Co-reporter:Gaoxin Zhou;Lushen Li;Jing Xing;Jin Cai
Journal of Sol-Gel Science and Technology 2017 Volume 82( Issue 2) pp:490-499
Publication Date(Web):2017 May
DOI:10.1007/s10971-017-4330-2
Mesoporous silica nanoparticle was expected to provide a versatile drug delivery platform with modification flexibility. To improve its hemocompatibility and realize controlled and/or targeting drug release, modification of bare mesoporous silica nanoparticles is essential. Herein, a novel method of coating mesoporous silica nanoparticles with lipid bilayers was developed. First, a homemade organosiloxane precursor was used for hydrophobic modification of mesoporous silica nanoparticles, then phospholipids (1,2-dihexadecanoyl-sn-glycero-3-phosphocholine and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)]) were coated by a filming-rehydration method based on hydrophobic interaction. The nanoparticle samples were characterized by transmission electron microscope, dynamic light scattering, nitrogen sorption, Fourier transform infrared, and thermal gravimetric analysis. Furthermore, the novel lipid bilayer coated mesoporous silica nanoparticles were compared with bare mesoporous silica nanoparticles in terms of suspension stability, drug release, hemolysis, and nonspecific protein absorption. Our data proved that lipid bilayer coated mesoporous silica nanoparticles had better hemobiocompatibility and controlled drug release properties than that of bare mesoporous silica nanoparticles.Open image in new window
Co-reporter:Xiquan Zhang;Meng Cao;Jing Xing;Fei Liu;Ping Dong
Medicinal Chemistry Research 2017 Volume 26( Issue 12) pp:3395-3406
Publication Date(Web):28 August 2017
DOI:10.1007/s00044-017-2032-5
To develop topoisomerase I targeted drug candidates with sophisticated liposolubility, a series of novel camptothecin derivatives were synthesized through structure-based molecular hybridization and prodrug design approach. The compounds were used as compositions in micellar emulsion preparations, and the antiproliferative efficacy of these preparations were evaluated in two cancer cell lines (A2780s and A549) in vitro. The designed molecules were afterwards optimized for better potency by modifications at the aliphatic chain, the linker and the camptothecin-yl group to reach the optimal structure 7c (TQ-B3203), an SN-38 (camptothecin derivative, 7-ethyl-camptothecin-10-yl) containing compound. 7c showed excellent capacity of inhibiting cell proliferation with IC50 value at nanomolar level, and the potency was further confirmed in other human cancer cell lines (HT-29 and HePG2) superior to the positive reference irinotecan. 7c can be a promising candidate as antitumor drug. Its micellar emulsion preparation has succeeded in the preclinical studies and is in process for investigational new drug(IND) application.
Co-reporter:Gaoxin Zhou, Lushen Li, Jing Xing, Shivakumar Jalde, Yan Li, Jin Cai, Junqing Chen, Peidang Liu, Ning Gu, Min Ji
Colloids and Surfaces B: Biointerfaces 2016 Volume 148() pp:518-525
Publication Date(Web):1 December 2016
DOI:10.1016/j.colsurfb.2016.09.033
•A novel redox-responsive cerasome for tumor-targeted drug delivery.•The CFL comprises a cleavable disulfide bond was synthesized and characterized.•The EE and DLC of DOX in RRC was 74.3% and 2.4%, respectively.•Accelerated DOX release in the presence of 10 mM GSH (from 34.4% to 79.8%, 48 h).•DOX/RRCs showed effective cytotoxicity against SMCC-7721 and MCF-7 cells.Cerasome is a freshly developped bilayer vehicle that resemble traditional liposome but has higher mophorlogical stability. In this study, a novel redox-responsive cerasome (RRC) was developed for tumor-targeting drug delivery. The cerasome-forming lipid (CFL) that comprise a cleavable disulfide bond as connector unit of the triethoxysilyl head and the hydrophobic alkyl double chain was synthesized and subsequently used to prepare cerasome through ethanol injection method. RRC that has liposome-resembling lipid bilayer structure was proved being outstanding at drug loading capacity as well as morphological stability as compared to conventional liposomes. In addition, in vitro drug release tests of DOX/RRCs showed a redox-responsive drug release profile: accelerated DOX releasing compared to reduction-insensitive cerasomes (RICs) in the presence of 10 mM of GSH. Under the same condition, the reduction sensibility of RRC was further proved by increased hydrodynamic diameter and destroying of integrity from DLS and SEM results. RRC showed non-toxic to human embryonic kidney 293 cells, indicating that this material has good biocompatibility. On the other hand, DOX/RRCs showed a resemble IC50 (half inhibitory concentration) value to that of free DOX to human hepatoma SMMC-7721 cells and breast cancer MCF-7 cells. IC50 values at 48 h were found to decrease in the following order: DOX/RIC > DOX/RRC > DOX. Taken together, the RRC developped in this study is of great potential to be utilized as a promising platform for intracellular anticancer drug delivery.
