Co-reporter:Na Song, Christopher J. Dares, Matthew V. Sheridan, and Thomas J. Meyer
The Journal of Physical Chemistry C 2016 Volume 120(Issue 42) pp:23984-23988
Publication Date(Web):October 4, 2016
DOI:10.1021/acs.jpcc.6b08176
The redox properties of a surface-bound phosphate flavin derivative (flavin mononucleotide, FMN) have been investigated on planar-FTO and nanoITO electrodes under acidic conditions in 1:1 CH3CN/H2O (V:V). On FTO, reversible 2e–/2H+ reduction of FTO|-FMN to FTO|-FMNH2 occurs with the pH and scan rate dependence expected for a 2e–/2H+ surface-bound couple. The addition of tetramethylbenzoquinone (Me4Q) results in rapid electrocatalyzed reduction to the hydroquinone by a pathway first order in quinone and first order in acid with kH = (2.6 ± 0.2) × 106 M–1 s–1. Electrocatalytic reduction of the quinone also occurs on derivatized, high surface area nanoITO electrodes with evidence for competitive rate-limiting diffusion of the quinone into the mesoporous nanostructure.
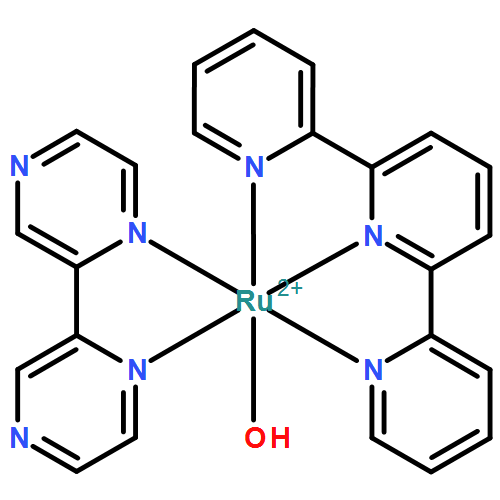
Co-reporter:Na Song, Ming-Tian Zhang, Robert A. Binstead, Zhen Fang, and Thomas J. Meyer
Inorganic Chemistry 2014 Volume 53(Issue 8) pp:4100-4105
Publication Date(Web):April 9, 2014
DOI:10.1021/ic500072e
Co-reporter:Akitaka Ito, Troy E. Knight, David J. Stewart, M. Kyle Brennaman, and Thomas J. Meyer
The Journal of Physical Chemistry A 2014 Volume 118(Issue 45) pp:10326-10332
Publication Date(Web):April 10, 2014
DOI:10.1021/jp5019873
Higher-energy emissions from the metal-to-ligand charge-transfer (MLCT) excited states of a series of polypyridyl Os(II) complexes were observed at the fluid-to-film transition in PEG-DMA550. The higher-energy excited states, caused by a “rigid medium effect” in the film, led to enhanced emission quantum yields and longer excited-state lifetimes. Detailed analyses of spectra and excited-state dynamics by Franck–Condon emission spectral analysis and application of the energy gap law for nonradiative excited-state decay reveal that the rigid medium effect arises from the inability of part of the local medium dielectric environment to respond to the change in charge distribution in the excited state during its lifetime. Enhanced excited-state lifetimes are consistent with qualitative and quantitative predictions of the energy gap law.
