Co-reporter:Emma E. M. Cating, Christopher W. Pinion, Joseph D. Christesen, Caleb A. Christie, Erik M. Grumstrup, James F. Cahoon, and John M. Papanikolas
Nano Letters October 11, 2017 Volume 17(Issue 10) pp:5956-5956
Publication Date(Web):September 12, 2017
DOI:10.1021/acs.nanolett.7b01876
Surface trap density in silicon nanowires (NWs) plays a key role in the performance of many semiconductor NW-based devices. We use pump–probe microscopy to characterize the surface recombination dynamics on a point-by-point basis in 301 silicon NWs grown using the vapor–liquid–solid (VLS) method. The surface recombination velocity (S), a metric of the surface quality that is directly proportional to trap density, is determined by the relationship S = d/4τ from measurements of the recombination lifetime (τ) and NW diameter (d) at distinct spatial locations in individual NWs. We find that S varies by as much as 2 orders of magnitude between NWs grown at the same time but varies only by a factor of 2 or three within an individual NW. Although we find that, as expected, smaller-diameter NWs exhibit shorter τ, we also find that smaller wires exhibit higher values of S; this indicates that τ is shorter both because of the geometrical effect of smaller d and because of a poorer quality surface. These results highlight the need to consider interwire heterogeneity as well as diameter-dependent surface effects when fabricating NW-based devices.Keywords: pump−probe microscopy; silicon nanowires; Surface recombination velocity; surface trap density;
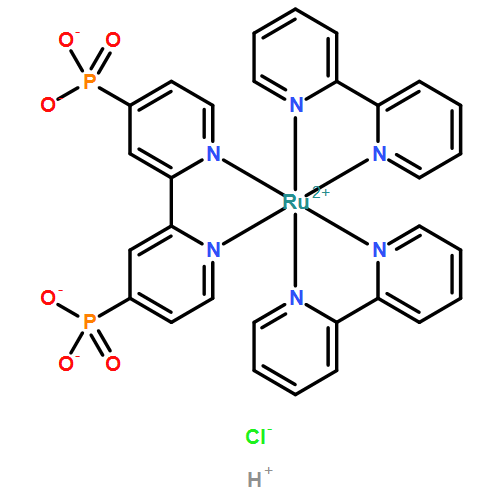
Co-reporter:Zachary A. Morseth, Toan V. Pho, Matthew V. Sheridan, Thomas J. Meyer, Kirk S. Schanze, John R. Reynolds, and John M. Papanikolas
ACS Applied Materials & Interfaces May 17, 2017 Volume 9(Issue 19) pp:16651-16651
Publication Date(Web):April 26, 2017
DOI:10.1021/acsami.7b02713
Photoinduced electron injection, intra-assembly electron transfer, and back-electron transfer are investigated in a single-site molecular assembly formed by covalently linking a phosphonated terthiophene (T3) chromophore to a Ru(terpyridine)(bipyridine)(L)2+ (L = MeCN or H2O) water oxidation catalyst adsorbed onto a mesoporous metal-oxide (MOx) film. Density functional theory calculations of the T3-trpy-Ru-L assembly indicate that the molecular components are strongly coupled with enhanced low-energy absorptions owing to the presence of an intraligand charge transfer (ILCT) transition between the T3 and trpy moieties. Ultrafast spectroscopy of the MOx//T3-trpy-Ru-L assemblies reveals that excitation of the surface-bound T3 chromophore results in ps–ns electron injection into the metal-oxide conduction band. Electron injection is followed by rapid (<35 ps) intra-assembly electron transfer from the RuII catalyst to regenerate the T3 chromophore with subsequent back-electron transfer on the microsecond time scale.Keywords: catalyst; photoinduced electron transfer; ruthenium; Solar fuels; thiophene; water oxidation;
Co-reporter:Robert J. Dillon, Leila Alibabaei, Thomas J. Meyer, and John M. Papanikolas
ACS Applied Materials & Interfaces August 16, 2017 Volume 9(Issue 32) pp:26786-26786
Publication Date(Web):July 21, 2017
DOI:10.1021/acsami.7b05856
The hole-injection and recombination photophysics for NiO sensitized with RuP ([RuII(bpy)2(4,4′-(PO3H2)2-bpy)]2+) are explored. Ultrafast transient absorption (TA) measurements performed with an external electrochemical bias reveal the efficiency for productive hole-injection, that is, quenching of the dye excited state that results in a detectable charge-separated electron–hole pair, is linearly dependent on the electronic occupation of intragap states in the NiO film. Population of these states via a negative applied potential increases the efficiency from 0% to 100%. The results indicate the primary loss mechanism for dye-sensitized NiO is rapid nongeminate recombination enabled by the presence of latent holes in the surface of the NiO film. Our findings suggest a new design paradigm for NiO photocathodes and devices centered on the avoidance of this recombination pathway.Keywords: applied bias; dye-sensitized nickel oxide NiO; hole-injection; photocathode; recombination; solar fuels; transient absorption;
Co-reporter:David F. Zigler, Zachary A. Morseth, Travis A. White, Theodore R. Canterbury, Hannah J. Sayre, José Á. Rodríguez-Corrales, M. Kyle Brennaman, Karen J. Brewer, John M. Papanikolas
Inorganica Chimica Acta 2017 Volume 454() pp:266-274
Publication Date(Web):1 January 2017
DOI:10.1016/j.ica.2016.06.034
The femtosecond transient absorption spectra (fsTA) and excited state kinetics for a series of six structurally related mixed-metal polypyridyl supramolecules are reported. Each complex consists of one or two light absorbers (LA) with Ru(II) or Os(II) centers attached to a Rh(III)-centered electron collector (EC) by an aromatic bridging ligand (BL). The resulting bimetallic and trimetallic complexes have LA-BL-EC and LA-BL-EC-BL-LA architectures, respectively. Excitation at 470 nm light populates metal-to-bridging ligand charge transfer states (MLCT), showing a transient absorption band near 380 nm due to π → π∗ transitions of a bridging ligand-localized radical anion and a transient bleach around 525 nm resulting from formal oxidation of the LA metal in the excited state. Loss of the ligand localized radical signal during the first 10 ps reflects conversion of the excited state population from an MLCT state into metal-to-metal (i.e. M(dπ)-to-Rh(dσ∗)) charge transfer states (MMCT). Each complex shares a similar ultrafast component, indicating that the kinetics governing MLCT → MMCT population transfer do not depend on the nature of the LA. Return to the ground state, however, is strongly LA dependent and controlled by the free-energy difference between the MMCT state and ground state, as well as an associated large reorganization energy.A series of photoexcited mixed-metal supramolecules are shown to undergo electron transfer from a metal-to-ligand charge transfer state (MLCT) to generate a metal-to-metal charge transfer state (MMCT) in ∼7 ps. Unlike the MLCT/MMCT free energy difference, the MMCT/ground state energy varies in the series, thus relaxation times span ∼20 ps to ∼30 ns.
