Co-reporter:Takahiro Masuya, Masatoshi Murai, Takeshi Ito, Shunsuke Aburaya, Wataru Aoki, and Hideto Miyoshi
Biochemistry August 15, 2017 Volume 56(Issue 32) pp:4279-4279
Publication Date(Web):July 14, 2017
DOI:10.1021/acs.biochem.7b00612
We previously showed that a bulky ring-strained cycloalkyne possessing a rhodamine fluorophore directly reacts (via strain-promoted click chemistry) with the azido group incorporated (via ligand-directed tosyl chemistry) into Asp160 in the 49 kDa subunit of complex I in bovine heart submitochondrial particles [Masuya, T., et al. (2014) Biochemistry 53, 7816–7823]. This two-step conjugation may be a promising technique for specific chemical modifications of the quinone-access channel in complex I by various molecular probes, which would lead to new methodologies for studying the enzyme. However, because the reactivities of ring-strained cycloalkynes are generally high, they also react with other nucleophilic amino acids in mitochondrial proteins, resulting in significant undesired side reactions. To minimize side reactions and achieve precise pinpoint chemical modification of 49 kDa Asp160, we investigated an optimal pair of chemical tags for the two-step conjugation reaction. We found that instead of strain-promoted click chemistry, Diels–Alder cycloaddition of a pair of cyclopropene incorporated into 49 kDa Asp160 (via ligand-directed tosyl chemistry) and externally added tetrazine is more efficient for the pinpoint modification. An excess of quinone-site inhibitors did not interfere with Diels–Alder cycloaddition between the cyclopropene and tetrazine. These results along with the previous findings (cited above) strongly suggest that in contrast to the predicted quinone-access channel modeled by X-ray crystallographic and single-particle cryo-electron microscopic studies, the channel is open or undergoes large structural rearrangements to allow bulky ligands into the proximity of 49 kDa Asp160.
Co-reporter:Masatoshi MuraiAyaka Okuda, Takenori Yamamoto, Yasuo Shinohara, Hideto Miyoshi
Biochemistry 2017 Volume 56(Issue 4) pp:
Publication Date(Web):January 4, 2017
DOI:10.1021/acs.biochem.6b01011
The role of the voltage-dependent anion channel (VDAC) as a metabolic gate of the mitochondrial outer membrane has been firmly established; however, its involvement in the regulation of mitochondrial permeability transition (PT) remains extremely controversial. Although some low-molecular-weight chemicals have been proposed to modulate the regulatory role of VDAC in the induction of PT, direct binding between these chemicals and VDAC has not yet been demonstrated. In the present study, we investigated whether the ubiquinone molecule directly binds to VDAC in Saccharomyces cerevisiae mitochondria through a photoaffinity labeling technique using two photoreactive ubiquinones (PUQ-1 and PUQ-2). The results of the labeling experiments demonstrated that PUQ-1 and PUQ-2 specifically bind to VDAC1 and that the labeled position is located in the C-terminal region Phe221–Lys234, connecting the 15th and 16th β-strand sheets. Mutations introduced in this region (R224A, Y225A, D228A, and Y225A/D228A) hardly affected the binding affinity of PUQ-1. PUQ-1 and PUQ-2 both significantly suppressed the Ca2+-induced mitochondrial PT (monitored by mitochondrial swelling) at the one digit μM level. Thus, the results of the present study provided, for the first time to our knowledge, direct evidence indicating that the ubiquinone molecule specifically binds to VDAC1 through its quinone-head ring.
Co-reporter:Kenji Okuda, Masatoshi Murai, Shunsuke Aburaya, Wataru Aoki, and Hideto Miyoshi
Biochemistry 2016 Volume 55(Issue 3) pp:470-481
Publication Date(Web):December 23, 2015
DOI:10.1021/acs.biochem.5b01090
We previously succeeded in site-specific chemical modifications of the inner part of the quinone binding pocket of bovine mitochondrial complex I through ligand-directed tosylate (LDT) chemistry using specific inhibitors as high-affinity ligands for the enzyme [Masuya, T., et al. (2014) Biochemistry 53, 2304–2317, 7816–7823]. To investigate whether a short-chain ubiquinone, in place of these specific inhibitors, serves as a ligand for LDT chemistry, we herein synthesized a LDT reagent QT possessing ubiquinone scaffold and performed LDT chemistry with bovine heart submitochondrial particles (SMP). Detailed proteomic analyses revealed that QT properly guides the tosylate group into the quinone binding pocket and transfers a terminal alkyne to nucleophilic amino acids His150 and Asp160 in the 49 kDa subunit. This result clearly indicates that QT occupies the inner part of the quinone binding pocket. Nevertheless, we noted that QT is a unique electron acceptor from complex I distinct from typical short-chain ubiquinones such as ubiquinone-1 (Q1) for several reasons; for example, QT reduction in NADH-QT oxidoreduction was almost completely insensitive to quinone-site inhibitors (such as bullatacin and piericidin A), and this reaction did not produce a membrane potential. On the basis of detailed comparisons of the electron transfer features between QT and typical short-chain quinones, we conclude that QT may accept electrons from an N2 cluster at a position different from that of typical short-chain quinones because of its unique side-chain structure; accordingly, QT reduction is unable to induce putative structural changes inside the quinone binding pocket, which are critical for driving proton translocation. Thus, QT is the first ubiquinone analogue, to the best of our knowledge, the catalytic reduction of which is decoupled from proton translocation through the membrane domain. Implications for mechanistic studies on QT are also discussed.
Co-reporter:Masatoshi Murai, Hiroyuki Inaoka, Takahiro Masuya, Shunsuke Aburaya, Wataru Aoki, and Hideto Miyoshi
Biochemistry 2016 Volume 55(Issue 23) pp:3189-3197
Publication Date(Web):May 23, 2016
DOI:10.1021/acs.biochem.6b00190
Asp160 in the 49 kDa subunit of bovine mitochondrial complex I, which is located in the inner part of the quinone binding cavity, is considered to be an essential residue for energy conversion of the enzyme. To elucidate the catalytic function of this residue, we attempted to specifically methylate 49 kDa Asp160 [Asp(COO)-CH3] through a ligand-directed tosyl (LDT) chemistry technique with an acetogenin derivative (ALM) as a high-affinity ligand. We confirmed the specific methylation of 49 kDa Asp160 through liquid chromatography–tandem mass spectrometry analysis of the tryptic digests of the 49 kDa subunit. The binding affinity of a quinazoline-type inhibitor ([125I]AzQ) occupying the quinone binding cavity was not affected by methylation, indicating that this chemical modification does not induce significant structural changes inside the quinone binding cavity. The methylation of 49 kDa Asp160 did not lead to the complete loss of catalytic activity; the modified enzyme retained partial electron transfer and proton translocation activities. These results along with the fact that 49 kDa Asp160 elicits a very strong nucleophilicity against various LDT reagents in the local protein environment strongly suggest that this residue is free from strict interactions (such as electrostatic interaction) arising from nearby residue(s) and is functionally important but not essential for the energy conversion of complex I.