Co-reporter:Hanlin Luo, Wenjing Song, Paul G. Hoertz, Kenneth Hanson, Rudresh Ghosh, Sylvie Rangan, M. Kyle Brennaman, Javier J. Concepcion, Robert A. Binstead, Robert Allen Bartynski, Rene Lopez, and Thomas J. Meyer
Chemistry of Materials 2013 Volume 25(Issue 2) pp:122
Publication Date(Web):December 11, 2012
DOI:10.1021/cm3027972
Orthorhombic Nb2O5 nanocrystalline films functionalized with [Ru(bpy)2(4,4′-(PO3H2)2bpy)]2+ were used as the photoanode in dye-sensitized photoelectrosynthesis cells (DSPEC) for hydrogen generation. A set of experiments to establish key properties—conduction band, trap state distribution, interfacial electron transfer dynamics, and DSPEC efficiency—were undertaken to develop a general protocol for future semiconductor evaluation and for comparison with other wide-band-gap semiconductors. We have found that, for a T-phase orthorhombic Nb2O5 nanocrystalline film, the conduction band potential is slightly positive (<0.1 eV), relative to that for anatase TiO2. Anatase TiO2 has a wide distribution of trap states including deep trap and band-tail trap states. Orthorhombic Nb2O5 is dominated by shallow band-tail trap states. Trap state distributions, conduction band energies, and interfacial barriers appear to contribute to a slower back electron transfer rate, lower injection yield on the nanosecond time scale, and a lower open-circuit voltage (Voc) for orthorhombic Nb2O5, compared to anatase TiO2. In an operating DSPEC, with the ethylenediaminetetraacetic tetra-anion (EDTA4–) added as a reductive scavenger, H2 quantum yield and photostability measurements show that Nb2O5 is comparable, but not superior, to TiO2.Keywords: conduction band; DSPEC; H2 evolution; Nb2O5; TiO2; trap states;
Co-reporter:Manuel A. Méndez, Leila Alibabaei, Javier J. Concepcion, and Thomas J. Meyer
ACS Catalysis 2013 Volume 3(Issue 8) pp:1850
Publication Date(Web):July 3, 2013
DOI:10.1021/cs4003595
A procedure is described for preparing and derivatizing novel, high surface area electrodes consisting of thin layers of nanostructured ITO (Sn(IV)-doped indium tin oxide, nanoITO) on reticulated vitreous carbon (RVC) to give RVC|nanoITO. The resulting hybrid electrodes are highly stabilized oxidatively. They were surface-derivatized by phosphonate binding of the electrocatalyst, [Ru(Mebimpy)(4,4′-((HO)2OPCH2)2bpy)(OH2)]2+ (Mebimpy = 2,6-bis(1-methylbenzimidazol-2-yl)pyridine; bpy = 2,2′-bipyridine) (1-PO3H2) to give RVC|nanoITO-RuII-OH22+. The redox properties of the catalyst are retained on the electrode surface. Electrocatalytic oxidation of benzyl alcohol to benzaldehyde occurs with a 75% Faradaic efficiency compared to 57% on nanoITO. Electrocatalytic water oxidation at 1.4 V vs SCE on derivatized RVC|nanoITO electrode with an internal surface area of 19.5 cm2 produced 7.3 μmoles of O2 in 70% Faradaic yield in 50 min.Keywords: electrocatalysis; high surface area; nanoITO; polypyridyl ruthenium complex; reticulated vitreous carbon; water oxidation
Co-reporter:Leila Alibabaei, Hanlin Luo, Ralph L. House, Paul G. Hoertz, Rene Lopez and Thomas J. Meyer
Journal of Materials Chemistry A 2013 vol. 1(Issue 13) pp:4133-4145
Publication Date(Web):28 Jan 2013
DOI:10.1039/C2TA00935H
Solar fuels hold great promise as a permanent, environmentally friendly, long-term renewable energy source, that would be readily available across the globe. In this account, an approach to solar fuels is described based on Dye Sensitized Photoelectrosynthesis Cells (DSPEC) that mimic the configuration used in Dye Sensitized Solar Cells (DSSC), but with the goal of producing oxygen and a high energy solar fuel in the separate compartments of a photoelectrochemical cell rather than a photopotential and photocurrent.
Co-reporter:Zhen Fang, Akitaka Ito, Shahar Keinan, Zuofeng Chen, Zoe Watson, Jason Rochette, Yosuke Kanai, Darlene Taylor, Kirk S. Schanze, and Thomas J. Meyer
Inorganic Chemistry 2013 Volume 52(Issue 15) pp:8511-8520
Publication Date(Web):July 16, 2013
DOI:10.1021/ic400520m
A ruthenium containing polymer featuring a short carbonyl-amino-methylene linker has been prepared by atom transfer radical polymerization (ATRP). The polymer was derived from ATRP of the N-hydroxysuccinimide (NHS) derivative of p-vinylbenzoic acid, followed by an amide coupling reaction of the NHS-polystyrene with Ru(II) complexes derivatized with aminomethyl groups (i.e., [Ru(bpy)2(CH3-bpy-CH2NH2)]2+ where bpy is 2,2′-bipyridine, and CH3-bpy-CH2NH2 is 4-methyl-4′-aminomethyl-2,2′-bipyridine). The Ru-functionalized polymer structure was confirmed by using nuclear magnetic resonance and infrared spectroscopy, and the results suggest that a high loading ratio of polypyridylruthenium chromophores on the polystyrene backbone was achieved. The photophysical properties of the polymer were characterized in solution and in rigid ethylene glycol glasses. In solution, emission quantum yield and lifetime studies reveal that the polymer’s metal-to-ligand charge transfer (MLCT) excited states are quenched relative to a model Ru complex chromophore. In rigid media, the MLCT-ground state band gap and lifetime are both increased relative to solution with time-resolved emission measurements revealing fast energy transfer hopping within the polymer. Molecular dynamics studies of the polymer synthesized here as well as similar model systems with various spatial arrangements of the pendant Ru complex chromophores suggest that the carbonyl-amino-methylene linker probed in our target polymer provides shorter Ru–Ru nearest-neighbor distances leading to an increased Ru*-Ru energy hopping rate, compared to those with longer linkers in counterpart polymers.