Co-reporter:Emma E. M. Cating, Christopher W. Pinion, Erika M. Van Goethem, Michelle M. Gabriel, James F. Cahoon, and John M. Papanikolas
Nano Letters 2016 Volume 16(Issue 1) pp:434-439
Publication Date(Web):December 2, 2015
DOI:10.1021/acs.nanolett.5b04075
Thermal management is an important consideration for most nanoelectronic devices, and an understanding of the thermal conductivity of individual device components is critical for the design of thermally efficient systems. However, it can be difficult to directly probe local changes in thermal conductivity within a nanoscale system. Here, we utilize the time-resolved and diffraction-limited imaging capabilities of ultrafast pump–probe microscopy to determine, in a contact-free configuration, the local thermal conductivity in individual Si nanowires (NWs). By suspending single NWs across microfabricated trenches in a quartz substrate, the properties of the same NW both on and off the substrate are directly compared. We find the substrate has no effect on the recombination lifetime or diffusion length of photogenerated charge carriers; however, it significantly impacts the thermal relaxation properties of the NW. In substrate-supported regions, thermal energy deposited into the lattice by the ultrafast laser pulse dissipates within ∼10 ns through thermal diffusion and coupling to the substrate. In suspended regions, the thermal energy persists for over 100 ns, and we directly image the time-resolved spatial motion of the thermal signal. Quantitative analysis of the transient images permits direct determination of the NW’s local thermal conductivity, which we find to be a factor of ∼4 smaller than in bulk Si. Our results point to the strong potential of pump–probe microscopy to be used as an all-optical method to quantify the effects of localized environment and morphology on the thermal transport characteristics of individual nanostructured components.
Co-reporter:David F. Zigler; Zachary A. Morseth; Li Wang; Dennis L. Ashford; M. Kyle Brennaman; Erik M. Grumstrup; Erinn C. Brigham; Melissa K. Gish; Robert J. Dillon; Leila Alibabaei; Gerald J. Meyer; Thomas J. Meyer
Journal of the American Chemical Society 2016 Volume 138(Issue 13) pp:4426-4438
Publication Date(Web):March 14, 2016
DOI:10.1021/jacs.5b12996
Interfacial electron transfer at titanium dioxide (TiO2) is investigated for a series of surface bound ruthenium-polypyridyl dyes whose metal-to-ligand charge-transfer state (MLCT) energetics are tuned through chemical modification. The 12 complexes are of the form RuII(bpy-A)(L)22+, where bpy-A is a bipyridine ligand functionalized with phosphonate groups for surface attachment to TiO2. Functionalization of ancillary bipyridine ligands (L) enables the potential of the excited state RuIII/* couple, E+/*, in 0.1 M perchloric acid (HClO4(aq)) to be tuned from −0.69 to −1.03 V vs NHE. Each dye is excited by a 200 fs pulse of light in the visible region of the spectrum and probed with a time-delayed supercontiuum pulse (350–800 nm). Decay of the MLCT excited-state absorption at 376 nm is observed without loss of the ground-state bleach, which is a clear signature of electron injection and formation of the oxidized dye. The dye-dependent decays are biphasic with time constants in the 3–30 and 30–500 ps range. The slower injection rate constant for each dye is exponentially distributed relative to E+/*. The correlation between the exponentially diminishing density of TiO2 sub-band acceptor levels and injection rate is well described using Marcus–Gerischer theory, with the slower decay components being assigned to injection from the thermally equilibrated state and the faster components corresponding to injection from higher energy states within the 3MLCT manifold. These results and detailed analyses incorporating molecular photophysics and semiconductor density of states measurements indicate that the multiexponential behavior that is often observed in interfacial injection studies is not due to sample heterogeneity. Rather, this work shows that the kinetic heterogeneity results from competition between excited-state relaxation and injection as the photoexcited dye relaxes through the 3MLCT manifold to the thermally equilibrated state, underscoring the potential for a simple kinetic model to reproduce the complex kinetic behavior often observed at the interface of mesoporous metal oxide materials.
Co-reporter:Zachary A. Morseth, Toan V. Pho, Alexander T. Gilligan, Robert J. Dillon, Kirk S. Schanze, John R. Reynolds, and John M. Papanikolas
The Journal of Physical Chemistry B 2016 Volume 120(Issue 32) pp:7937-7948
Publication Date(Web):July 19, 2016
DOI:10.1021/acs.jpcb.6b05589
Ultrafast energy and electron transfer (EnT and ET, respectively) are characterized in a light-harvesting assembly based on a π-conjugated polymer (poly(fluorene)) functionalized with broadly absorbing pendant organic isoindigo (iI) chromophores using a combination of femtosecond transient absorption spectroscopy and large-scale computer simulation. Photoexcitation of the π-conjugated polymer leads to near-unity quenching of the excitation through a combination of EnT and ET to the iI pendants. The excited pendants formed by EnT rapidly relax within 30 ps, whereas recombination of the charge-separated state formed following ET occurs within 1200 ps. A computer model of the excited-state processes is developed by combining all-atom molecular dynamics simulations, which provides a molecular-level view of the assembly structure, with a kinetic model that accounts for the multiple excited-state quenching pathways. Direct comparison of the simulations with experimental data reveals that the underlying structure has a dramatic effect on the partitioning between EnT and ET in the polymer assembly, where the distance and orientation of the pendants in relation to the backbone serve to direct the dominant quenching pathway.
