8':4',8''-Ter-2H-1-benzopyran]-3,3',3'',5,5',5'',7,7',7''-nonol,2,2',2''-tris(3,4-dihydroxyphenyl)-3,3',3'',4,4',4''-hexahydro-,(2R,2'R,2''R,3R,3'R,3''R,4R,4'S)-](http://img.cochemist.com/ccimg/37100/37064-30-5.png)
![[4,8':4',8''-Ter-2H-1-benzopyran]-3,3',3'',5,5',5'',7,7',7''-nonol,2,2',2''-tris(3,4-dihydroxyphenyl)-3,3',3'',4,4',4''-hexahydro-,(2R,2'R,2''R,3R,3'R,3''R,4R,4'S)-](http://img.cochemist.com/ccimg/37100/37064-30-5_b.png)

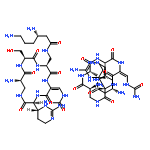

![(e)-1-(2,4-dihydroxyphenyl)-3-[4-[(2s,3r,4s,5s,6r)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxyphenyl]prop-2-en-1-one](http://img.cochemist.com/ccimg/5100/5041-81-6.png)
![(e)-1-(2,4-dihydroxyphenyl)-3-[4-[(2s,3r,4s,5s,6r)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxyphenyl]prop-2-en-1-one](http://img.cochemist.com/ccimg/5100/5041-81-6_b.png)





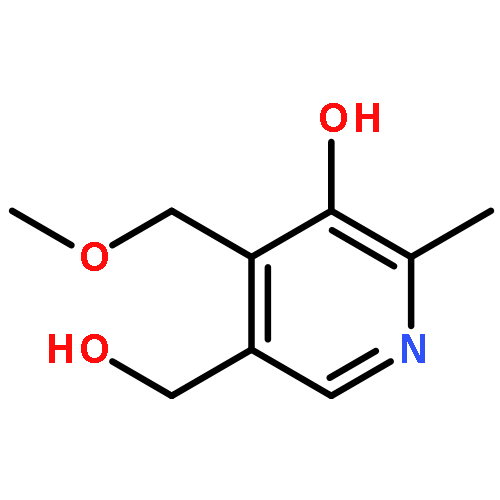
![(2s)-7-hydroxy-2-[4-[(2s,3r,4s,5s,6r)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxyphenyl]-2,3-dihydrochromen-4-one](http://img.cochemist.com/ccimg/600/551-15-5.png)
![(2s)-7-hydroxy-2-[4-[(2s,3r,4s,5s,6r)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxyphenyl]-2,3-dihydrochromen-4-one](http://img.cochemist.com/ccimg/600/551-15-5_b.png)
![2-Naphthalenemethanol, decahydro-α,α,4a-trimethyl-8-methylene-, [2R-(2α,4aα,8aβ)]-](http://img.cochemist.com/ccimg/500/473-15-4.png)
![2-Naphthalenemethanol, decahydro-α,α,4a-trimethyl-8-methylene-, [2R-(2α,4aα,8aβ)]-](http://img.cochemist.com/ccimg/500/473-15-4_b.png)