Co-reporter:Michael R. Norris ; Javier J. Concepcion ; Christopher R. K. Glasson ; Zhen Fang ; Alexander M. Lapides ; Dennis L. Ashford ; Joseph L. Templeton
Inorganic Chemistry 2013 Volume 52(Issue 21) pp:12492-12501
Publication Date(Web):October 21, 2013
DOI:10.1021/ic4014976
Water-stable, surface-bound chromophores, catalysts, and assemblies are an essential element in dye-sensitized photoelectrosynthesis cells for the generation of solar fuels by water splitting and CO2 reduction to CO, other oxygenates, or hydrocarbons. Phosphonic acid derivatives provide a basis for stable chemical binding on metal oxide surfaces. We report here the efficient synthesis of 4,4′-bis(diethylphosphonomethyl)-2,2′-bipyridine and 4,4′-bis(diethylphosphonate)-2,2′-bipyridine, as well as the mono-, bis-, and tris-substituted ruthenium complexes, [Ru(bpy)2(Pbpy)]2+, [Ru(bpy)(Pbpy)2]2+, [Ru(Pbpy)3]2+, [Ru(bpy)2(CPbpy)]2+, [Ru(bpy)(CPbpy)2]2+, and [Ru(CPbpy)3]2+ [bpy = 2,2′-bipyridine; Pbpy = 4,4′-bis(phosphonic acid)-2,2′-bipyridine; CPbpy = 4,4′-bis(methylphosphonic acid)-2,2′-bipyridine].
Co-reporter:David R. Weinberg, Christopher J. Gagliardi, Jonathan F. Hull, Christine Fecenko Murphy, Caleb A. Kent, Brittany C. Westlake, Amit Paul, Daniel H. Ess, Dewey Granville McCafferty, and Thomas J. Meyer
Chemical Reviews 2012 Volume 112(Issue 7) pp:4016
Publication Date(Web):June 18, 2012
DOI:10.1021/cr200177j
Co-reporter:Aaron K. Vannucci ; Jonathan F. Hull ; Zuofeng Chen ; Robert A. Binstead ; Javier J. Concepcion
Journal of the American Chemical Society 2012 Volume 134(Issue 9) pp:3972-3975
Publication Date(Web):February 6, 2012
DOI:10.1021/ja210718u
Four distinct intermediates, RuIV═O2+, RuIV(OH)3+, RuV═O3+, and RuV(OO)3+, formed by oxidation of the catalyst [Ru(Mebimpy)(4,4′-((HO)2OPCH2)2bpy)(OH2)]2+ [Mebimpy = 2,6-bis(1-methylbenzimidazol-2-yl) and 4,4′-((HO)2OPCH2)2bpy = 4,4′-bismethylenephosphonato-2,2′-bipyridine] on nanoITO (1-PO3H2) have been identified and utilized for electrocatalytic benzyl alcohol oxidation. Significant catalytic rate enhancements are observed for RuV(OO)3+ (∼3000) and RuIV(OH)3+ (∼2000) compared to RuIV═O2+. The appearance of an intermediate for RuIV═O2+ as the oxidant supports an O-atom insertion mechanism, and H/D kinetic isotope effects support net hydride-transfer oxidations for RuIV(OH)3+ and RuV(OO)3+. These results illustrate the importance of multiple reactive intermediates under catalytic water oxidation conditions and possible control of electrocatalytic reactivity on modified electrode surfaces.