Co-reporter:Takeshi Ito, Masatoshi Murai, Hironobu Morisaka, and Hideto Miyoshi
Biochemistry 2015 Volume 54(Issue 23) pp:3677-3686
Publication Date(Web):May 26, 2015
DOI:10.1021/acs.biochem.5b00385
We previously demonstrated that amilorides bind to the quinone binding pocket of bovine mitochondrial complex I, not to the hitherto suspected Na+/H+ antiporter-like subunits (ND2, ND4, and ND5) [Murai, M., et al. (2015) Biochemistry 54, 2739–2746]. To characterize the binding position of amilorides within the pocket in more detail, we conducted specific chemical labeling [alkynylation (−C≡CH)] of complex I via ligand-directed tosyl (LDT) chemistry using a newly synthesized amide-type amiloride AAT as a LDT chemistry reagent. The inhibitory potency of AAT, in terms of its IC50 value, was markedly higher (∼1000-fold) than that of prototypical guanidine-type amilorides such as commercially available EIPA and benzamil. Detailed proteomic analyses in combination with click chemistry revealed that the chemical labeling occurred at Asp160 of the 49 kDa subunit (49 kDa Asp160). This labeling was significantly suppressed in the presence of an excess amount of other amilorides or ordinary inhibitors such as quinazoline and acetogenin. Taking into consideration the fact that 49 kDa Asp160 was also specifically labeled by LDT chemistry reagents derived from acetogenin [Masuya, T., et al. (2014) Biochemistry 53, 2307–2317, 7816–7823], we found this aspartic acid to elicit very strong nucleophilicity in the local protein environment. The structural features of the quinone binding pocket in bovine complex I are discussed on the basis of this finding.
Co-reporter:Masatoshi Murai, Sonomi Murakami, Takeshi Ito, and Hideto Miyoshi
Biochemistry 2015 Volume 54(Issue 17) pp:2739-2746
Publication Date(Web):April 7, 2015
DOI:10.1021/acs.biochem.5b00187
Amilorides, well-known inhibitors of Na+/H+ antiporters, were previously shown to inhibit bacterial and mitochondrial NADH-quinone oxidoreductase (complex I) but were markedly less active for complex I. Because membrane subunits ND2, ND4, and ND5 of bovine complex I are homologous to Na+/H+ antiporters, amilorides have been thought to bind to any or all of the antiporter-like subunits; however, there is currently no direct experimental evidence that supports this notion. To identify the binding site of amilorides in bovine complex I, we synthesized two photoreactive amilorides (PRA1 and PRA2), which have a photoreactive azido (-N3) group and terminal alkyne (-C≡CH) group at the opposite ends of the molecules, respectively, and conducted photoaffinity labeling with bovine heart submitochondrial particles. The terminal alkyne group allows various molecular tags to covalently attach to it via Cu+-catalyzed click chemistry, thereby allowing purification and/or detection of the labeled peptides. Proteomic analyses revealed that PRA1 and PRA2 label none of the antiporter-like subunits; they specifically label the accessory subunit B14.5a and core subunit 49 kDa (N-terminal region of Thr25–Glu115), respectively. Suppressive effects of ordinary inhibitors (bullatacin, fenpyroximate, and quinazoline), which bind to the putative quinone binding pocket, on labeling were fairly different between the B14.5a and 49 kDa subunits probably because the binding positions of the three inhibitors differ within the pocket. The results of this study clearly demonstrate that amilorides inhibit complex I activity by occupying the quinone binding pocket rather than directly blocking translocation of protons through the antiporter-like subunits (ND2, ND4, and ND5). The accessory subunit B14.5a may be located adjacent to the N-terminal region of the 49 kDa subunits. The structural features of the quinone binding pocket in bovine complex I were discussed on the basis of these results.
Co-reporter:Masato Abe, Masaaki Nakano, Ayumi Kosaka, Hideto Miyoshi
Tetrahedron Letters 2015 Volume 56(Issue 17) pp:2258-2261
Publication Date(Web):22 April 2015
DOI:10.1016/j.tetlet.2015.03.056
•We synthesized three photoreactive cardiolipins (pCLs), that possess an unstable diazirine ring at different positions.•For synthesis of phosphoramidite intermediate, we use a catalytic amount of 1H-tetrazole and MS 4A to get a good yield.•The pCLs-reconstituted liposomes induced significantly high peroxidase activities of cyt c.•We confirmed that the pCLs synthesized in this study are promising CL mimics for photoaffinity labeling experiments.The photoaffinity labeling technique using photoreactive cardiolipin (CL) is a powerful means for investigating the molecular mechanism of the formation of a specific cytochrome c–cardiolipin complex. Using phosphoramidite chemistry, we synthesized three photoreactive CLs, that possess an unstable diazirine ring at different positions; that is, the acyl chain in the sn-1 or sn-2 position and the central glycerol moiety.