Co-reporter:Peng Wang, Jin Cai, Junqing Chen, Min Ji
European Journal of Medicinal Chemistry 2015 Volume 93() pp:1-8
Publication Date(Web):26 March 2015
DOI:10.1016/j.ejmech.2015.01.056
•New ceritinib analogs by extensive functionalization of the tail piperidine ring were synthesized.•All compounds were evaluated in H2228 cell line.•Compound 9 exhibited robust tumor growth inhibition in vitro and vivo.A series of new ceritinib analogs by extensive functionalization of the tail piperidine ring with various phosphamides and carbamates have been synthesized. All the ceritinib derivatives were evaluated for their cytotoxic activities against H2228 cell line. From the activity profile obtained, three of the tested compounds (compounds 4, 7 and 9) showed significant cytotoxic effects. Among these derivatives compound 9 was found to possess cytotoxicity that is better than standard drug ceritinib (IC50 = 24 nM). Moreover, compound 9 demonstrated robust tumor growth inhibition in vivo model.
Co-reporter:Jin Cai, Ligang Liu, Kwon Ho Hong, Peng Wang, Lushen Li, Meng Cao, Chunlong Sun, Xiaoqing Wu, Xi Zong, Junqing Chen, Min Ji
Bioorganic & Medicinal Chemistry 2015 Volume 23(Issue 4) pp:657-667
Publication Date(Web):15 February 2015
DOI:10.1016/j.bmc.2015.01.003
A series of phenoxybutanoic acid derivatives were synthesized and tested for their antagonistic activity on the contraction of the rat thoracic aortic ring induced by endothelin-1. Preliminary screening results showed that 6e and 6g with benzoheterocycles demonstrated significant antagonistic activities when compared to the reference compound BQ123. The results from additional assays for the binding affinity and selectivity for endothelin receptors showed that 6e was a selective ETA antagonist with a nanomolar IC50. Moreover, 6e was effective in relieving hypoxia-induced pulmonary arterial hypertension and right ventricular weight ratio. Therefore, 6e may have potential for further development as a therapeutic agent for the treatment of cardiovascular diseases.
Co-reporter:Xi Zong, Jin Cai, Junqing Chen, Chunlong Sun, Lushen Li and Min Ji
RSC Advances 2015 vol. 5(Issue 32) pp:24814-24823
Publication Date(Web):13 Feb 2015
DOI:10.1039/C5RA02576A
In the present study, twenty-five pyrazole–quinoxaline derivatives (4a–4y) were designed and synthesized, and their biological activity as potential EGFR or HER-2 kinase inhibitors was evaluated. Among them, compound 4l displayed better antiproliferative activity against A549 and MCF-7 cell lines than Eroltinib. Further kinase inhibitory activity assay results indicated that compound 4l demonstrated the most potent enzyme inhibitory activity. Docking simulations were then performed to position compounds 4l and 4x into the active binding site of EGFR to determine the probable binding model. 3D-QSAR models were built to aid in the effective design of the presently studied and future EGFR inhibitors. These discoveries suggested that the title compounds are potential EGFR/HER-2 dual inhibitors and compound 4l may be a promising lead compound for the development of novel antitumor agents, potentially via inhibiting EGFR/HER-2.