Co-reporter:Na Song ; Christopher J. Gagliardi ; Robert A. Binstead ; Ming-Tian Zhang ; Holden Thorp
Journal of the American Chemical Society 2012 Volume 134(Issue 45) pp:18538-18541
Publication Date(Web):November 1, 2012
DOI:10.1021/ja308700t
Benzoquinone/hydroquinone redox interconversion by the reversible Os(dmb)33+/2+ couple over an extended pH range with added acids and bases has revealed the existence of seven discrete pathways. Application of spectrophotometric monitoring with stopped-flow mixing has been used to explore the role of PCET. The results have revealed a role for phosphoric acid and acetate as proton donor and acceptor in the concerted electron–proton transfer reduction of benzoquinone and oxidation of hydroquinone, respectively.
Co-reporter:Akitaka Ito and Thomas J. Meyer
Physical Chemistry Chemical Physics 2012 vol. 14(Issue 40) pp:13731-13745
Publication Date(Web):21 Jun 2012
DOI:10.1039/C2CP41658A
Time-dependent perturbation theory and application of the Golden Rule have been shown to be quantitatively applicable to electron transfer in the inverted region, energy transfer, and excited-state decay based on spectroscopic measurements on dπ6 polypyridyl complexes of Ru(II), Os(II), and Re(I).
Co-reporter:Zuofeng Chen;Xiangqian Hu;Xiangsong Lin;Weitao Yang;Shubin Liu;Javier J. Concepcion
PNAS 2012 Volume 109 (Issue 39 ) pp:
Publication Date(Web):2012-09-25
DOI:10.1073/pnas.1118344109
Water oxidation is a linchpin in solar fuels formation, and catalysis by single-site ruthenium complexes has generated significant
interest in this area. Combining several theoretical tools, we have studied the entire catalytic cycle of water oxidation
for a single-site catalyst starting with [RuII(tpy)(bpm)(OH2)]2+ (i.e., [RuII-OH2]2+; tpy is 2,2′∶6′,2′′-terpyridine and bpm is 2,2′-bypyrimidine) as a representative example of a new class of single-site catalysts. The redox
potentials and pKa calculations for the first two proton-coupled electron transfers (PCETs) from [RuII-OH2]2+ to [RuIV = O]2+ and the following electron-transfer process to [RuV = O]3+ suggest that these processes can proceed readily in acidic or weakly basic conditions. The subsequent water splitting process
involves two water molecules, [RuV = O]3+ to generate [RuIII-OOH]2+, and H3O+ with a low activation barrier (∼10 kcal/mol). After the key O---O bond forming step in the single-site Ru catalysis, another
PECT process oxidizes [RuIII-OOH]2+ to [RuIV-OO]2+ when the pH is lower than 3.7. Two possible forms of [RuIV-OO]2+, open and closed, can exist and interconvert with a low activation barrier (< 7 kcal/mol) due to strong spin-orbital coupling
effects. In Pathway 1 at pH = 1.0, oxygen release is rate-limiting with an activation barrier ∼12 kcal/mol while the electron-transfer
step from [RuIV-OO]2+ to [RuV - OO]3+ becomes rate-determining at pH = 0 (Pathway 2) with Ce(IV) as oxidant. The results of these theoretical studies with atomistic
details have revealed subtle details of reaction mechanisms at several stages during the catalytic cycle. This understanding
is helpful in the design of new catalysts for water oxidation.
Co-reporter:Christopher J. Gagliardi ; Robert A. Binstead ; H. Holden Thorp
Journal of the American Chemical Society 2011 Volume 133(Issue 49) pp:19594-19597
Publication Date(Web):October 27, 2011
DOI:10.1021/ja207379n
Tryptophan is unique among the redox-active amino acids owing to its weakly acidic indolic proton (pKa ≈ 16) compared to the −O–H proton of tyrosine (pKa = 10.1) or the −S–H proton of cysteine (pKa = 8.2). Stopped-flow and electrochemical measurements have been used to explore the roles of proton-coupled electron transfer and concerted electron–proton transfer (EPT) in tryptophan oxidation. The results of these studies have revealed a role for OH– as a proton acceptor base in EPT oxidation of N-acetyl-tryptophan but not for other common bases. The reorganizational barrier for (N-acetyl-tryptophan)+/• self-exchange is also estimated.