Co-reporter:Takahiro Masuya, Masatoshi Murai, Kentaro Ifuku, Hironobu Morisaka, and Hideto Miyoshi
Biochemistry 2014 Volume 53(Issue 14) pp:
Publication Date(Web):March 24, 2014
DOI:10.1021/bi500205x
The site-specific chemical modification of NADH-quinone oxidoreductase (complex I) by various functional probes such as fluorophores and microbeads, without affecting the enzyme activity, may allow single-molecule analyses of putative dynamic conformational changes in the enzyme. In an attempt to address this challenge, we performed site-specific alkynylation of complex I in bovine heart submitochondrial particles by means of a ligand-directed tosylate (LDT) chemistry strategy with synthetic acetogenin ligand 1, which has an alkynylated tosylate in the tail moiety, as a high-affinity ligand against the enzyme. The terminal alkyne was chosen as the tag to be incorporated into the enzyme because this functional group can serve as a “footing” for subsequent diverse chemical modifications via so-called click chemistry (i.e., azide–alkyne [3+2] cycloaddition in water). To identify the position alkynylated by ligand 1, fluorescent tetramethylrhodamine was covalently attached to the incorporated alkyne by click chemistry after the solubilization of complex I. Detailed proteomic analyses revealed that alkynylation occurred at Asp160 in the 49 kDa subunit, which may be located in the inner part of the putative quinone-binding cavity. The alkynylation was completely suppressed in the presence of an excess of other inhibitors such as bullatacin and quinazoline. While the reaction yield of the alkynylation step via LDT chemistry was estimated to be ∼50%, the alkynylation unfortunately resulted in the almost complete inhibition of enzyme activity. Nevertheless, the results of this study demonstrate that complex I can be site-specifically alkynylated through LDT chemistry, providing a clue about the diverse chemical modifications of the enzyme in combination with click chemistry.
Co-reporter:Masatoshi Murai, Kohei Matsunobu, Sawako Kudo, Kentaro Ifuku, Makoto Kawamukai, and Hideto Miyoshi
Biochemistry 2014 Volume 53(Issue 24) pp:
Publication Date(Web):May 28, 2014
DOI:10.1021/bi500347s
Mitochondrial Coq10 is a ubiquinone (UQ)-binding protein that is a member of the steroidogenic acute regulatory protein (StAR)-related lipid transfer (START) domain superfamily. Deletion of the COQ10 gene was previously shown to cause a marked respiratory defect in Saccharomyces cerevisiae and Schizosaccharomyces pombe, which indicated that Coq10 may support efficient electron transfer between the respiratory complexes; however, its physiological role remains elusive. To elucidate the role of Coq10, we attempted to identify the binding site of UQ in recombinant S. pombe Coq10 expressed in an Escherichia coli cell membrane through photoaffinity labeling with the photoreactive UQ probe, UQ-1, in combination with biotinylation of the labeled peptide by means of the so-called click chemistry. Comprehensive proteomic analyses revealed that the quinone-head ring of UQ-1 specifically binds to the N-terminal region of Phe39–Lys45 of Coq10, which corresponds to the ligand-binding pocket of many proteins containing the START domain. The labeling was completely suppressed in the presence of an excess amount of artificial short-chain UQ analogues, such as UQ2. In the Phe39Ala and Pro41Ala mutants, the extents of labeling were ∼40 and ∼60%, respectively, of that of wild-type Coq10. While Coq10 has been thought to bind UQ, our work first provides the direct evidence of Coq10 accommodating the quinone-head ring of UQ in its START domain. On the basis of these results, the physiological role of Coq10 has been discussed.
Co-reporter:Takahiro Masuya, Masatoshi Murai, Hironobu Morisaka, and Hideto Miyoshi
Biochemistry 2014 Volume 53(Issue 49) pp:
Publication Date(Web):November 24, 2014
DOI:10.1021/bi501342w
Through a ligand-directed tosyl (LDT) chemistry strategy using the synthetic acetogenin ligand AL1, we succeeded in the pinpoint alkynylation (−C≡CH) of Asp160 in the 49 kDa subunit of bovine complex I, which may be located in the inner part of the putative quinone binding cavity of the enzyme [Masuya, T., et al. (2014) Biochemistry, 53, 2307–2317]. This study provided a promising technique for diverse chemical modifications of complex I. To further improve this technique for its adaptation to intact complex I, we here synthesized the new acetogenin ligand AL2, possessing an azido (−N3) group in place of the terminal alkyne in AL1, and attempted the pinpoint azidation of complex I in bovine heart submitochondrial particles. Careful proteomic analyses revealed that, just as in the case of AL1, azidation occurred at 49 kDa Asp160 with a reaction yield of ∼50%, verifying the high site specificity of our LDT chemistry using acetogenin ligands. This finding prompted us to speculate that a reactivity of the azido group incorporated into Asp160 (Asp160-N3) against externally added chemicals can be employed to characterize the structural features of the quinone/inhibitor binding cavity. Consequently, we used a ring-strained cycloalkyne possessing a rhodamine fluorophore (TAMRA-DIBO), which can covalently attach to an azido group via so-called click chemistry without Cu1+ catalysis, as the reaction partner of Asp160-N3. We found that bulky TAMRA-DIBO is capable of reacting directly with Asp160-N3 in intact complex I. Unexpectedly, the presence of an excess amount of short-chain ubiquinones as well as some strong inhibitors (e.g., quinazoline and fenpyroximate) did not interfere with the reaction between TAMRA-DIBO and Asp160-N3; nevertheless, bullatacin, a member of the natural acetogenins, markedly interfered with this reaction. Taking the marked bulkiness of TAMRA-DIBO into consideration, it appears to be difficult to reconcile these results with the proposal that only a narrow entry point accessing to the quinone/inhibitor binding cavity exists in complex I [Baradaran, R., et al. (2013) Nature, 494, 443–448]; rather, they suggest that there may be another access path for TAMRA-DIBO to the cavity.
Co-reporter:Masatoshi Murai
Journal of Bioenergetics and Biomembranes 2014 Volume 46( Issue 4) pp:313-321
Publication Date(Web):2014 August
DOI:10.1007/s10863-014-9562-z
Studies on chemical modifications of bacterial and mitochondrial complex I by synthetic chemical probes as well as endogenous chemicals have provided useful information on the structural and functional aspects of this enzyme. We herein reviewed recent studies that investigated chemical modifications of complex I by endogenous chemicals (e.g. Cys-S-nitrosation, Cys-S-glutathionylation, and Ser-O-phosphorylation) and synthetic reagents (e.g. Cys-SH modification by SH-reagents and the cross-linking of nearby subunits by bifunctional cross-linkers). We also reviewed recent photoaffinity labeling studies using complex I inhibitors, which can be recognized as “site-specific modification” by synthetic chemicals. In addition, we discussed the possibility of site-specific modification by various functional probes via ligand-directed tosylate (LDT) chemistry as a promising approach for unique biophysical studies on complex I.
