Co-reporter:Fotini Liepouri, Giovanni Bernasconi, and Nicos A. Petasis
Organic Letters 2015 Volume 17(Issue 7) pp:1628-1631
Publication Date(Web):March 19, 2015
DOI:10.1021/acs.orglett.5b00024
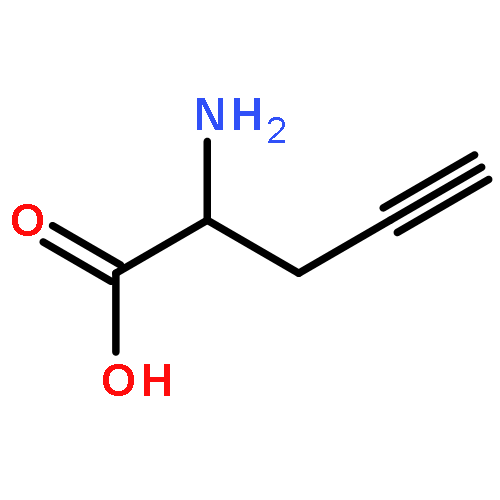
A one-step, three-component condensation of allenyl boronic acids or allenyl pinacolboronates with amines and aldehydes affords α-allenyl or α-propargyl α-amino acids and anti-β-amino alcohols. This process gives the allenyl or propargyl product depending on the amine and boron components. Secondary amines generate exclusively α-allenyl α-amino acids, while primary aliphatic amines lead to α-propargyl α-amino acids. Secondary aliphatic amines react with chiral α-hydroxy aldehydes and allenyl boron derivatives to form stereoselectively allenyl anti-β-amino alcohol products.
Co-reporter:Stephen J. Glynn, Kevin J. Gaffney, Marcos A. Sainz, Stan G. Louie and Nicos A. Petasis
Organic & Biomolecular Chemistry 2015 vol. 13(Issue 13) pp:3887-3899
Publication Date(Web):30 Jan 2015
DOI:10.1039/C4OB02512A
The green tea polyphenol epigallocatechin-3-gallate (EGCG) was reported to effectively antagonize the ability of Bortezomib (BZM) to induce apoptosis in cancer cells. This interaction was attributed to the formation of a covalent adduct between a phenolic moiety of EGCG with the boronic acid group of Bortezomib. However, the structural details of this boron adduct and the molecular factors that contribute to its formation and its ability to inhibit Bortezomib's activity remain unclear. This paper describes the use of NMR spectroscopy and cell assays to characterize the structures and properties of the boron adducts of EGCG and related polyphenols. The observed boron adducts included both boronate and borate derivatives, and their structural characteristics were correlated with cell-based evaluation of the ability of EGCG and other phenols to antagonize the anticancer activity of Bortezomib. The enhanced stability of the BZM/EGCG adduct was attributed to electronic and steric reasons, and a newly identified intramolecular interaction of the boron atom of BZM with the adjacent amide bond. The reported approach provides a useful method for determining the potential ability of polyphenols to form undesired adducts with boron-based drugs and interfere with their actions.
Co-reporter:Jeremy W. Winkler, Jasim Uddin, Charles N. Serhan, and Nicos A. Petasis
Organic Letters 2013 Volume 15(Issue 7) pp:1424-1427
Publication Date(Web):March 19, 2013
DOI:10.1021/ol400484u
The first total synthesis of stereochemically pure resolvin D3 and aspirin-triggered resolvin D3 is reported. These enzymatic metabolites of docosahexaenoic acid (DHA) have potent anti-inflammatory and pro-resolving actions. The convergent synthetic strategy is based on enantiomerically pure starting materials, and it is highly stereocontrolled.