Co-reporter:Jin Cai, Hongtao Wei, Kwon Ho Hong, Xiaoqing Wu, Xi Zong, Meng Cao, Peng Wang, Lushen Li, Chunlong Sun, Bo Chen, Gaoxing Zhou, Junqing Chen, Min Ji
Bioorganic & Medicinal Chemistry 2015 Volume 23(Issue 13) pp:3457-3471
Publication Date(Web):1 July 2015
DOI:10.1016/j.bmc.2015.04.028
In our study, three series of hydroxamate, 2-aminobenzamide, and trifluoromethyl ketone analogues have been designed and synthesized. The synthesized compounds were investigated for their in vitro antiproliferative activities using the MTT-based assay against three human cancer cell lines including A549, NCI-H661, and U937. Most analogues exhibited higher antiproliferative activities against human acute myeloid leukemia cell U937 than the other two human lung cancer cell lines. Furthermore, the compounds were examined against HDAC1, 2, and 8 isoforms. Docking study of compounds 6h, 9b, and 10a suggested that they might bind tightly to the binding pocket of HDAC2 and/or HDAC8. The results suggest that these compounds might have potential as lead compounds for the development of anti-tumor drugs with HDACs inhibitory activities.
Co-reporter:Xi Zong, Jin Cai, Junqing Chen, Peng Wang, Gaoxin Zhou, Bo Chen, Wei Li, Min Ji
Bioorganic & Medicinal Chemistry Letters 2015 Volume 25(Issue 16) pp:3147-3150
Publication Date(Web):15 August 2015
DOI:10.1016/j.bmcl.2015.06.006
Twenty-five novel imidazole N–H substituted Daclatasvir (BMS-790052, DCV) analogues (8a–8y) were designed and synthesized as potential prodrugs. Structure modifications were performed in order to improve potency and pharmacokinetic (PK) properties. All target compounds were evaluated in a hepatitis C virus (HCV) genotype 1b replicon, and the 2-oxoethyl acetate substituted compound 8t showed similar anti-HCV activity (EC50 = 0.08 nM) to that of the lead compound Daclatasvir. Moreover, the utility of prodrug 8t was demonstrated through similar exposure of the parent compound when the prodrugs were dosed in vivo. PK studies showed that prodrug 8t was an ideal candidate for a slower and sustained release form of Daclatasvir.A design for DCV prodrugs.
Co-reporter:Jin Cai;Yong Li;Junqing Chen;Peng Wang
Research on Chemical Intermediates 2015 Volume 41( Issue 1) pp:1-9
Publication Date(Web):2015 January
DOI:10.1007/s11164-013-1162-8
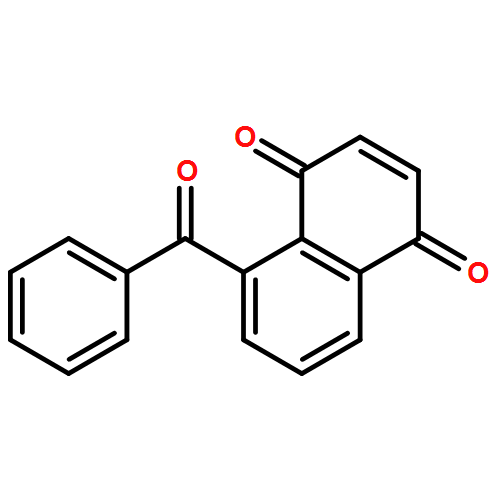
A novel and convenient method for the preparation of 5-benzoyl-1,4-naphthoquinone is reported for the first time. The process starts with condensation of malonic acid, followed by Diers–Alder cyclization, methylation, aromatization, hydrolysis, Friedel–Crafts acylation, and oxidation. The new synthesis procedure with high yields does not require special conditions and column chromatographic purification. The method involves readily available reagents and has been demonstrated to be suitable for large-scale preparation.
Co-reporter:Jin Cai, Hongtao Wei, Kwon Ho Hong, Xiaoqing Wu, Meng Cao, Xi Zong, Lushen Li, Chunlong Sun, Junqing Chen, Min Ji
European Journal of Medicinal Chemistry 2015 96() pp: 1-13
Publication Date(Web):
DOI:10.1016/j.ejmech.2015.04.002
Co-reporter:Jin Cai, Lili Li, Kwon Ho Hong, Xiaoqing Wu, Junqing Chen, Peng Wang, Meng Cao, Xi Zong, Min Ji
Bioorganic & Medicinal Chemistry 2014 Volume 22(Issue 21) pp:5813-5823
Publication Date(Web):1 November 2014
DOI:10.1016/j.bmc.2014.09.029
Two series of 20 novel 4-aminoquinazoline—urea derivatives have been designed and synthesized. The entire target compounds were investigated for their in vitro antiproliferative activity against six human cancer cell lines (K562, U937, A549, NCI-H661, HT29 and LoVo) using the MTT-based assay. Most compounds showed significant antiproliferative activities against four solid tumor cell lines, but no or poor activities against two leukemia cell lines. Furthermore, the target compounds were screened for Aurora A/B kinases inhibitory activity. Among them, 7c, 7d, 8c, and 8d are more potent against Aurora A kinase than ZM447439. Docking study of compounds 7d and ZM447439 revealed that they bound strongly to the ATP-binding sites of Aurora A and B. Thus, they may be promising lead compounds for the development of novel anti-tumor drug potentially via inhibiting Aurora kinases.Two series of 20 novel 4-aminoquinazoline—urea derivatives were designed, synthesized and investigated for their antiproliferative activities and Aurora kinases inhibitory activities.