Co-reporter:Naoto Kojima, Masato Abe, Yuki Suga, Kazufumi Ohtsuki, Tetsuaki Tanaka, Hiroki Iwasaki, Masayuki Yamashita, Hideto Miyoshi
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 5) pp:1217-1219
Publication Date(Web):1 March 2013
DOI:10.1016/j.bmcl.2013.01.018
C34-epi and C34-epi-C35-trifluoro analogues of solamin, a mono-THF annonaceous acetogenin, were synthesized. Their inhibitory activity, along with previously synthesized analogues (C35-fluoro, C35-difluoro, and C35-trifluorosolamins), against bovine mitochondrial NADH–ubiquinone oxidoreductase (complex I) was determined. The present study revealed that the methyl group on the γ-lactone moiety is critical to the potent inhibition of complex I by natural acetogenins.
Co-reporter:Yusuke Shiraishi, Masatoshi Murai, Naoto Sakiyama, Kentaro Ifuku, and Hideto Miyoshi
Biochemistry 2012 Volume 51(Issue 9) pp:1953
Publication Date(Web):February 21, 2012
DOI:10.1021/bi300047h
Using a photoaffinity labeling technique, Nakamaru-Ogiso et al. demonstrated that fenpyroximate, a strong inhibitor of bovine heart mitochondrial NADH-ubiquinone oxidoreductase (complex I), binds to the ND5 subunit [Nakamaru-Ogiso, E., et al. (2003) Biochemistry 42, 746–754]. Considering that the main body of the ND5 subunit composed of transmembrane helixes 1–15 is located at the distal end of the membrane domain [Efremov, R. G., et al. (2010) Nature 465, 441–445], however, their result may be questionable. Because establishing the number and location of inhibitors and/or quinone binding sites in the membrane domain is necessary to elucidate the function of the enzyme, it is critical to clarify whether there is an additional inhibitor and/or quinone binding site besides the interface between the hydrophilic and membrane domains. We therefore performed photoaffinity labeling experiments using two newly synthesized fenpyroximate derivatives [[125I]-4-azidophenyl fenpyroximate ([125I]APF) and [125I]-3-azido-5-iodobenzyl fenpyroximate ([125I]AIF)] possessing a photoreactive azido group at and far from the pharmacophoric core moiety, respectively. Doubled sodium dodecyl sulfate–polyacrylamide gel electrophoresis revealed that [125I]APF and [125I]AIF bind to the PSST and 49 kDa subunits, respectively. Careful examination of the fragmentation patterns of the labeled PSST and 49 kDa subunits generated by limited proteolysis indicated that the residues labeled by [125I]APF and [125I]AIF are located in the Ser43–Arg66 (PSST) and Asp160–Arg174 (49 kDa) regions, respectively, which face the supposed quinone-binding pocket formed at the interface of the PSST, 49 kDa, and ND1 subunits. We conclude that fenpyroximate does not bind to the distal end of the membrane domain but rather resides at the interface between the two domains in a manner such that the pharmacophoric pyrazole ring and side chain of the inhibitor orient toward the PSST and 49 kDa subunits, respectively. This study answers a critical question relating to complex I.
Co-reporter:Masato Abe, Ryota Niibayashi, Shinya Koubori, Ikuko Moriyama, and Hideto Miyoshi
Biochemistry 2011 Volume 50(Issue 39) pp:
Publication Date(Web):August 30, 2011
DOI:10.1021/bi2010202
Induction of the peroxidase activity of cytochrome c (cyt c) by cardiolipin (CL) and H2O2 in mitochondria is suggested to be a key event in early apoptosis. Although electrostatic interaction between the positively charged cyt c and negatively charged CL is a predominant force behind the formation of a specific cyt c–CL complex and sequential induction of the peroxidase activity, molecular mechanisms of hydrophobic interactions involving the fatty acyl chains of CL remain to be investigated. To elucidate the function of the acyl chains, particularly the role of the double bond, we synthesized a variety of CL analogues and examined their peroxidase inducing activity. Irrespective of the number of double bonds in the acyl chains, the peroxidase activity of cyt c induced by liposomes composed of 1,2-dioleoyl-sn-glycero-3-phosphatidylcholine (DOPC) and a different CL (9:1 molar ratio) was similar, except for that of 1,1′,2,2′-tetrastearoylcardiolipin (TSCL, C18:0)-containing liposomes. The peroxidase inducing activity of TSCL-containing liposomes was 3–4-fold greater than that of other CL-containing liposomes. The peroxidase activity induced by all CL-containing liposomes was much lower at high ionic strengths than that at low ionic strengths because of diminution of the electrostatic interaction. The peroxidase inducing effects of various CL-containing liposomes were related well to their ability to associate with cyt c. Thus, our results revealed that at low CL levels, the saturated acyl chain of CL is favorable for the activation of peroxidase activity of CL-bound cyt c and the proposed critical role of the double bond is not a general feature of the cyt c–CL interaction. The polarity of the membrane surface of TSCL-containing liposomes was slightly, but significantly, lower than that of other CL-containing liposomes, suggesting that the higher activating ability of TSCL-containing liposomes may be due to a reduced level of hydration of the polar head region reflecting tighter packing of the fully saturated acyl chains. Moreover, using CL analogues in which a central glycerol head moiety was modified, we revealed that the natural structure of the head moiety is not critical for the formation of the active cyt c–CL complex. The effects of the CL content of the liposomal membrane on the cyt c–CL interaction are discussed.
Co-reporter:Masatoshi Murai, Yuko Mashimo, Judy Hirst, and Hideto Miyoshi
Biochemistry 2011 Volume 50(Issue 32) pp:
Publication Date(Web):July 1, 2011
DOI:10.1021/bi200883c
Quinazolines are strong inhibitors of NADH-ubiquinone oxidoreductase (complex I) from bovine heart mitochondria. Using a photoreactive quinazoline, [125I]AzQ, and bovine heart submitochondrial particles (SMPs), we demonstrated previously that [125I]AzQ binds at the interface of the 49 kDa and ND1 subunits in complex I; it labeled a site in the N-terminal (Asp41–Arg63) region of the 49 kDa subunit, suggesting that this region contacts the ND1 subunit [Murai, M., et al. (2009) Biochemistry 48, 688–698]. The labeled region of ND1 could not be identified because it is highly hydrophobic, and the SMPs did not yield sufficient amounts of labeled protein. Here, we describe how photoaffinity labeling of isolated complex I by [125I]AzQ yielded sufficient material for identification of the labeled region of the ND1 subunit. The inhibition of the isolated enzyme by AzQ is comparable to that of SMPs. Our results reveal that the labeled site in ND1 is between Asp199 and Lys262, mostly likely in the third matrix loop that connects the fifth and sixth transmembrane helices. Thus, our results reveal new information about the interface between the hydrophilic and hydrophobic domains of complex I, a region that is thought to be important for ubiquinone reduction and energy transduction.