Co-reporter:Christopher J. Gagliardi ; Jonah W. Jurss ; H. Holden Thorp
Inorganic Chemistry 2011 Volume 50(Issue 6) pp:2076-2078
Publication Date(Web):February 8, 2011
DOI:10.1021/ic102524f
Dramatic rate enhancements are observed for the oxidation of phenols, including tyrosine, at indium−tin oxide electrodes modified by the addition of the electron-transfer relays [MII(bpy)2(4,4′-(HO)2P(O)CH2)2bpy)]2+ (M = Ru, Os) with clear evidence for the importance of proton-coupled electron transfer and concerted electron−proton transfer.
Co-reporter:Thomas J. Meyer;John M. Papanikolas;Catherine M. Heyer
Catalysis Letters 2011 Volume 141( Issue 1) pp:1-7
Publication Date(Web):2011 January
DOI:10.1007/s10562-010-0495-9
The UNC Energy Frontier Research Center: “Solar Fuels and Next Generation Photovoltaics” is funded by a $17.5 M grant from the US Department of Energy. Its mission is to conduct basic research that will enable a revolution in the collection and conversion of sunlight into storable solar fuels and electricity.
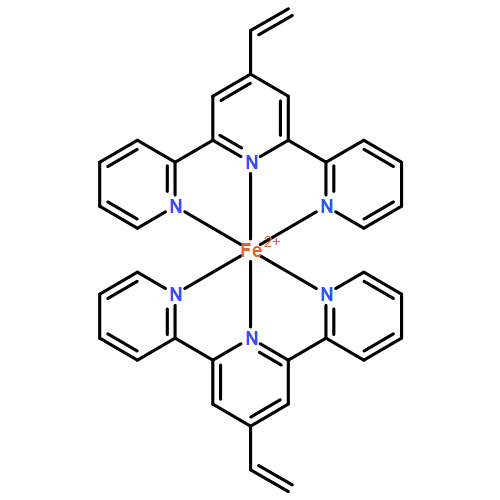
Co-reporter:Troy E. Knight, Anna P. Goldstein, M. Kyle Brennaman, Thomas Cardolaccia, Ashish Pandya, Joseph M. DeSimone, and Thomas J. Meyer
The Journal of Physical Chemistry B 2011 Volume 115(Issue 1) pp:64-70
Publication Date(Web):December 14, 2010
DOI:10.1021/jp107077t
Photophysical properties of the salts [Ru(bpy)3](p-Tos)2, [Ru(dmb)3](PF6)2, [Ru(vbpy)3](PF6)2, and [Ru(phen)3](p-Tos)2 (bpy = 2,2′-bipyridine, dmb = 4,4′-dimethyl-2,2′-bipyridine, vbpy = 4-methyl-4′-vinyl-2,2′-bipyridine, phen = 1,10-phenanthroline, and p-Tos = p-toluene sulfonate) in fluid and film polyethylene glycol dimethacrylate containing nine ethylene glycol spacers (PEG-DMA550) are reported. MLCT absorption energies and bandshapes are similar in fluid and film PEG-DMA550 pointing to similar local dielectric environments, presumably dominated by the polar acrylate groups. Emission energies and excited-to-ground state 0−0 energy gaps (E0), determined by emission spectral fitting, are blue-shifted, and band-widths-at-half height (Δv̅0,1/2) decreased, due to an expected “rigid medium effect” in PEG-DMA550 film. The extent of loss of medium dipole reorientation in the rigid environment, and the increased emission energies in the film, resulted in enhanced emission quantum yields and excited state lifetimes in accordance with the energy gap law. The “rigid medium effect” in PEG-DMA550 is less pronounced than in films of poly(methyl methacrylate) (PMMA) pointing to a more fluid-like local environment presumably arising from the ethylene glycol linker spacers in PEG-DMA550. Comparison of the absorption, emission, emission spectral fitting, and emission lifetime results for [Ru(dmb)3](PF6)2 and [Ru(vbpy)3](PF6)2 shows that the vinyl groups of vbpy copolymerize with PEG-DMA550 covalently incorporating Ru(vbpy)32+ as a cross-linker into the polymer network. The most dramatic effect of the fluid-to-film transition is seen in the emission lifetime data for [Ru(phen)3](p-Tos)2, with an increase of ∼3 in the PEG-DMA550 film. Ru(phen)32+ cations appear to occupy a low symmetry site in the films probably close to the polar acrylate groups in a structurally confined environment.