Co-reporter:Peng Wang, Rui Zhang, Jin Cai, Jun-Qing Chen, Min Ji
Chinese Chemical Letters 2014 Volume 25(Issue 4) pp:549-552
Publication Date(Web):April 2014
DOI:10.1016/j.cclet.2014.01.017
An efficient synthesis of substituted 1,3-dihydroisobenzofurans is developed. In this novel route, o-aroylbenzaldehydes, as key intermediates, can be obtained by lead tetraacetate oxidation of N-aroylhydrazones of salicylaldehydes. The mild and general strategy enables the synthesis of various substituted 1,3-dihydroisobenzofurans in high yields. Moreover, this method can be applied to efficiently synthesize escitalopram.An efficient synthesis of substituted 1,3-dihydroisobenzofurans is developed.
Co-reporter:Meng Cao;Hua-You Hu;Hu-Cheng Zhao;Xi-Quan Zhang;Hong-Mei Gu
Chemical Papers 2014 Volume 68( Issue 10) pp:1408-1414
Publication Date(Web):2014 October
DOI:10.2478/s11696-014-0581-3
In the current study a facile synthetic route for preparing antineoplastic drug GDC-0449 is investigated. Starting with pyridine-1-oxide and 1-iodo-3-nitrobenzene, the intermediate product 2-(2-chloro-5-nitrophenyl) pyridine was prepared by cross-coupling, deoxidation and halogenation. The final compound was then synthesised by reduction of the nitro group followed by amidation. This synthetic route avoids the use of unstable organometallic or organic boride compounds; it employs relatively inexpensive and bench-stable reagents, involves readily controllable reaction conditions, and achieves a relatively high yield.
Co-reporter:Jin Cai, Min Sun, Xiaoqing Wu, Junqing Chen, Peng Wang, Xi Zong, Min Ji
European Journal of Medicinal Chemistry 2013 Volume 63() pp:702-712
Publication Date(Web):May 2013
DOI:10.1016/j.ejmech.2013.03.013
•Novel Dasatinib derivatives have been designed and synthesized.•The cytotoxic activities of the compounds against cancer cell lines were evaluated.•The compounds were screened for EGFR, SRC and ABL kinase inhibitory activity.•The compounds may be promising leads to be developed as an alternative for CML.Three series of novel 4-benzothiazole amino quinazolines Dasatinib derivatives have been designed and synthesized. The entire target compounds were investigated for their in vitro cytotoxic activity by the MTT-based assay against 6 human cancer cell lines. Compared with the parental Dasatinib, most of the new compounds, especially 2, 4, 6-trimethylaniline series (3), demonstrated significant inhibitory activities against six cell lines. Furthermore, the target compounds were screened for Src and Abl kinase inhibitory activity. Among them, 1a, 1f and 3a–3f are more potential dual Src/Abl kinase inhibitors. Thus they may be promising lead compounds to be developed as an alternative for current Dasatinib therapy or for Imatinib-resistant patients, potentially via simultaneously blocking multiple RTK signaling pathways.Three series of novel 4-benzothiazole amino quinazolines Dasatinib derivatives have been designed, synthesized and investigated for their cytotoxicity and kinase inhibitory activities.