Co-reporter:Shuhei Yamamoto, Masato Abe, Sayo Nakanishi, Masatoshi Murai, Hideto Miyoshi
Tetrahedron Letters 2011 Volume 52(Issue 24) pp:3090-3093
Publication Date(Web):15 June 2011
DOI:10.1016/j.tetlet.2011.03.149
Acetogenins are valuable inhibitor probes to get an insight into the structural and functional properties of mitochondrial NADH-ubiquinone oxidoreductase (complex I). We synthesized a photoreactive acetogenin mimic ([125I]DANA) which retained a strong inhibitory activity. The preliminary photoaffinity labeling with bovine heart submitochondrial particles revealed that [125I]DANA binds to the ND1 subunit among 45 different subunits with high specificity.Figure optionsDownload full-size imageDownload as PowerPoint slide
Co-reporter:Masatoshi Murai, Tetsuo Yamashita, Mai Senoh, Yuko Mashimo, Michihiko Kataoka, Hiroaki Kosaka, Akemi Matsuno-Yagi, Takao Yagi and Hideto Miyoshi
Biochemistry 2010 Volume 49(Issue 13) pp:
Publication Date(Web):March 1, 2010
DOI:10.1021/bi100005j
The Ndi1 enzyme found in the mitochondrial membrane of Saccharomyces cerevisiae is an NDH-2-type alternative NADH-quinone oxidoreductase. As Ndi1 is expected to be a possible remedy for complex I defects of mammalian mitochondria, a detailed biochemical characterization of the enzyme is needed. To identify the ubiquinone (UQ) binding site in Ndi1, we conducted photoaffinity labeling using a photoreactive biotinylated UQ mimic (compound 2) synthesized following a concept of the least possible modification of the substituents on the quinone ring. Cleavage with CNBr of Ndi1 cross-linked by 2 revealed the UQ ring of 2 to be specifically cross-linked to the Phe281−Met410 region (130 amino acids). Digestion of the CNBr fragment with V8 protease and lysylendopeptidase (Lys-C) gave ∼8 and ∼4 kDa peptides, respectively. The ∼8 kDa V8 digest was identified as the Thr329−Glu399 region (71 amino acids) by an N-terminal sequence analysis. Although the ∼4 kDa Lys-C digest could not be identified by N-terminal sequence analysis, the band was thought to cover the Gly374−Lys405 region (32 amino acids). Taken together, the binding site of the Q ring of 2 must be located in a common region of the V8 protease, and Lys-C digests Gly374−Glu399 (26 amino acids). Superimposition of the Ndi1 sequence onto a three-dimensional structural model of NDH-2 from Escherichia coli suggested that the C-terminal portion of this region is close to the isoalloxazine ring of FAD.
Co-reporter:Nobuyuki Kakutani, Masatoshi Murai, Naoto Sakiyama and Hideto Miyoshi
Biochemistry 2010 Volume 49(Issue 23) pp:
Publication Date(Web):May 11, 2010
DOI:10.1021/bi100454b
Biochemical characterization of the inhibition mechanism of Δlac-acetogenins synthesized in our laboratory indicated that they are a new type of inhibitor of bovine heart mitochondrial NADH−ubiquinone oxidoreductase (complex I) [Murai, M., et al. (2006) Biochemistry 45, 9778−9787]. To identify the binding site of Δlac-acetogenins with a photoaffinity labeling technique, we synthesized a photoreactive Δlac-acetogenin ([125I]diazinylated Δlac-acetogenin, [125I]DAA) which has a small photoreactive diazirine group attached to a pharmacophore, the bis-THF ring moiety. Characterization of the inhibitory effects of DAA on bovine complex I revealed unique features specific to, though not completely the same as those of, the original Δlac-acetogenin. Using [125I]DAA, we carried out photoaffinity labeling with bovine heart submitochondrial particles. Analysis of the photo-cross-linked protein by Western blotting and immunoprecipitation revealed that [125I]DAA binds to the membrane subunit ND1 with high specificity. The photo-cross-linking to the ND1 subunit was suppressed by an exogenous short-chain ubiquinone (Q2) in a concentration-dependent manner. Careful examination of the fragmentation patterns of the cross-linked ND1 generated by limited proteolysis using lysylendopeptidase, endoprotease Asp-N, or trypsin and their changes in the presence of the original Δlac-acetogenin strongly suggested that the cross-linked residues are located at two different sites in the third matrix-side loop connecting the fifth and sixth transmembrane helices.
Co-reporter:Masato Abe, Shigeki Kitsuda, Shunji Ohyama, Shinya Koubori, Masatoshi Murai, Hideto Miyoshi
Tetrahedron Letters 2010 Volume 51(Issue 15) pp:2071-2073
Publication Date(Web):14 April 2010
DOI:10.1016/j.tetlet.2010.02.071
Production of a wide variety of cardiolipin (CL) analogues is critical for studying molecular mechanism of diverse biological functions of CL in mitochondria. We describe a concise procedure for the synthesis of CL using a phosphoramidite approach, which allows for the production of diverse CL analogues bearing linoleic acid(s) at any position on the glycerol backbone.