Co-reporter:Melissa K. Gish, Alexander M. Lapides, M. Kyle Brennaman, Joseph L. Templeton, Thomas J. Meyer, and John M. Papanikolas
The Journal of Physical Chemistry Letters 2016 Volume 7(Issue 24) pp:5297-5301
Publication Date(Web):December 2, 2016
DOI:10.1021/acs.jpclett.6b02388
Interfacial dynamics are investigated in SnO2/TiO2 core/shell films derivatized with a Ru(II)-polypyridyl chromophore ([RuII(bpy)2(4,4′-(PO3H2)2bpy)]2+, RuP) using transient absorption methods. Electron injection from the chromophore into the TiO2 shell occurs within a few picoseconds after photoexcitation. Loss of the oxidized dye through recombination occurs across time scales spanning 10 orders of magnitude. The majority (60%) of charge recombination events occur shortly after injection (τ = 220 ps), while a small fraction (≤20%) of the oxidized chromophores persists for milliseconds. The lifetime of long-lived charge-separated states (CSS) depends exponentially on shell thickness, suggesting that the injected electrons reside in the SnO2 core and must tunnel through the TiO2 shell to recombine with oxidized dyes. While the core/shell architecture extends the lifetime in a small fraction of the CSS, making water oxidation possible, the subnanosecond recombination process has profound implications for the overall efficiencies of dye-sensitized photoelectrosynthesis cells (DSPECs).
Co-reporter:Zachary A. Morseth, Li Wang, Egle Puodziukynaite, Gyu Leem, Alexander T. Gilligan, Thomas J. Meyer, Kirk S. Schanze, John R. Reynolds, and John M. Papanikolas
Accounts of Chemical Research 2015 Volume 48(Issue 3) pp:818
Publication Date(Web):February 3, 2015
DOI:10.1021/ar500382u
The use of sunlight to make chemical fuels (i.e., solar fuels) is an attractive approach in the quest to develop sustainable energy sources. Using nature as a guide, assemblies for artificial photosynthesis will need to perform multiple functions. They will need to be able to harvest light across a broad region of the solar spectrum, transport excited-state energy to charge-separation sites, and then transport and store redox equivalents for use in the catalytic reactions that produce chemical fuels. This multifunctional behavior will require the assimilation of multiple components into a single macromolecular system.A wide variety of different architectures including porphyrin arrays, peptides, dendrimers, and polymers have been explored, with each design posing unique challenges. Polymer assemblies are attractive due to their relative ease of production and facile synthetic modification. However, their disordered nature gives rise to stochastic dynamics not present in more ordered assemblies. The rational design of assemblies requires a detailed understanding of the energy and electron transfer events that follow light absorption, which can occur on time scales ranging from femtoseconds to hundreds of microseconds, necessitating the use of sophisticated techniques. We have used a combination of time-resolved absorption and emission spectroscopies with observation times that span 9 orders of magnitude to follow the excited-state evolution within polymer-based molecular assemblies. We complement experimental observations with molecular dynamics simulations to develop a microscopic view of these dynamics.This Account provides an overview of our work on polymers decorated with pendant Ru(II) chromophores, both in solution and on surfaces. We have examined site-to-site energy transport among the Ru(II) complexes, and in systems incorporating π-conjugated polymers, we have observed ultrafast formation of a long-lived charge-separated state. When attached to TiO2, these assemblies exhibit multifunctional behavior in which photon absorption is followed by energy transport to the surface and electron injection to produce an oxidized metal complex. The oxidizing equivalent is then transferred to the conjugated polymer, giving rise to a long-lived charge-separated state.
Co-reporter:Erik M. Grumstrup, Michelle M. Gabriel, Emma E.M. Cating, Erika M. Van Goethem, John M. Papanikolas
Chemical Physics 2015 Volume 458() pp:30-40
Publication Date(Web):8 September 2015
DOI:10.1016/j.chemphys.2015.07.006
•Diffraction limited pump–probe microscopy methods are described.•Spatial variation in dynamical phenomena across single structures.•Direct observation of carrier motion in individual nanostructures.Excited state dynamics at the nanoscale provide important insight into the influence of structural features such as interfaces, defects, and surfaces on material properties. Pump–probe microscopy combines the spatial resolution of far-field optical microscopy with the temporal resolution of ultrafast spectroscopy, and has emerged as a powerful technique for characterizing spatial variation in dynamical phenomena across nanometer length scales. It has helped correlate dynamical phenomena with specific structural features in a variety of materials, shedding light on how excited state behaviors can dramatically differ from one member of the ensemble to the next, and even at different points within a single structure. It has also enabled direct imaging of transport phenomena such as free carrier diffusion, exciton migration and plasmon propagation in nanostructures. This ability to observe individual objects provides unique insight into complex materials where heterogeneous behavior makes it difficult, if not impossible, to reach clear and quantitative conclusions.
Co-reporter:Erik M. Grumstrup, Michelle M. Gabriel, Christopher W. Pinion, James K. Parker, James F. Cahoon, and John M. Papanikolas
Nano Letters 2014 Volume 14(Issue 11) pp:6287-6292
Publication Date(Web):September 26, 2014
DOI:10.1021/nl5026166
Strain-induced changes to the electronic structure of nanoscale materials provide a promising avenue for expanding the optoelectronic functionality of semiconductor nanostructures in device applications. Here we use pump–probe microscopy with femtosecond temporal resolution and submicron spatial resolution to characterize charge–carrier recombination and transport dynamics in silicon nanowires (NWs) locally strained by bending deformation. The electron–hole recombination rate increases with strain for values above a threshold of ∼1% and, in highly strained (∼5%) regions of the NW, increases 6-fold. The changes in recombination rate are independent of NW diameter and reversible upon reduction of the applied strain, indicating the effect originates from alterations to the NW bulk electronic structure rather than introduction of defects. The results highlight the strong relationship between strain, electronic structure, and charge–carrier dynamics in low-dimensional semiconductor systems, and we anticipate the results will assist the development of strain-enabled optoelectronic devices with indirect-bandgap materials such as silicon.