Co-reporter:Ming Zheng;Youguang Zheng;Yunsheng Xue;Yi Liu;Lin An;Ling Zhang;Bai Xue;Xuan Wu;Xuedong Gong;Ning Gu;Xi Zhan
Chemical Biology & Drug Design 2013 Volume 81( Issue 3) pp:399-407
Publication Date(Web):
DOI:10.1111/cbdd.12089
Nine novel 4-aminoquinazoline derivatives were designed and synthesized. Biochemical and cellular analyses demonstrated that most of the derivatives exhibited a strong activity to inhibit Aurora A and B kinases and to suppress the proliferation of a panel of human tumor cell lines (U937, K562, A549, LoVo, and HT29). Quantum chemical studies were also carried out to determine the structural features of these compounds engaged in the inhibition of Aurora kinases.
Co-reporter:Peng Wang, Jin Cai, Jiabin Yang, Chunlong Sun, Lushen Li, Huayou Hu, Min Ji
Tetrahedron Letters 2013 Volume 54(Issue 6) pp:533-535
Publication Date(Web):6 February 2013
DOI:10.1016/j.tetlet.2012.11.076
Efficient oxidation of primary and secondary alcohols to the corresponding carbonyl compounds can be carried out in acetonitrile, using chloramine-T in the presence of catalytic zinc(II) salts. Primary alcohols are selectively oxidized to aldehydes without carboxylic acid byproduct.
Co-reporter:Cheng-Liang Feng, Shu-Guang Zhang, Jun-Qing Chen, Jin Cai, Min Ji
Chinese Chemical Letters 2013 Volume 24(Issue 8) pp:767-769
Publication Date(Web):August 2013
DOI:10.1016/j.cclet.2013.04.039
The first total synthesis of isoquinolinone alkaloid marinamide 1 and its methyl ester 2 was described. The key steps involved a regioselective Friedel–Crafts reaction of 1-benzyl-1H-pyrrole to form the intermediate 8.The first total synthesis of marinamide 1 and its methyl ester 2 was described.
Co-reporter:Youguang Zheng, Ming Zheng, Xin Ling, Yi Liu, Yunsheng Xue, Lin An, Ning Gu, Min Jin
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 12) pp:3523-3530
Publication Date(Web):15 June 2013
DOI:10.1016/j.bmcl.2013.04.039
Novel pyrazole–benzimidazole derivatives have been designed and synthesized. The entire target compounds were determined against cancer cell lines U937, K562, A549, LoVo and HT29 and were screened for Aurora A/B kinase inhibitory activity in vitro. The compounds 7a, 7b, 7i, 7k and 7l demonstrated significant cancer cell lines and Aurora A/B kinase inhibitory activities. Molecular modeling studies suggested the derivatives have bound in the active site of Aurora A kinase through the formation of four hydrogen bonds. Quantum chemical studies were carried out on these compounds to understand the structural features essential for activity. The cellular activity of 7k was also tested by immunofluorescence.
Co-reporter:Youguang Zheng, Ming Zheng, Xin Ling, Yi Liu, Yunsheng Xue, Lin An, Ning Gu, Min Ji
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 15) pp:4471
Publication Date(Web):1 August 2013
DOI:10.1016/j.bmcl.2013.05.084
Co-reporter:Ning Gu;Jiabin Yang;Peng Wang;Lushen Li;Yang Chen
Research on Chemical Intermediates 2013 Volume 39( Issue 7) pp:3105-3110
Publication Date(Web):2013 September
DOI:10.1007/s11164-012-0822-4
The Wittig–Horner reaction is a classic method to get alkenes by reaction phosphonates with carbonyl compounds. In this study, it was used for the synthesis of the anticancer drug neratinib. In this method, ethyl diethoxyphosphinylacetate and dimethylaminoacetaldehyde diethylacetal, replacing (E)-4-(dimethylamino)but-2-enoyl acid hydrochloride and oxalyl chloride, were used to synthesize the 6-position side chain of neratinib.
Co-reporter:Jin Cai, Shaoning Zhang, Ming Zheng, Xiaoqing Wu, Junqing Chen, Min Ji
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 2) pp:806-810
Publication Date(Web):15 January 2012
DOI:10.1016/j.bmcl.2011.12.070
Two series of novel Dasatinib derivatives have been designed and synthesized, with their in vitro cytostatic effect screened on human chronic myeloid leukemia cell line K562 and human myeloid leukemia cell line U937. Some target compounds demonstrated significant inhibitory activities against both cell lines. Compared to the contrast drug Dasatinib, 1b, 1c, 1d, 1e and 1f were found to demonstrate more potent antitumor activities. The structures of all the newly synthesized compounds were determined by 1H NMR and 13C NMR.Two series of novel Dasatinib derivatives have been designed and synthesized, with their in vitro cytostatic effect screened on human chronic myeloid leukemia cell line K562 and human myeloid leukemia cell line U937. Compared to the contrast drug Dasatinib, several compounds were found to demonstrate more potent antitumor activities.