Co-reporter:Masatoshi Murai, Koji Sekiguchi, Takaaki Nishioka and Hideto Miyoshi
Biochemistry 2009 Volume 48(Issue 4) pp:
Publication Date(Web):January 7, 2009
DOI:10.1021/bi8019977
The diverse inhibitors of bovine heart mitochondrial complex I (NADH−ubiquinone oxidoreductase) are believed to share a common large binding domain with partially overlapping sites, though it remains unclear how these binding sites relate to each other. To obtain new insight into the inhibitor binding domain in complex I, we synthesized a photoreactive azidoquinazoline {[125I]-6-azido-4-(4-iodophenethylamino)quinazoline, [125I]AzQ}, in which a photolabile azido group was introduced into the toxophoric quinazoline ring to allow specific cross-linking, and carried out a photoaffinity labeling study using bovine heart submitochondrial particles. Analysis of the photo-cross-linked proteins by peptide mass fingerprinting and immunoblotting revealed that [125I]AzQ specifically binds to the 49 kDa and ND1 subunits with a frequency of ∼4:1. The cross-linking was completely blocked by excess amounts of other inhibitors such as acetogenin and fenpyroximate. Considerable cross-linking was also detected in the ADP/ATP carrier and 3-hydroxybutyrate dehydrogenase, though it was not associated with dysfunction of the two proteins. The partial proteolysis of the [125I]AzQ-labeled 49 kDa subunit by V8-protease and N-terminal sequencing of the resulting peptides revealed that the amino acid residue cross-linked by [125I]AzQ is within the sequence region Thr25−Glu143 (118 amino acids). Furthermore, examination of fragment patterns generated by exhaustive digestion of the [125I]AzQ-labeled 49 kDa subunit by V8-protease, lysylendopeptidase, or trypsin strongly suggested that the cross-linked residue is located within the region Asp41−Arg63 (23 amino acids). The present study has revealed, for the first time, the inhibitor binding site in complex I at the sub-subunit level.
Co-reporter:Naoya Ichimaru, Masatoshi Murai, Nobuyuki Kakutani, Junko Kako, Atsushi Ishihara, Yoshiaki Nakagawa, Takaaki Nishioka, Takao Yagi and Hideto Miyoshi
Biochemistry 2008 Volume 47(Issue 40) pp:
Publication Date(Web):September 10, 2008
DOI:10.1021/bi8010362
The mode of action of Δlac-acetogenins, strong inhibitors of bovine heart mitochondrial complex I, is different from that of traditional inhibitors such as rotenone and piericidin A [Murai, M., et al. (2007) Biochemistry 46, 6409−6416]. As further exploration of these unique inhibitors might provide new insights into the terminal electron transfer step of complex I, we drastically modified the structure of Δlac-acetogenins and characterized their inhibitory action. In particular, on the basis of structural similarity between the bis-THF and the piperazine rings, we here synthesized a series of piperazine derivatives. Some of the derivatives exhibited very potent inhibition at nanomolar levels. The hydrophobicity of the side chains and their balance were important structural factors for the inhibition, as is the case for the original Δlac-acetogenins. However, unlike in the case of the original Δlac-acetogenins, (i) the presence of two hydroxy groups is not crucial for the activity, (ii) the level of superoxide production induced by the piperazines is relatively high, (iii) the inhibitory potency for the reverse electron transfer is remarkably weaker than that for the forward event, and (iv) the piperazines efficiently suppressed the specific binding of a photoaffinity probe of natural-type acetogenins ([125I]TDA) to the ND1 subunit. We therefore conclude that the action mechanism of the piperazine series differs from that of the original Δlac-acetogenins. The photoaffinity labeling study using a newly synthesized photoreactive piperazine ([125I]AFP) revealed that this compound binds to the 49 kDa subunit and an unidentified subunit, not ND1, with a frequency of ∼1:3. A variety of traditional complex I inhibitors as well as Δlac-acetogenins suppressed the specific binding of [125I]AFP to the subunits. The apparent competitive behavior of inhibitors that seem to bind to different sites may be due to structural changes at the binding site, rather than occupying the same site. The meaning of the occurrence of diverse inhibitors exhibiting different mechanisms of action is discussed in light of the functionality of the membrane arm of complex I.
Co-reporter:Masato Abe, Akina Kubo, Shuhei Yamamoto, Yoshinori Hatoh, Masatoshi Murai, Yasunao Hattori, Hidefumi Makabe, Takaaki Nishioka and Hideto Miyoshi
Biochemistry 2008 Volume 47(Issue 23) pp:
Publication Date(Web):May 14, 2008
DOI:10.1021/bi800506s
Studies of the action mechanism of acetogenins, the most potent and structurally unique inhibitors of bovine heart mitochondrial complex I (NADH-ubiquinone oxidoreductase), are valuable in characterizing the inhibitor binding site in this enzyme. Our previous study deepened our understanding of the dynamic function of the spacer region of bis-THF acetogenins [Abe, M., et al. (2005) Biochemistry 44, 14898−14906] but, at the same time, posed new important questions. First, while the two toxophores (i.e., the hydroxylated THF and the γ-lactone rings) span a distance shorter than that of the extended 13 carbon atoms [-(CH2)13-], what is the apparent optimal length of the spacer for the inhibition of 13 carbon atoms? In other words, what is the functional role of the additional methylene groups? Second, why was the inhibitory potency of the mono-THF derivative, but not the bis-THF derivative, drastically reduced by hardening the spacer covering 10 carbon atoms into a rodlike shape [-CH2-(C≡C)4-CH2-]? This study was designed not only to answer these questions but also to further disclose the dynamic functions of the spacer. We here synthesized systematically designed acetogenins, including mono- and bis-THF derivatives, and evaluated their inhibitory effects on bovine complex I. With regard to the first question, we demonstrated that the additional methylenes enhance the hydrophobicity of the spacer region, which may be thermodynamically advantageous for bringing the polar γ-lactone ring into the membrane-embedded segment of complex I. With regard to the second question, we observed that a decrease in the flexibility of the spacer region is more adverse to the action of the mono-THF series than that of the bis-THF series. As a cause of this difference, we suggest that for bis-THF derivatives, one of the two THF rings, being adjacent to the spacer, is capable of working as a pseudospacer to overcome the remarkable decrease in the conformational freedom and/or the length of the spacer. Moreover, using photoresponsive acetogenins that undergo drastic and reversible conformational changes with alternating UV−vis irradiation, we provided further evidence that the spacer region is free from steric congestion arising from the putative binding site probably because there is no receptor wall for the spacer region.