Co-reporter:Michelle M. Gabriel, Erik M. Grumstrup, Justin R. Kirschbrown, Christopher W. Pinion, Joseph D. Christesen, David F. Zigler, Emma E. M. Cating, James F. Cahoon, and John M. Papanikolas
Nano Letters 2014 Volume 14(Issue 6) pp:3079-3087
Publication Date(Web):May 27, 2014
DOI:10.1021/nl5012118
Silicon nanowires incorporating p-type/n-type (p-n) junctions have been introduced as basic building blocks for future nanoscale electronic components. Controlling charge flow through these doped nanostructures is central to their function, yet our understanding of this process is inferred from measurements that average over entire structures or integrate over long times. Here, we have used femtosecond pump–probe microscopy to directly image the dynamics of photogenerated charge carriers in silicon nanowires encoded with p-n junctions along the growth axis. Initially, motion is dictated by carrier–carrier interactions, resulting in diffusive spreading of the neutral electron–hole cloud. Charge separation occurs at longer times as the carrier distribution reaches the edges of the depletion region, leading to a persistent electron population in the n-type region. Time-resolved visualization of the carrier dynamics yields clear, direct information on fundamental drift, diffusion, and recombination processes in these systems, providing a powerful tool for understanding and improving materials for nanotechnology.
Co-reporter:Stephanie E. Bettis, Kenneth Hanson, Li Wang, Melissa K. Gish, Javier J. Concepcion, Zhen Fang, Thomas J. Meyer, and John M. Papanikolas
The Journal of Physical Chemistry A 2014 Volume 118(Issue 45) pp:10301-10308
Publication Date(Web):April 15, 2014
DOI:10.1021/jp411139j
Femtosecond transient absorption spectroscopy is used to characterize the first photoactivation step in a chromophore/water oxidation catalyst assembly formed through a “layer-by-layer” approach. Assemblies incorporating both chromophores and catalysts are central to the function of dye-sensitized photoelectrosynthesis cells (DSPECs) for generating solar fuels. The chromophore, [RuaII]2+ = [Ru(pbpy)2(bpy)]2+, and water oxidation catalyst, [RubII-OH2]2+ = [Ru(4,4′-(CH2PO3H2)2bpy)(Mebimpy)(H2O)]2+, where bpy = 2,2′-bipyridine, pbpy = 4,4′-(PO3H2)2bpy, and Mebimpy = 2,6-bis(1-methylbenzimidazol-2-yl)pyridine), are arranged on nanocrystalline TiO2 via phosphonate-Zr(IV) coordination linkages. Analysis of the transient spectra of the assembly (denoted TiO2-[RuaII-Zr-RubII-OH2]4+) reveal that photoexcitation initiates electron injection, which is then followed by the transfer of the oxidative equivalent from the chromophore to the catalyst with a rate of kET = 5.9 × 109 s–1 (τ = 170 ps). While the assembly, TiO2-[RuaII-Zr-RubII-OH2]4+, has a near-unit efficiency for transfer of the oxidative equivalent to the catalyst, the overall efficiency of the system is only 43% due to nonproductive photoexcitation of the catalyst and nonunit efficiency for electron injection. The modular nature of the layer-by-layer system allows for variation of the light-harvesting chromophore and water oxidation catalyst for future studies to increase the overall efficiency.
Co-reporter:Zhuo Chen, Erik M. Grumstrup, Alexander T. Gilligan, John M. Papanikolas, and Kirk S. Schanze
The Journal of Physical Chemistry B 2014 Volume 118(Issue 1) pp:372-378
Publication Date(Web):December 3, 2013
DOI:10.1021/jp411565p
Energy transfer along a nonconjugated polymer chain is studied with a polystyrene-based copolymer of oligo(phenylene-ethynylene) (OPE) donor and thiophene-benzothiadiazole (TBT) acceptor pendants. The graft copolymers are prepared from reversible addition–fragmentation transfer polymerization (RAFT) and copper(I)-catalyzed azide–alkyne “click” reaction. The singlet energy transfer from donor to accept is studied via fluorescence emission and ultrafast transient absorption spectroscopy. Near unity quenching of the OPE excited state by the TBT moiety occurs on multiple time scales (2–50 ps) dependent on where the initial exciton is formed on the polymer.
Co-reporter:Gyu Leem ; Zachary A. Morseth ; Egle Puodziukynaite ; Junlin Jiang ; Zhen Fang ; Alexander T. Gilligan ; John R. Reynolds ; John M. Papanikolas ;Kirk S. Schanze
The Journal of Physical Chemistry C 2014 Volume 118(Issue 49) pp:28535-28541
Publication Date(Web):November 17, 2014
DOI:10.1021/jp5113558
This paper describes the photophysical and photoelectrochemical characterization of a light harvesting polychromophore array featuring a polyfluorene backbone with covalently attached Ru(II) polypyridyl complexes (PF-Ru-A), adsorbed on the surface of mesostructured TiO2 (PF-Ru-A//TiO2). The surface adsorbed polymer is characterized by transmission electron microscopy (TEM), scanning electron microscopy (SEM), and attenuated total reflectance-Fourier transform infrared (ATR-FTIR) spectroscopy, providing evidence for the morphology of the surface adsorbed polymer and the mode of binding. Photoexcitation of the Ru(II) complexes bound to the metal oxide surface (proximal) results in electron injection into the conduction band of TiO2, which is then followed by ultrafast hole transfer to the polymer to form oxidized polyfluorene (PF+). More interestingly, chromophores that are not directly bound to the TiO2 interface (distal) that are excited participate in site-to-site energy transfer processes that transport the excited state to surface bound chromophores where charge injection occurs, underscoring the antenna-like nature of the polymer assembly. The charge separated state is long-lived and persists for >100 μs, a consequence of the increased separation between the hole and injected electron.