Co-reporter:Xiaoqing Wu;Mingdong Li;Wenhua Tang;Youguang Zheng;Jiqin Lian;Liang Xu
Chemical Biology & Drug Design 2011 Volume 78( Issue 6) pp:932-940
Publication Date(Web):
DOI:10.1111/j.1747-0285.2011.01234.x
Here, we describe the design and synthesis of two series of 4-pyrrylamino quinazolines as new analogs of the epidermal growth factor receptor inhibitor gefitinib. In vitro antitumor activity of these novel compounds against pancreatic (Miapaca2) and prostate (DU145) cancer cell lines was evaluated. Compared with the parental gefitinib, all 18 derivatives show a greatly increased cytotoxicity to cancer cells. In vitro kinase inhibitory activity on epidermal growth factor receptor was also investigated. Among them, compounds GI-6, GII-4, GII-6, GII-8, and GII-9 are more potential receptor tyrosine kinase (RTK) inhibitors. Based on these results, we propose simple structure–activity relationship to provide information for designing and developing more potent antitumor agents.
Co-reporter:Junqing Chen;Min Sun;Jin Cai;Meng Cao;Wen Zhou
Journal of Chemical Crystallography 2011 Volume 41( Issue 4) pp:519-522
Publication Date(Web):2011 April
DOI:10.1007/s10870-010-9912-6
(4α,8β,13β,16β)-13-methyl-16,18-diol-17-norkaurane was synthesized by esterification and reduction of isosteviol, respectively. The structure of the title compound was established by spectral analysis and X-ray diffraction studies. The compound crystallizes in the orthorhombic space group P212121 with unit cell parameters: a = 7.3705 (14) Å, b = 13.508 (3) Å, c = 20.139 (4) Å, V = 2005.1 (7) Å3, Z = 4. The conformation of rings A and B is chair, whereas the conformation of ring C is unsymmetrical distorted chair. The stereochemistry of the A/B and B/C ring junctions is trans, while the five-membered ring D adopts an envelope conformation.
Co-reporter:Min Sun, Xiaoqing Wu, Junqing Chen, Jin Cai, Meng Cao, Min Ji
European Journal of Medicinal Chemistry 2010 Volume 45(Issue 6) pp:2299-2306
Publication Date(Web):June 2010
DOI:10.1016/j.ejmech.2010.02.005
Two series of novel diaryl ureas have been designed and synthesized, with their in vitro antitumor effect screened on human non-small cell lung cancer (NSCLC) cell line A549 and human breast cancer cell line MDA-MB-231. Some target compounds demonstrated significant inhibitory activities against both cell lines. Compared to contrast drug Sorafenib, 1b, 1d, 1f, 1i were found to demonstrate more potent antitumor activities. The structures of all the newly synthesized compounds were determined by 1H, 13C NMR, MS, IR and elementary analysis.Two series of diaryl ureas derivatives were designed, synthesized, and evaluated for their antitumor activities.
Co-reporter:Xiaoqing Wu, Mingdong Li, Yang Qu, Wenhua Tang, Youguang Zheng, Jiqin Lian, Min Ji, Liang Xu
Bioorganic & Medicinal Chemistry 2010 Volume 18(Issue 11) pp:3812-3822
Publication Date(Web):1 June 2010
DOI:10.1016/j.bmc.2010.04.046
There is an urgent need to design and develop new and more potent EGFR inhibitors with improved anti-tumor activity. Here we describe the design and synthesis of two series of 4-benzothienyl amino quinazolines as new analogues of the EGFR inhibitor Gefitinib. The anti-tumor activity of these novel Gefitinib analogues in 6 human cancer cell lines was examined. Compared with the parental Gefitinib, most of the new compounds show a markedly increased cytotoxicity to cancer cells. Furthermore, several of the series B compounds that side chains at position 7 contain either a methyl or ethyl group are potent pan-RTK inhibitors. Two representative compounds in this class, 15 and 17, have an enhanced capability to inhibit cancer cell growth and induce apoptosis in vitro and inhibit tumor formation in vivo in human cancer cells with high HER-2, as compared with the parental Gefitinib. Thus they may be promising lead compounds to be developed as an alternative for current Gefitinib therapy or for Gefitinb-resistant patients, potentially via simultaneously blocking multiple RTK signaling pathways.Two series of 4-benzothienyl amino quinazolines were designed and synthesized as new analogues of the EGFR inhibitor Gefitinib, with improved anti-tumor and pro-apoptotic activity.