Co-reporter:Sayo Nakanishi, Masato Abe, Shuhei Yamamoto, Masatoshi Murai, Hideto Miyoshi
Biochimica et Biophysica Acta (BBA) - Bioenergetics (September 2011) Volume 1807(Issue 9) pp:1170-1176
Publication Date(Web):September 2011
DOI:10.1016/j.bbabio.2011.05.012
Co-reporter:Koji Sekiguchi, Masatoshi Murai, Hideto Miyoshi
Biochimica et Biophysica Acta (BBA) - Bioenergetics (September 2009) Volume 1787(Issue 9) pp:
Publication Date(Web):1 September 2009
DOI:10.1016/j.bbabio.2009.02.016
125I-labeled (trifluoromethyl)phenyldiazirinyl acetogenin, [125I]TDA, a photoaffinity labeling probe of acetogenin, photo-cross-links to the ND1 subunit of bovine heart mitochondrial NADH–ubiquinone oxidoreductase (complex I) with high specificity [M. Murai, A. Ishihara, T. Nishioka, T. Yagi, and H. Miyoshi, (2007) The ND1 subunit constructs the inhibitor binding domain in bovine heart mitochondrial complex I, Biochemistry 46 6409–6416.]. To identify the binding site of [125I]TDA in the ND1 subunit, we carried out limited proteolysis of the subunit cross-linked by [125I]TDA using various proteases and carefully analyzed the fragmentation patterns. Our results revealed that the cross-linked residue is located within the region of the 4th to 5th transmembrane helices (Val144–Glu192) of the subunit. It is worth noting that an excess amount of short-chain ubiquinones such as ubiquinone-2 (Q2) and 2-azido-Q2 suppressed the cross-linking by [125I]TDA in a concentration-dependent way. Although the question of whether the binding sites for ubiquinone and different inhibitors in complex I are identical remains to be answered, the present study provided, for the first time, direct evidence that an inhibitor (acetogenin) and ubiquinone competitively bind to the enzyme. Considering the present results along with earlier photoaffinity labeling studies, we propose that not all inhibitors acting at the terminal electron transfer step of complex I necessarily bind to the ubiquinone binding site itself.
Co-reporter:Masatoshi Murai, Hideto Miyoshi
Biochimica et Biophysica Acta (BBA) - Bioenergetics (July 2016) Volume 1857(Issue 7) pp:884-891
Publication Date(Web):July 2016
DOI:10.1016/j.bbabio.2015.11.009

![3-Pyrrolidinesulfonic acid,
1-[3-[[2-[[5-[(3aS,4S,6aR)-hexahydro-2-oxo-1H-thieno[3,4-d]imidazol-4
-yl]-1-oxopentyl]amino]ethyl]dithio]-1-oxopropoxy]-2,5-dioxo-,
monosodium salt](http://img.cochemist.com/ccimg/325200/325143-98-4.png)
![3-Pyrrolidinesulfonic acid,
1-[3-[[2-[[5-[(3aS,4S,6aR)-hexahydro-2-oxo-1H-thieno[3,4-d]imidazol-4
-yl]-1-oxopentyl]amino]ethyl]dithio]-1-oxopropoxy]-2,5-dioxo-,
monosodium salt](http://img.cochemist.com/ccimg/325200/325143-98-4_b.png)
![2(5H)-Furanone,3-[(2R,13R)-2,13-dihydroxy-13-[(2R,2'R,5R,5'R)-octahydro-5'-[(1R)-1-hydroxyundecyl][2,2'-bifuran]-5-yl]tridecyl]-5-methyl-,(5S)-](http://img.cochemist.com/ccimg/103000/102989-24-2.png)
![2(5H)-Furanone,3-[(2R,13R)-2,13-dihydroxy-13-[(2R,2'R,5R,5'R)-octahydro-5'-[(1R)-1-hydroxyundecyl][2,2'-bifuran]-5-yl]tridecyl]-5-methyl-,(5S)-](http://img.cochemist.com/ccimg/103000/102989-24-2_b.png)





![1,3-Dioxolane, 4-[[(4-methoxyphenyl)methoxy]methyl]-2,2-dimethyl-,(4S)-](http://img.cochemist.com/ccimg/109800/109786-73-4.png)
![1,3-Dioxolane, 4-[[(4-methoxyphenyl)methoxy]methyl]-2,2-dimethyl-,(4S)-](http://img.cochemist.com/ccimg/109800/109786-73-4_b.png)
![3,5,9-Trioxa-4-phosphaheptacos-18-en-1-aminium,4-hydroxy-N,N,N-trimethyl-10-oxo-7-[(1-oxohexadecyl)oxy]-, inner salt, 4-oxide,(7R,18Z)-](http://img.cochemist.com/ccimg/59500/59491-62-2.png)
![3,5,9-Trioxa-4-phosphaheptacos-18-en-1-aminium,4-hydroxy-N,N,N-trimethyl-10-oxo-7-[(1-oxohexadecyl)oxy]-, inner salt, 4-oxide,(7R,18Z)-](http://img.cochemist.com/ccimg/59500/59491-62-2_b.png)
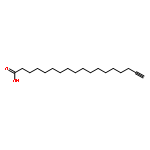
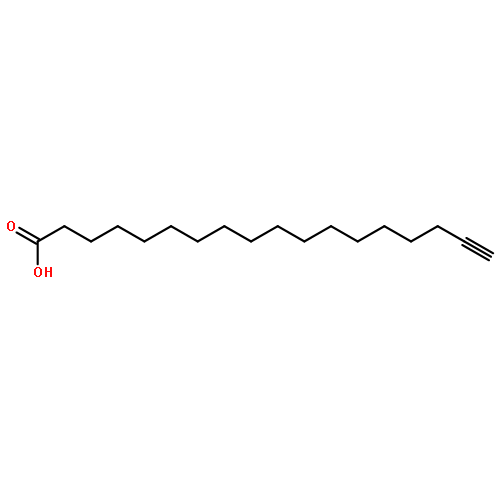