Co-reporter:Javier J. Concepcion ; Ming-Kang Tsai ; James T. Muckerman
Journal of the American Chemical Society 2010 Volume 132(Issue 5) pp:1545-1557
Publication Date(Web):January 19, 2010
DOI:10.1021/ja904906v
The mechanism of Ce(IV) water oxidation catalyzed by [Ru(tpy)(bpm)(OH2)]2+ (tpy = 2,2′:6′,2′′-terpyridine; bpm = 2,2′-bipyrimidine) and related single-site catalysts has been determined by a combination of mixing and stopped-flow experiments with spectrophotometric monitoring. The mechanism features O---O coupling by water attack on RuV═O3+ and three peroxidic intermediates that have been characterized by a combination of spectroscopy and DFT calculations.
Co-reporter:Daniel H. Ess ; Cynthia K. Schauer
Journal of the American Chemical Society 2010 Volume 132(Issue 46) pp:16318-16320
Publication Date(Web):October 27, 2010
DOI:10.1021/ja106403e
Quantum mechanical analysis reveals that carbonyl reduction of aldehydes and ketones by the imine-based reductant cis-[OsIII(tpy)(Cl)(NH═CHCH3)(NSAr)] (2), which is accessible by reduction of the analogous nitrile, occurs by hydride-proton transfer (HPT) involving both the imine and sulfilimido ligands. In carbonyl reduction, water or alcohol is necessary to significantly lower the barrier for proton shuttling between ligands. The −N(H)SAr group activates the carbonyl group through hydrogen bonding while the −NC(H)CH3 ligand delivers the hydride.
Co-reporter:Jonah W. Jurss ; Javier C. Concepcion ; Michael R. Norris ; Joseph L. Templeton
Inorganic Chemistry 2010 Volume 49(Issue 9) pp:3980-3982
Publication Date(Web):April 8, 2010
DOI:10.1021/ic100469x
Single-electron activation of multielectron catalysis has been shown to be viable in catalytic water oxidation with stepwise proton-coupled electron transfer, leading to high-energy catalytic precursors. For the blue dimer, cis,cis-[(bpy)2(H2O)RuIIIORuIII(H2O)(bpy)2]4+, the first well-defined molecular catalyst for water oxidation, stepwise 4e−/4H+ oxidation occurs to give the reactive precursor [(O)RuVORuV(O)]4+. This key intermediate is kinetically inaccessible at an unmodified metal oxide surface, where the only available redox pathway is electron transfer. We report here a remarkable surface activation of indium−tin oxide (In2O3:Sn) electrodes toward catalytic water oxidation by the blue dimer at electrodes derivatized by surface phosphonate binding of [Ru(4,4′-((HO)2P(O)CH2)2bpy)2(bpy)]2+. Surface binding dramatically improves the rate of surface oxidation of the blue dimer and induces water oxidation catalysis.
Co-reporter:Javier J. Concepcion;Zuofeng Chen;Weitao Yang;Paul G. Hoertz;Xiangqian Hu
PNAS 2010 Volume 107 (Issue 16 ) pp:7225-7229
Publication Date(Web):2010-04-20
DOI:10.1073/pnas.1001132107
As the terminal step in photosystem II, and a potential half-reaction for artificial photosynthesis, water oxidation (2H2O → O2 + 4e- + 4H+) is key, but it imposes a significant mechanistic challenge with requirements for both 4e-/4H+ loss and O—O bond formation. Significant progress in water oxidation catalysis has been achieved recently by use of single-site
Ru metal complex catalysts such as [Ru(Mebimpy)(bpy)(OH2)]2+ [Mebimpy = 2,6-bis(1-methylbenzimidazol-2-yl)pyridine; bpy = 2,2′-bipyridine]. When oxidized from to RuV = O3+, these complexes undergo O—O bond formation by O-atom attack on a H2O molecule, which is often the rate-limiting step. Microscopic details of O—O bond formation have been explored by quantum
mechanical/molecular mechanical (QM/MM) simulations the results of which provide detailed insight into mechanism and a strategy
for enhancing catalytic rates. It utilizes added bases as proton acceptors and concerted atom–proton transfer (APT) with O-atom
transfer to the O atom of a water molecule in concert with proton transfer to the base (B). Base catalyzed APT reactivity
in water oxidation is observed both in solution and on the surfaces of oxide electrodes derivatized by attached phosphonated
metal complex catalysts. These results have important implications for catalytic, electrocatalytic, and photoelectrocatalytic
water oxidation.