Co-reporter:Erik M. Grumstrup ; Emma M. Cating ; Michelle M. Gabriel ; Christopher W. Pinion ; Joseph D. Christesen ; Justin R. Kirschbrown ; Ernest L. Vallorz ; III; James F. Cahoon
The Journal of Physical Chemistry C 2014 Volume 118(Issue 16) pp:8626-8633
Publication Date(Web):March 21, 2014
DOI:10.1021/jp501079b
Ultrafast carrier dynamics in silicon nanowires with average diameters of 40, 50, 60, and 100 nm were studied with transient absorption spectroscopy. After 388 nm photoexcitation near the direct band gap of silicon, broadband spectra from 400 to 800 nm were collected between 200 fs and 1.3 ns. The transient spectra exhibited both absorptive and bleach features that evolved on multiple time scales, reflecting contributions from carrier thermalization and recombination as well as transient shifts of the ground-state absorption spectrum. The initially formed “hot” carriers relaxed to the band edge within the first ∼300 fs, followed by recombination over several hundreds of picoseconds. The charge carrier lifetime progressively decreased with decreasing diameter, a result consistent with a surface-mediated recombination process. Recombination dynamics were quantitatively modeled using the diameter distribution measured from each sample, and this analysis yielded a consistent surface recombination velocity of ∼2 × 104 cm/s across all samples. The results indicate that transient absorption spectroscopy, which interrogates thousands of individual nanostructures simultaneously, can be an accurate probe of material parameters in inhomogeneous semiconductor samples when geometrical differences within the ensemble are properly analyzed.
Co-reporter:Erik M. Grumstrup ; Michelle M. Gabriel ; Emma M. Cating ; Christopher W. Pinion ; Joseph D. Christesen ; Justin R. Kirschbrown ; Ernest L. Vallorz ; III; James F. Cahoon
The Journal of Physical Chemistry C 2014 Volume 118(Issue 16) pp:8634-8640
Publication Date(Web):March 21, 2014
DOI:10.1021/jp502737e
Ultrafast charge carrier dynamics in silicon nanowires (NWs) grown by a vapor–liquid–solid mechanism were interrogated with optical pump–probe microscopy. The high time and spatial resolutions achieved by the experiments provide insight into the charge carrier dynamics of single nanostructures. Individual NWs were excited by a femtosecond pump pulse focused to a diffraction-limited spot, producing photogenerated carriers (electrons and holes) in a localized region of the structure. Photoexcited carriers undergo both electron–hole recombination and diffusional migration away from the excitation spot on similar time scales. The evolution of the carrier population is monitored by a delayed probe pulse that is also focused to a diffraction-limited spot. When the pump and probe are spatially overlapped, the transient signal reflects both recombination and carrier migration. Diffusional motion is directly observed by spatially separating the pump and probe beams, enabling carriers to be generated in one location and detected in another. Quantitative analysis of the signals yields a statistical distribution of carrier lifetimes from a large number of individual NWs. On average, the lifetime was found to be linearly proportional to the diameter, consistent with a surface-mediated recombination mechanism. These results highlight the capability of pump–probe microscopy to quantitatively evaluate key recombination characteristics in semiconductor nanostructures, which are important for their implementation in nanotechnologies.
Co-reporter:Stephanie E. Bettis ; Derek M. Ryan ; Melissa K. Gish ; Leila Alibabaei ; Thomas J. Meyer ; Marcey L. Waters
The Journal of Physical Chemistry C 2014 Volume 118(Issue 12) pp:6029-6037
Publication Date(Web):March 4, 2014
DOI:10.1021/jp410646u
We report a detailed kinetic analysis of ultrafast interfacial and intra-assembly electron transfer following excitation of an oligoproline scaffold functionalized by chemically linked light-harvesting chromophore [Ru(pbpy)2(bpy)]2+ (pbpy = 4,4′-(PO3H2)2-2,2′-bipyridine, bpy = 2,2′-bipyridine) and water oxidation catalyst [Ru(Mebimpy)(bpy)OH2]2+ (Mebimpy = 2,6-bis(1-methylbenzimidazol-2-yl)pyridine). The oligoproline scaffold approach is appealing due to its modular nature and helical tertiary structure. They allow for the control of electron transfer distances in chromophore–catalyst assemblies for applications in dye-sensitized photoelectrosynthesis cells (DSPECs). The proline chromophore–catalyst assembly was loaded onto nanocrystalline TiO2 with the helical structure of the oligoproline scaffold maintaining the controlled relative positions of the chromophore and catalyst. Ultrafast transient absorption spectroscopy was used to analyze the kinetics of the first photoactivation step for oxidation of water in the assembly. A global kinetic analysis of the transient absorption spectra reveals that photoinduced electron injection occurs in 18 ps and is followed by intra-assembly oxidative activation of the water oxidation catalyst on the hundreds of picoseconds time scale (kET = 2.6 × 109 s–1; τ = 380 ps). The first photoactivation step in the water oxidation cycle of the chromophore–catalyst assembly anchored to TiO2 is complete within 380 ps.
Co-reporter:Michelle M. Gabriel, Justin R. Kirschbrown, Joseph D. Christesen, Christopher W. Pinion, David F. Zigler, Erik M. Grumstrup, Brian P. Mehl, Emma E. M. Cating, James F. Cahoon, and John M. Papanikolas
Nano Letters 2013 Volume 13(Issue 3) pp:1336-1340
Publication Date(Web):February 19, 2013
DOI:10.1021/nl400265b
We have developed a pump–probe microscope capable of exciting a single semiconductor nanostructure in one location and probing it in another with both high spatial and temporal resolution. Experiments performed on Si nanowires enable a direct visualization of the charge cloud produced by photoexcitation at a localized spot as it spreads along the nanowire axis. The time-resolved images show clear evidence of rapid diffusional spreading and recombination of the free carriers, which is consistent with ambipolar diffusion and a surface recombination velocity of ∼104 cm/s. The free carrier dynamics are followed by trap carrier migration on slower time scales.
Co-reporter:Da Ma ; Stephanie E. Bettis ; Kenneth Hanson ; Maria Minakova ; Leila Alibabaei ; William Fondrie ; Derek M. Ryan ; Garegin A. Papoian ; Thomas J. Meyer ; Marcey L. Waters
Journal of the American Chemical Society 2013 Volume 135(Issue 14) pp:5250-5253
Publication Date(Web):March 20, 2013
DOI:10.1021/ja312143h
Solid-phase peptide synthesis has been applied to the preparation of phosphonate-derivatized oligoproline assemblies containing two different RuII polypyridyl chromophores coupled via “click” chemistry. In water or methanol the assembly adopts the polyproline II (PPII) helical structure, which brings the chromophores into close contact. Excitation of the assembly on ZrO2 at the outer RuII in 0.1 M HClO4 at 25 °C is followed by rapid, efficient intra-assembly energy transfer to the inner RuII (kEnT = 3.0 × 107 s–1, implying 96% relative efficiency). The comparable energy transfer rate constants in solution and on nanocrystalline ZrO2 suggest that the PPII structure is retained when bound to ZrO2. On nanocrystalline films of TiO2, excitation at the inner RuII is followed by rapid, efficient injection into TiO2. Excitation of the outer RuII is followed by rapid intra-assembly energy transfer and then by electron injection. The oligoproline/click chemistry approach holds great promise for the preparation of interfacial assemblies for energy conversion based on a family of assemblies having controlled compositions and distances between key functional groups.