![9-Octadecenoic acid(9Z)-, 1,1'-[(1S)-1-(hydroxymethyl)-1,2-ethanediyl] ester](http://img.cochemist.com/ccimg/24600/24529-88-2.png)
![9-Octadecenoic acid(9Z)-, 1,1'-[(1S)-1-(hydroxymethyl)-1,2-ethanediyl] ester](http://img.cochemist.com/ccimg/24600/24529-88-2_b.png)










![4-Pyridinol,2-[(2E,5E,7E,9R,10R,11E)-10-hydroxy-3,7,9,11-tetramethyl-2,5,7,11-tridecatetraen-1-yl]-5,6-dimethoxy-3-methyl-](http://img.cochemist.com/ccimg/2800/2738-64-9.png)
![4-Pyridinol,2-[(2E,5E,7E,9R,10R,11E)-10-hydroxy-3,7,9,11-tetramethyl-2,5,7,11-tridecatetraen-1-yl]-5,6-dimethoxy-3-methyl-](http://img.cochemist.com/ccimg/2800/2738-64-9_b.png)

![PYRAZINECARBOXAMIDE, 3,5-DIAMINO-6-CHLORO-N-[IMINO[(2-PHENYLETHYL)AMINO]METHYL]-](http://img.cochemist.com/ccimg/1200/1163-46-8.png)
![PYRAZINECARBOXAMIDE, 3,5-DIAMINO-6-CHLORO-N-[IMINO[(2-PHENYLETHYL)AMINO]METHYL]-](http://img.cochemist.com/ccimg/1200/1163-46-8_b.png)
![Pyrazinecarboxamide,3-amino-N-(aminoiminomethyl)-6-chloro-5-[(2-methylpropyl)amino]-](http://img.cochemist.com/ccimg/1200/1151-55-9.png)
![Pyrazinecarboxamide,3-amino-N-(aminoiminomethyl)-6-chloro-5-[(2-methylpropyl)amino]-](http://img.cochemist.com/ccimg/1200/1151-55-9_b.png)
![3,5,9-Trioxa-4-phosphaheptacosa-18,21-dien-1-aminium,4-hydroxy-N,N,N-trimethyl-10-oxo-7-[[(9Z,12Z)-1-oxo-9,12-octadecadien-1-yl]oxy]-,inner salt, 4-oxide, (7R,18Z,21Z)-](http://img.cochemist.com/ccimg/1000/998-06-1.png)
![3,5,9-Trioxa-4-phosphaheptacosa-18,21-dien-1-aminium,4-hydroxy-N,N,N-trimethyl-10-oxo-7-[[(9Z,12Z)-1-oxo-9,12-octadecadien-1-yl]oxy]-,inner salt, 4-oxide, (7R,18Z,21Z)-](http://img.cochemist.com/ccimg/1000/998-06-1_b.png)


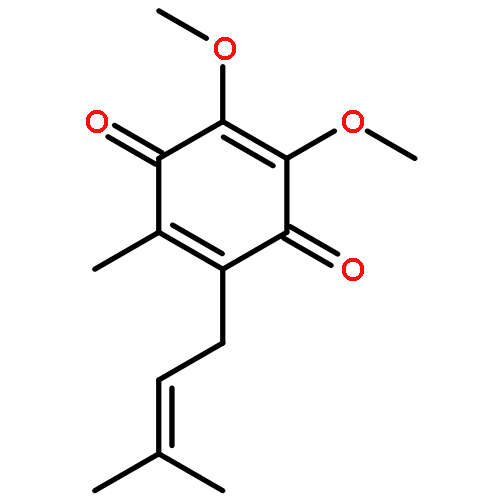
![2,5-Cyclohexadiene-1,4-dione,2-[(2E)-3,7-dimethyl-2,6-octadien-1-yl]-5,6-dimethoxy-3-methyl-](http://img.cochemist.com/ccimg/700/606-06-4.png)
![2,5-Cyclohexadiene-1,4-dione,2-[(2E)-3,7-dimethyl-2,6-octadien-1-yl]-5,6-dimethoxy-3-methyl-](http://img.cochemist.com/ccimg/700/606-06-4_b.png)

![2(5H)-Furanone,3-[(2R,13R)-2,13-dihydroxy-13-[(2R,2'R,5R,5'R)-octahydro-5'-[(1S)-1-hydroxyundecyl][2,2'-bifuran]-5-yl]tridecyl]-5-methyl-,(5S)-](http://img.cochemist.com/ccimg/123200/123123-32-0.png)
![2(5H)-Furanone,3-[(2R,13R)-2,13-dihydroxy-13-[(2R,2'R,5R,5'R)-octahydro-5'-[(1S)-1-hydroxyundecyl][2,2'-bifuran]-5-yl]tridecyl]-5-methyl-,(5S)-](http://img.cochemist.com/ccimg/123200/123123-32-0_b.png)
![1,2-Propanediol, 3-[(4-methoxyphenyl)methoxy]-, (R)-](http://img.cochemist.com/ccimg/109800/109786-74-5.png)
![1,2-Propanediol, 3-[(4-methoxyphenyl)methoxy]-, (R)-](http://img.cochemist.com/ccimg/109800/109786-74-5_b.png)
![2-Pyrazinecarboxamide,3-amino-N-(aminoiminomethyl)-6-chloro-5-[methyl(2-methylpropyl)amino]-](http://img.cochemist.com/ccimg/96900/96861-65-3.png)
![2-Pyrazinecarboxamide,3-amino-N-(aminoiminomethyl)-6-chloro-5-[methyl(2-methylpropyl)amino]-](http://img.cochemist.com/ccimg/96900/96861-65-3_b.png)
![3,5,9-Trioxa-4-phosphaheptacos-18-en-1-aminium,4-hydroxy-N,N,N-trimethyl-10-oxo-7-[[(9Z)-1-oxo-9-octadecen-1-yl]oxy]-, innersalt, 4-oxide, (7R,18Z)-](http://img.cochemist.com/ccimg/4300/4235-95-4.png)
![3,5,9-Trioxa-4-phosphaheptacos-18-en-1-aminium,4-hydroxy-N,N,N-trimethyl-10-oxo-7-[[(9Z)-1-oxo-9-octadecen-1-yl]oxy]-, innersalt, 4-oxide, (7R,18Z)-](http://img.cochemist.com/ccimg/4300/4235-95-4_b.png)
![2-Pyrazinecarboxamide,3,5-diamino-6-chloro-N-[imino[(phenylmethyl)amino]methyl]-](http://img.cochemist.com/ccimg/2900/2898-76-2.png)
![2-Pyrazinecarboxamide,3,5-diamino-6-chloro-N-[imino[(phenylmethyl)amino]methyl]-](http://img.cochemist.com/ccimg/2900/2898-76-2_b.png)
![2-Pyrazinecarboxamide,3-amino-N-(aminoiminomethyl)-6-chloro-5-[ethyl(1-methylethyl)amino]-](http://img.cochemist.com/ccimg/1200/1154-25-2.png)
![2-Pyrazinecarboxamide,3-amino-N-(aminoiminomethyl)-6-chloro-5-[ethyl(1-methylethyl)amino]-](http://img.cochemist.com/ccimg/1200/1154-25-2_b.png)