Co-reporter:Javier J. Concepcion;Joseph L. Templeton;Jonah W. Jurss
PNAS 2008 Volume 105 (Issue 46 ) pp:17632-17635
Publication Date(Web):2008-11-18
DOI:10.1073/pnas.0807153105
Light-driven water oxidation occurs in oxygenic photosynthesis in photosystem II and provides redox equivalents directed to
photosystem I, in which carbon dioxide is reduced. Water oxidation is also essential in artificial photosynthesis and solar
fuel-forming reactions, such as water splitting into hydrogen and oxygen (2 H2O + 4 hν → O2 + 2 H2) or water reduction of CO2 to methanol (2 H2O + CO2 + 6 hν → CH3OH + 3/2 O2), or hydrocarbons, which could provide clean, renewable energy. The “blue ruthenium dimer,” cis,cis-[(bpy)2(H2O)RuIIIORuIII(OH2)(bpy)2]4+, was the first well characterized molecule to catalyze water oxidation. On the basis of recent insight into the mechanism,
we have devised a strategy for enhancing catalytic rates by using kinetically facile electron-transfer mediators. Rate enhancements
by factors of up to ≈30 have been obtained, and preliminary electrochemical experiments have demonstrated that mediator-assisted
electrocatalytic water oxidation is also attainable.
Co-reporter:Konstantinos D. Demadis, Dana M. Dattelbaum, Edward M. Kober, Javier J. Concepcion, Jared J. Paul, Thomas J. Meyer, Peter S. White
Inorganica Chimica Acta 2007 Volume 360(Issue 3) pp:1143-1153
Publication Date(Web):15 February 2007
DOI:10.1016/j.ica.2006.08.029
Structural changes between [OsIIL3]2+ and [OsIIIL3]3+ (L: 2,2′-bipyridine; 1,10-phenanthroline) and molecular and electronic structures of the OsIII complexes [OsIII(bpy)3]3+ and [OsIII(phen)3]3+ are discussed in this paper. Mid-infrared spectra in the ν(bpy) and ν(phen) ring stretching region for [OsII(bpy)3](PF6)2, [OsIII(bpy)3](PF6)3, [OsII(phen)3](PF6)2, and [OsIII(phen)3](PF6)3 are compared, as are X-ray crystal structures. Absorption spectra in the UV region for [OsIII(bpy)3](PF6)3 and [OsIII(phen)3](PF6)3 are dominated by very intense absorptions (ε = 40 000–50 000 M−1 cm−1) due to bpy and phen intra-ligand π → π∗ transitions. In the visible region, relatively narrow bands with vibronic progressions of ∼1500 cm−1 appear, and have been assigned to bpy or phen-based, spin–orbit coupling enhanced, 1π → 3π∗ electronic transitions. Also present in the visible region are ligand-to-metal charge transfer bands (LMCT) arising from π(bpy) → t2g(OsIII) or π(phen) → t2g(OsIII) transitions. In the near infrared, two broad absorption features appear for oxidized forms [OsIII(bpy)3](PF6)3 and [OsIII(phen)3](PF6)3 arising from dπ–dπ interconfigurational bands characteristic of dπ5OsIII. They are observed at 4580 and 5090 cm−1 for [OsIII(bpy)3](PF6)3 and at 4400 and 4990 cm−1 for [OsIII(phen)3](PF6)3. The bpy and phen infrared vibrational bands shift to higher energy upon oxidation of Os(II) to Os(III). In the cation structure in [OsIII(bpy)3](PF6)3, the OsIII atom resides at a distorted octahedral site, as judged by ∠N–Os–N, which varies from 78.78(22)° to 96.61(22)°. Os–N bond lengths are also in general longer for [OsIII(bpy)3](PF6)3 compared to [OsII(bpy)3](PF6)2 (0.010 Å), and for [OsIII(phen)3](PF6)3 compared to [OsII(phen)3](PF6)2 (0.014 Å). Structural changes in the ligands between oxidation states are discussed as originating from a combination of dπ(OsII) → π∗ (bpy or phen) backbonding and charge redistribution on the ligands as calculated by natural population analysis.[Os(bpy)3]3+/2+ and [Os(phen)3]3+/2+ structural changes (bpy is 2,2′-bipyridine; phen is 1,10-phenanthroline) and electronic structures of the OsIII complexes [OsIII(bpy)3]3+ and [OsIII(phen)3]3+ are discussed. Average Os–N bond distances increase for OsIII compared to OsII because of it loss in backbonding. Structural changes in the ligands between oxidation states are discussed as originating from a combination of dπ(OsII) → π∗ (bpy or phen) backbonding and charge redistribution on the ligands.