Co-reporter:Li Wang, Dennis L. Ashford, David W. Thompson, Thomas J. Meyer, and John M. Papanikolas
The Journal of Physical Chemistry C 2013 Volume 117(Issue 46) pp:24250-24258
Publication Date(Web):October 29, 2013
DOI:10.1021/jp410571x
This paper examines the ultrafast dynamics of the initial photoactivation step in a molecular assembly consisting of a chromophore (denoted [RuaII]2+) and a water-splitting catalyst (denoted [RubII]2+) anchored to TiO2. Photoexcitation of the chromophore is followed by rapid electron injection from the Ru(II) metal-to-ligand charge-transfer (MLCT) excited state. The injection process was followed via the decay of the bpy radical anion absorption at 375 nm. Injection is ∼95% efficient and exhibits multiple kinetic components with decay times ranging from <250 fs to 250 ps. Electron injection is followed by the transfer of the oxidative equivalent from the chromophore to the catalyst (ΔG = −0.28 eV) with a transfer time of 145 ps. In the absence of subsequent photoexcitation events, the charge-separated state undergoes electron-transfer recombination on the microsecond time scale.
Co-reporter:Erik M. Grumstrup, Zhuo Chen, Ryan P. Vary, Andrew M. Moran, Kirk S. Schanze, and John M. Papanikolas
The Journal of Physical Chemistry B 2013 Volume 117(Issue 27) pp:8245-8255
Publication Date(Web):June 12, 2013
DOI:10.1021/jp404498u
A Raman-pump frequency modulation scheme and an automated signal-processing algorithm are developed for improved collection of time-resolved femtosecond stimulated Raman spectra. Together, these two advancements remove the broad background signals endemic to FSRS measurements and retrieve signals with high sensitivity. We apply this frequency-modulated femtosecond stimulated Raman spectroscopy (FM-FSRS) to the characterization of ultrafast energy transport in a copolymer comprised of polystyrene linked oligo(phenylene–ethynylene) donor and thiophene–benzothiadiazole acceptor chromophores. After photoexcitation of the donor, ultrafast energy transfer is monitored by the decay of donor vibrational modes and simultaneous growth of acceptor modes. The FM-FSRS method shows clear advantages in signal-to-noise levels, mitigation of artifact features, and ease of data processing over the conventional FSRS technique.
Co-reporter:Brian P. Mehl, Justin R. Kirschbrown, Michelle M. Gabriel, Ralph L. House, and John M. Papanikolas
The Journal of Physical Chemistry B 2013 Volume 117(Issue 16) pp:4390-4398
Publication Date(Web):October 23, 2012
DOI:10.1021/jp307089h
Femtosecond pump–probe microscopy is used to investigate the charge recombination dynamics at different points within a single needle-shaped ZnO rod. Recombination in the tips of the rod occurs through an excitonic or electron–hole plasma state, taking place on a picosecond time scale. Photoexcitation in the larger diameter sections of the interior exhibit dramatically slower recombination that occurs primarily through defects sites, i.e., trap mediated recombination. Transient absorption imaging shows that the spatial variation in the dynamics is also influenced by the cavity resonances supported within the hexagonal cross section of the rod. Finite element simulations suggest that these optical resonator modes produce qualitatively different intensity patterns in the two different locations. Near the end of the rod, the intensity pattern has significant standing-wave character, which leads to the creation of photoexcited carriers in the core of the structure. The larger diameter regions, on the other hand, exhibit intensity distributions in which the whispering gallery (WG) mode character dominates. At these locations, the photoexcited carriers are produced in subsurface depletion zone, where the internal fields separate the electrons and holes and lead to a greater degree of trap recombination on longer time scales.
Co-reporter:Justin R. Kirschbrown, Ralph L. House, Brian P. Mehl, James K. Parker, and John M. Papanikolas
The Journal of Physical Chemistry C 2013 Volume 117(Issue 20) pp:10653-10660
Publication Date(Web):April 16, 2013
DOI:10.1021/jp4011907
Two-photon emission microscopy is used to investigate the photoluminescence properties of individual ZnO rods. The rods are 10–20 μm in length with a tapered cross section that varies from 1 to 2 μm at the midpoint to several hundred nanometers at the ends. The tapered shape and hexagonal cross section result in complex optical resonator modes that lead to periodic patterns in the two-photon emission image. Finite-difference frequency domain methods using a series of excitation sources, including focused Gaussian, point dipole, and plane wave, suggest that resonator modes have both standing wave (Fabry-Pérot) and whispering gallery mode character, whose relative contributions vary along the rod axis.