Co-reporter:Jesmond Dalli, Jeremy W. Winkler, Romain A. Colas, Hildur Arnardottir, Chien-Yee C. Cheng, Nan Chiang, Nicos A. Petasis, Charles N. Serhan
Chemistry & Biology 2013 Volume 20(Issue 2) pp:188-201
Publication Date(Web):21 February 2013
DOI:10.1016/j.chembiol.2012.11.010
Resolvins are a family of n-3 lipid mediators initially identified in resolving inflammatory exudates that temper inflammatory responses to promote catabasis. Here, temporal metabololipidomics with self-limited resolving exudates revealed that resolvin (Rv) D3 has a distinct time frame from other lipid mediators, appearing late in the resolution phase. Using synthetic materials prepared by stereocontrolled total organic synthesis and metabololipidomics, we established complete stereochemistry of RvD3 and its aspirin-triggered 17R-epimer (AT-RvD3). Both synthetic resolvins potently regulated neutrophils and mediators, reducing murine peritonitis and dermal inflammation. RvD3 and AT-RvD3 displayed leukocyte-directed actions, e.g., blocking human neutrophil transmigration and enhancing macrophage phagocytosis and efferocytosis. These results position RvD3 uniquely within the inflammation-resolution time frame to vantage and contribute to the beneficial actions of aspirin and essential n-3 fatty acids.Graphical AbstractFigure optionsDownload full-size imageDownload high-quality image (133 K)Download as PowerPoint slideHighlights► RvD3 accumulates in late inflammation resolution ► Aspirin triggers biosynthesis of RvD3 17R-epimer (AT-RvD3) that is proresolving ► Complete stereochemistry of RvD3 and AT-RvD3 are established ► RvD3 and AT-RvD3 govern leukocyte functions and local inflammatory responses
Co-reporter:Nicos A. Petasis, Rong Yang, Jeremy W. Winkler, Min Zhu, Jasim Uddin, Nicolas G. Bazan, Charles N. Serhan
Tetrahedron Letters 2012 Volume 53(Issue 14) pp:1695-1698
Publication Date(Web):4 April 2012
DOI:10.1016/j.tetlet.2012.01.032
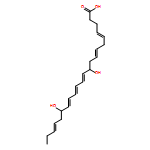
Neuroprotectin D1/Protectin D1, a potent anti-inflammatory, proresolving, and neuroprotective lipid mediator derived biosynthetically from docosahexaenoic acid, was prepared in an enantiomerically pure form via total organic synthesis. The synthetic strategy is highly stereocontrolled and convergent, featuring epoxide opening of glycidol starting materials for the introduction of the 10(R) and 17(S) hydroxyl groups. The desired alkene Z geometry was secured via the cis-reduction of alkyne precursors, while the conjugated E,E,Z triene was introduced at the end, in order to minimize Z/E isomerization. The same strategy, was also employed for the total synthesis of aspirin-triggered neuroprotectin D1/protectin D1 having the 17(R)-stereochemistry. Synthetic compounds obtained with the reported method were matched with endogenously derived materials, and helped establish their complete stereochemistry.
Co-reporter:Shili Xu;Roppei Yamada;Alexey N. Butkevich;Ebrahim Zandi;Roger Duncan;Bikash Debnath;Yu Zhou;Nouri Neamati
PNAS 2012 Volume 109 (Issue 40 ) pp:16348-16353
Publication Date(Web):2012-10-02
DOI:10.1073/pnas.1205226109
Protein disulfide isomerase (PDI), an endoplasmic reticulum chaperone protein, catalyzes disulfide bond breakage, formation,
and rearrangement. The effect of PDI inhibition on ovarian cancer progression is not yet clear, and there is a need for potent,
selective, and safe small-molecule inhibitors of PDI. Here, we report a class of propynoic acid carbamoyl methyl amides (PACMAs)
that are active against a panel of human ovarian cancer cell lines. Using fluorescent derivatives, 2D gel electrophoresis,
and MS, we established that PACMA 31, one of the most active analogs, acts as an irreversible small-molecule inhibitor of
PDI, forming a covalent bond with the active site cysteines of PDI. We also showed that PDI activity is essential for the
survival and proliferation of human ovarian cancer cells. In vivo, PACMA 31 showed tumor targeting ability and significantly
suppressed ovarian tumor growth without causing toxicity to normal tissues. These irreversible small-molecule PDI inhibitors
represent an important approach for the development of targeted anticancer agents for ovarian cancer therapy, and they can
also serve as useful probes for investigating the biology of PDI-implicated pathways.