Co-reporter:Akitaka Ito and Thomas J. Meyer
Physical Chemistry Chemical Physics 2012 - vol. 14(Issue 40) pp:NaN13745-13745
Publication Date(Web):2012/06/21
DOI:10.1039/C2CP41658A
Time-dependent perturbation theory and application of the Golden Rule have been shown to be quantitatively applicable to electron transfer in the inverted region, energy transfer, and excited-state decay based on spectroscopic measurements on dπ6 polypyridyl complexes of Ru(II), Os(II), and Re(I).
Co-reporter:Leila Alibabaei, Hanlin Luo, Ralph L. House, Paul G. Hoertz, Rene Lopez and Thomas J. Meyer
Journal of Materials Chemistry A 2013 - vol. 1(Issue 13) pp:NaN4145-4145
Publication Date(Web):2013/01/28
DOI:10.1039/C2TA00935H
Solar fuels hold great promise as a permanent, environmentally friendly, long-term renewable energy source, that would be readily available across the globe. In this account, an approach to solar fuels is described based on Dye Sensitized Photoelectrosynthesis Cells (DSPEC) that mimic the configuration used in Dye Sensitized Solar Cells (DSSC), but with the goal of producing oxygen and a high energy solar fuel in the separate compartments of a photoelectrochemical cell rather than a photopotential and photocurrent.
.jpg)











![Phosphonic acid, (4-[2,2':6',2''-terpyridin]-4'-ylphenyl)-](http://img.cochemist.com/ccimg/194900/194800-57-2.png)
![Phosphonic acid, (4-[2,2':6',2''-terpyridin]-4'-ylphenyl)-](http://img.cochemist.com/ccimg/194900/194800-57-2_b.png)


![2,2'-[(2,3,4,5,6-pentafluorophenyl)methylene]bis-1H-Pyrrole](http://img.cochemist.com/ccimg/167500/167482-91-9.png)
![2,2'-[(2,3,4,5,6-pentafluorophenyl)methylene]bis-1H-Pyrrole](http://img.cochemist.com/ccimg/167500/167482-91-9_b.png)


![2-[4-[1-(2-PHOSPHONOETHYL)PYRIDIN-1-IUM-4-YL]PYRIDIN-1-IUM-1-YL]ETHYLPHOSPHONIC ACID](/data/chemimg/537000/143951-41-1.png)
![2-[4-[1-(2-PHOSPHONOETHYL)PYRIDIN-1-IUM-4-YL]PYRIDIN-1-IUM-1-YL]ETHYLPHOSPHONIC ACID](/data/chemimg/537000/143951-41-1_b.png)

















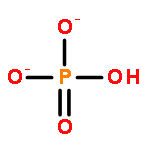
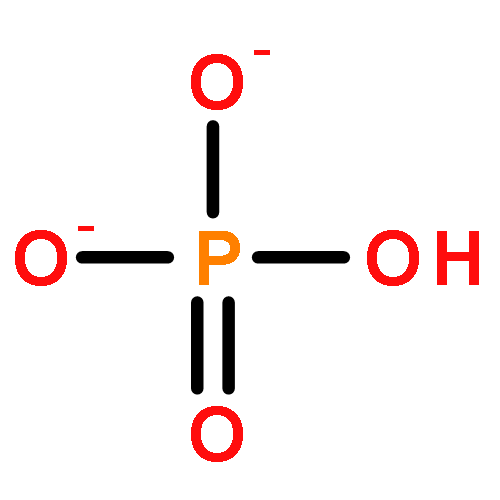




![[2,2'-Bipyridine]-6,6'-dicarboxylic acid](/data/chemimg/52900/4479-74-7.png)
![[2,2'-Bipyridine]-6,6'-dicarboxylic acid](/data/chemimg/52900/4479-74-7_b.png)