Co-reporter:Li Wang, Egle Puodziukynaite, Erik M. Grumstrup, Aaron C. Brown, Shahar Keinan, Kirk S. Schanze, John R. Reynolds, and John M. Papanikolas
The Journal of Physical Chemistry Letters 2013 Volume 4(Issue 14) pp:2269-2273
Publication Date(Web):June 26, 2013
DOI:10.1021/jz401089v
A light-harvesting macromolecular assembly (PT-Ru) consisting of a poly(3-hexylthiophene) (P3HT) scaffold and pendant Ru(II) polypyridyl complexes that exhibits rapid and efficient formation of a long-lived charge-separated state is described here. Photoinduced electron transfer from the polymer backbone to Ru(II) was investigated by femtosecond transient absorption spectroscopy. Photoexcitation at 388 nm results in the excitation of both the polymer backbone and Ru(II) complexes, with relative excitation probabilities of 60 and 40%, respectively. The dominant pathway (∼85%) for decay of the polymer excited state is direct electron transfer from the polymer scaffold to Ru(II), forming a positive polaron and a reduced complex [RuII(L)2(L–)]+, denoted Ru(I). The charge-separated state PT+•-Ru(I) is long-lived, persisting for 20–60 μs, and is attributed to the high mobility of holes on the polymer backbone, which facilitates spatial separation of the electron and hole, delaying recombination. The remaining 15% of the polymer excited states undergo an alternate deactivation mechanism, possibly energy transfer to Ru(II), forming Ru(II)*. Ru(II)* formed by either direct excitation or energy transfer undergoes back energy transfer to the scaffold, forming the low-lying polymer triplet state on the nanosecond time scale.Keywords: electron transfer; energy transfer; femtosecond; P3HT; pump−probe;
Co-reporter:Dale J. Wilger, Stephanie E. Bettis, Christopher K. Materese, Maria Minakova, Garegin A. Papoian, John M. Papanikolas, and Marcey L. Waters
Inorganic Chemistry 2012 Volume 51(Issue 21) pp:11324-11338
Publication Date(Web):June 8, 2012
DOI:10.1021/ic300669t
Herein we report energy transfer studies in a series of Ru(II) and Os(II) linked coiled-coil peptides in which the supramolecular scaffold controls the functional properties of the assembly. A general and convergent method for the site-specific incorporation of bipyridyl Ru(II) and Os(II) complexes using solid-phase peptide synthesis and the copper-catalyzed azide–alkyne cycloaddition is reported. Supramolecular assembly positions the chromophores for energy transfer. Using time-resolved emission spectroscopy we measured position-dependent energy transfer that can be varied through changes in the sequence of the peptide scaffold. High level molecular dynamics simulations were used in conjunction with the spectroscopic techniques to gain molecular-level insight into the observed trends in energy transfer. The most efficient pair of Ru(II) and Os(II) linked peptides as predicted by molecular modeling also exhibited the fastest rate of energy transfer (with kEnT = 2.3 × 107 s–1 (42 ns)). Additionally, the emission quenching for the Ru(II) and Os(II) peptides can be fit to binding models that agree with the dissociation constants determined for the peptides via chemical denaturation.
Co-reporter:Brittany C. Westlake, Jared J. Paul, Stephanie E. Bettis, Shaun D. Hampton, Brian P. Mehl, Thomas J. Meyer, and John M. Papanikolas
The Journal of Physical Chemistry B 2012 Volume 116(Issue 51) pp:14886-14891
Publication Date(Web):November 28, 2012
DOI:10.1021/jp308505p
Excited-state proton-transfer dynamics between 7-hydroxy-4-(trifluoromethyl)coumarin and 1-methylimidazole base in toluene were studied using ultrafast pump–probe and time-resolved emission methods. Charge-transfer excitation of the hydroxycoumarin shifts electron density from the hydroxyl group to the carbonyl, resulting in an excited state where proton transfer to the base is highly favored. In addition to its the photoacid characteristics, the shift in the hydroxycoumarin electronic distribution gives it characteristics of a photobase as well. The result is a tautomerization process occurring on the picosecond time scale in which the 1-methylimidazole base acts as a proton-transfer shuttle from the hydroxyl group to the carbonyl.
Co-reporter:Li Wang, Egle Puodziukynaite, Ryan P. Vary, Erik M. Grumstrup, Ryan M. Walczak, Olga Y. Zolotarskaya, Kirk S. Schanze, John R. Reynolds, and John M. Papanikolas
The Journal of Physical Chemistry Letters 2012 Volume 3(Issue 17) pp:2453-2457
Publication Date(Web):August 15, 2012
DOI:10.1021/jz300979j
This Letter describes the synthesis and photophysical characterization of a Ru(II) assembly consisting of metal polypyridyl complexes linked together by a polyfluorene scaffold. Unlike many scaffolds incorporating saturated linkages, the conjugated polymer in this system acts as a functional light-harvesting component. Conformational disorder breaks the conjugation in the polymer backbone, resulting in a chain composed of many chromophore units, whose relative energies depend on the segment lengths. Photoexcitation of the polyfluorene by a femtosecond laser pulse results in the excitation of polyfluorene, which then undergoes direct energy transfer to the pendant Ru(II) complexes, producing Ru(II)* excited states within 500 fs after photoexcitation. Femtosecond transient absorption data show the presence of electron transfer from PF* to Ru(II) to form charge-separated (CS) products within 1–2 ps. The decay of the oxidized and reduced products, PF+• and Ru(I), through back electron transfer are followed using picosecond transient absorption methods.Keywords: metal polypyridyl complexes; organic chromophore; photoinduced energy transfer; polymer assembly; redox separation; spectroscopy;
Co-reporter:Brian P. Mehl, Justin R. Kirschbrown, Ralph L. House, and John M. Papanikolas
The Journal of Physical Chemistry Letters 2011 Volume 2(Issue 14) pp:1777-1781
Publication Date(Web):June 30, 2011
DOI:10.1021/jz200809c
Pump–probe transient absorption microscopy was used to follow the electron–hole (e–h) recombination dynamics at different points within individual needle-shaped ZnO rods to characterize spatial differences in dynamical behavior. The results from pump–probe experiments are correlated with spatially resolved ultrafast emission measurements, and scanning electron microscopy provides structural details. Dramatically different e–h recombination dynamics are observed in the narrow tips compared to the interior, with the ends exhibiting a greater propensity for electron–hole plasma (EHP) formation and faster recombination of carriers across the band gap that stem from a physical confinement of the charge carriers. In the interior of the rod, a greater fraction of the e–h recombination is trap-mediated and occurs on a significantly longer time scale.Keywords: electron−hole plasma; imaging; microscopy; pump−probe; transient absorption; ZnO;
Co-reporter:Brittany C. Westlake;M. Kyle Brennaman;Javier J. Concepcion;Jared J. Paul;Stephanie E. Bettis;Shaun D. Hampton;Stephen A. Miller;Natalia V. Lebedeva;Malcolm D. E. Forbes;Andrew M. Moran;Thomas J. Meyer
PNAS 2011 108 (21 ) pp:8554-8558
Publication Date(Web):2011-05-24
DOI:10.1073/pnas.1104811108
The simultaneous, concerted transfer of electrons and protons—electron-proton transfer (EPT)—is an important mechanism utilized
in chemistry and biology to avoid high energy intermediates. There are many examples of thermally activated EPT in ground-state
reactions and in excited states following photoexcitation and thermal relaxation. Here we report application of ultrafast
excitation with absorption and Raman monitoring to detect a photochemically driven EPT process (photo-EPT). In this process,
both electrons and protons are transferred during the absorption of a photon. Photo-EPT is induced by intramolecular charge-transfer
(ICT) excitation of hydrogen-bonded-base adducts with either a coumarin dye or 4-nitro-4′-biphenylphenol. Femtosecond transient
absorption spectral measurements following ICT excitation reveal the appearance of two spectroscopically distinct states having
different dynamical signatures. One of these states corresponds to a conventional ICT excited state in which the transferring
H+ is initially associated with the proton donor. Proton transfer to the base (B) then occurs on the picosecond time scale.