Co-reporter:Charles N. Serhan and Nicos A. Petasis
Chemical Reviews 2011 Volume 111(Issue 10) pp:5922
Publication Date(Web):July 18, 2011
DOI:10.1021/cr100396c
Co-reporter:Nicos A. Petasis, Alexey N. Butkevich
Journal of Organometallic Chemistry 2009 694(11) pp: 1747-1753
Publication Date(Web):
DOI:10.1016/j.jorganchem.2008.11.050
Co-reporter:Nicos A. Petasis, Raquel Keledjian, Yee-Ping Sun, Kalyan C. Nagulapalli, Eric Tjonahen, Rong Yang, Charles N. Serhan
Bioorganic & Medicinal Chemistry Letters 2008 Volume 18(Issue 4) pp:1382-1387
Publication Date(Web):15 February 2008
DOI:10.1016/j.bmcl.2008.01.013
A new class of chemically and metabolically stable lipoxin analogs featuring a replacement of the tetraene unit of native LXA4 with a substituted benzo-fused ring system have been designed and studied. These molecules were readily synthesized via a convergent synthetic route involving iterative palladium-mediated cross-coupling, and exhibit enhanced chemical stability, as well as resistance to metabolic inactivation via eicosanoid oxido-reductase. These new LX analogs were evaluated in a model of acute inflammation and were shown to exhibit potent anti-inflammatory properties, significantly decreasing neutrophil infiltration in vivo. The most potent among these was compound 9 (o-[9,12]-benzo-15-epi-LXA4 methyl ester. Taken together, these findings help identify a new class of stable and easily prepared LX analogs that may serve as novel tools and as promising leads for new anti-inflammatory agents with improved therapeutic profile.New types of benzo-lipoxin A4 analogs, such as 9, were synthesized and shown to have potent anti-inflammatory properties by suppressing neutrophil infiltration in vivo.
Co-reporter:Stephen J. Glynn, Kevin J. Gaffney, Marcos A. Sainz, Stan G. Louie and Nicos A. Petasis
Organic & Biomolecular Chemistry 2015 - vol. 13(Issue 13) pp:NaN3899-3899
Publication Date(Web):2015/01/30
DOI:10.1039/C4OB02512A
The green tea polyphenol epigallocatechin-3-gallate (EGCG) was reported to effectively antagonize the ability of Bortezomib (BZM) to induce apoptosis in cancer cells. This interaction was attributed to the formation of a covalent adduct between a phenolic moiety of EGCG with the boronic acid group of Bortezomib. However, the structural details of this boron adduct and the molecular factors that contribute to its formation and its ability to inhibit Bortezomib's activity remain unclear. This paper describes the use of NMR spectroscopy and cell assays to characterize the structures and properties of the boron adducts of EGCG and related polyphenols. The observed boron adducts included both boronate and borate derivatives, and their structural characteristics were correlated with cell-based evaluation of the ability of EGCG and other phenols to antagonize the anticancer activity of Bortezomib. The enhanced stability of the BZM/EGCG adduct was attributed to electronic and steric reasons, and a newly identified intramolecular interaction of the boron atom of BZM with the adjacent amide bond. The reported approach provides a useful method for determining the potential ability of polyphenols to form undesired adducts with boron-based drugs and interfere with their actions.
.jpg)



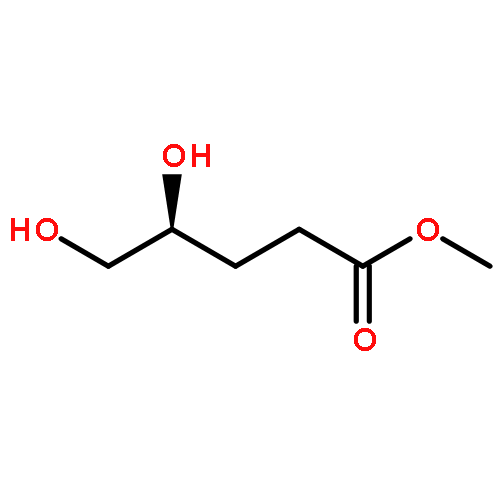
![Pentanoic acid, 4,5-bis[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, methyl ester, (4S)-](/data/chemimg/137500/196080-41-8.png)
![Pentanoic acid, 4,5-bis[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, methyl ester, (4S)-](/data/chemimg/137500/196080-41-8_b.png)


































![3-[[2-(ACETYLOXY)ETHYL][4-[(4-NITROPHENYL)AZO]PHENYL]AMINO]PROPIONONITRILE](http://img.cochemist.com/ccimg/76800/76726-92-6.png)
![3-[[2-(ACETYLOXY)ETHYL][4-[(4-NITROPHENYL)AZO]PHENYL]AMINO]PROPIONONITRILE](http://img.cochemist.com/ccimg/76800/76726-92-6_b.png)