The other state is an ICT-EPT photoproduct. Upon excitation it forms initially in the nuclear configuration of the ground
state by application of the Franck–Condon principle. However, due to the change in electronic configuration induced by the
transition, excitation is accompanied by proton transfer with the protonated base formed with a highly elongated +H─B bond. Coherent Raman spectroscopy confirms the presence of a vibrational mode corresponding to the protonated base in
the optically prepared state.
Co-reporter:Ralph L. House ; Justin R. Kirschbrown ; Brian P. Mehl ; Michelle M. Gabriel ; Joseph A. Puccio ; James K. Parker
The Journal of Physical Chemistry C 2011 Volume 115(Issue 43) pp:21436-21442
Publication Date(Web):September 22, 2011
DOI:10.1021/jp207830h
We have used two-photon emission microscopy to characterize the charge carrier dynamics at different locations within a single ZnO rod. Photoexcitation by a focused laser produces carriers (electrons and holes) in a localized region. Emission is detected using both time-integrated and time-resolved methods. Results show that the electron–hole plasma (EHP) state plays a larger role at the end of the rod compared to other points within the structure, where electron–hole recombination proceeds through an excitonic state. The origin of this spatial dependence is attributed to the physical confinement at the end of the structure that prevents an expansion of the photoexcited electron–hole cloud through processes such as carrier diffusion. Whispering gallery modes are identified as contributing to a periodic emission pattern along the length of the structure.
Co-reporter:Ralph L. House ; Brian P. Mehl ; Justin R. Kirschbrown ; Scott C. Barnes
The Journal of Physical Chemistry C 2011 Volume 115(Issue 21) pp:10806-10816
Publication Date(Web):May 5, 2011
DOI:10.1021/jp1118426
Zinc oxide has emerged as an attractive candidate for a variety of optoelectronic and photonic applications, due in part to a large second-order nonlinear susceptibility, its wide band gap, and large exciton binding energy. We have used time-resolved nonlinear two-photon emission microscopy to characterize the excited-state dynamics of individual ZnO rods. Photoluminescence images reveal a rich structure in the spatial distribution of both the band-edge and trap emission, and spectra recorded following excitation at a specific point in the structure show the characteristic band-edge and defect emission. Time-resolved emission spectra reveal a dynamic red shift of the trap emission band in the as-grown structures. Our results suggest that the trap emission is composed of at least two overlapping emission bands. The higher-energy band is assigned to e–h recombination between a conduction band electron and photogenerated hole bound to an acceptor defect lying within the band gap. The lower-energy band is attributed to a donor–acceptor pair (DAP) transition in which an electron localized on a donor defect recombines with a nearby hole-bound acceptor. The DAP transition energy and recombination rate depend upon the spatial proximity of the two traps, with higher-energy transitions corresponding to closely spaced pairs and occurring more rapidly, resulting in the dynamic red shift. Reduction in the trap density following annealing suppresses the DAP emission and its signature time-dependent red shift. The blue shift of the static photoluminescence spectrum is attributed to the donor defects being preferentially annealed out of the structure, resulting in a larger contribution of the higher-energy band.
Co-reporter:Caleb A. Kent ; Brian P. Mehl ; Liqing Ma ; John M. Papanikolas ; Thomas J. Meyer ;Wenbin Lin
Journal of the American Chemical Society 2010 Volume 132(Issue 37) pp:12767-12769
Publication Date(Web):August 25, 2010
DOI:10.1021/ja102804s
Isomorphous metal−organic frameworks (MOFs) based on {M[4,4′-(HO2C)2-bpy]2bpy}2+ building blocks (where M = Ru or Os) were designed and synthesized to study the classic Ru to Os energy transfer process that has potential applications in light-harvesting with supramolecular assemblies. The crystalline nature of the MOFs allows precise determination of the distances between metal centers by X-ray diffraction, thereby facilitating the study of the Ru→Os energy transfer process. The mixed-metal MOFs with 0.3, 0.6, 1.4, and 2.6 mol % Os doping were also synthesized in order to study the energy transfer dynamics with a two-photon excitation at 850 nm. The Ru lifetime at 620 nm decreases from 171 ns in the pure Ru MOF to 29 ns in the sample with 2.6 mol % Os doping. In the mixed-metal samples, energy transfer was observed with an initial growth in Os emission corresponding with the rate of decay of the Ru excited state. These results demonstrate rapid, efficient energy migration and long distance transfer in isomorphous MOFs.
Co-reporter:Brian P. Mehl, Ralph L. House, Abhineet Uppal, Amanda J. Reams, Chuan Zhang, Justin R. Kirschbrown, and John M. Papanikolas
The Journal of Physical Chemistry A 2010 Volume 114(Issue 3) pp:1241-1246
Publication Date(Web):July 9, 2009
DOI:10.1021/jp9009614
Images of second harmonic generation (SHG) in needle-shaped ZnO rods obtained from individual structures show areas of enhanced second harmonic intensity along the longitudinal axis of the rod that are periodically distributed and symmetrically situated relative to the rod midpoint. The spatial modulation is a direct consequence of the fundamental optical field coupling into standing wave resonator modes of the ZnO structure, leading to an enhanced backscattered second harmonic condition that cannot be achieved in bulk ZnO. A more complicated second harmonic image is observed when excitation is below the band gap, which is attributed to whispering gallery modes. This physical phenomenon, which extends beyond just ZnO to many other optical materials, could pave the way to new applications that exploit the nonlinear optical properties of individual structures.
.jpg)