Co-reporter:Min Sun;Junqing Chen;Jin Cai;Meng Cao;Shuangqing Yin
Chemical Biology & Drug Design 2010 Volume 75( Issue 5) pp:494-505
Publication Date(Web):
DOI:10.1111/j.1747-0285.2010.00958.x
Considering the fact that majority of support vector regression models have not been fully optimized in the realm of quantitative structure-activity relationship, an idea of simultaneous optimization has been proposed and evaluated on a set of novel kinase insert domain receptor/vascular endothelial growth factor receptor-2 inhibitors including naphthalene and indazole-based compounds in this study. After the powerful feature searching process using genetic algorithm, the final support vector regression model was constructed on an optimal set of six descriptors, based on which simultaneous optimization was carried out. Specifically, the global optimum is grid searched in the joint parametric space defined by cost (C), γ and ε, where performance of support vector regression using each combination of (C, γ, ε) is evaluated and recorded, resulting in bulky information. Based on the data decomposition strategies provided in the main paper, the best performance was achieved for C = 1.2, γ = 0.15 and ε = 0.065. As a comparison, a linear model based on genetic algorithm-multiple linear regression has also been developed and compared. Performances of these models are rigorously validated using both leave-one-out cross-validation and also external validation. The significant higher R2 (0.908, 0.837) and lower root-mean-square error (0.237, 0.311) for 45 training and 16 test samples compared to that of genetic algorithm-multiple linear regression (0.764, 0.700 and 0.402, 0.421) confirm the superior performance of genetic algorithm-support vector regression. Robustness and predictive ability of this model is further prudently evaluated. The resulting models introduced not only the idea of simultaneous optimization in support vector regression, but also an efficient strategy for estimating the vascular endothelial growth factor receptor-2 inhibitory activity of novel naphthalene and indazole-based compounds. Moreover, some insights into the structural features related to the biological activity of these compounds have also been provided, which might be of great help for further designing novel vascular endothelial growth factor receptor-2/kinase insert domain receptor inhibitors with potent activity.
Co-reporter:Youguang Zheng;Xiaoqing Wu;Bai Xue;Mingdong Li
Chemical Biology & Drug Design 2010 Volume 76( Issue 3) pp:285-290
Publication Date(Web):
DOI:10.1111/j.1747-0285.2010.01008.x
Several novel quinazolino [3, 4-a] thieno [3, 2-d] pyrimidin-8-one derivatives were synthesized. All of the compounds were determined against MiaPaCa2 and DU145 cells in vitro, and the crystal structures of analog 8 and 20 in the active site of the EGFR complexes were presented. The entire compounds had been identified by 1HNMR, 13CNMR, IR, MS and EA.
Co-reporter:Min Sun;Junqing Chen;Hongtao Wei;Shuangqing Yin;Yan Yang
Chemical Biology & Drug Design 2009 Volume 73( Issue 6) pp:644-654
Publication Date(Web):
DOI:10.1111/j.1747-0285.2009.00814.x
Quantitative structure–activity relationship analysis has been carried out for 74 diaryl ureas including aminobenzoisoxazole ureas, aminoindazole ureas, aminopyrazolopyridine ureas against vascular endothelial growth factor receptor-2 kinase using both linear and non-linear models. Considering simplicity and predictivity, multivariate linear regression was first employed in combination with various variable selection methods, including forward selection, genetic algorithm and enhanced replacement method based on descriptors generated by e-dragon software. Another model using support vector regression has also been constructed and compared. Performances of these models are rigorously validated by leave-one-out cross-validation, fivefold cross-validation and external validation. The enhanced replacement method model significantly outperforms the others with R 2 = 0.813 and = 0.809. Robustness and predictive ability of this model is prudently evaluated. Moreover, to find out the most significant features associated with the difference between highly active compounds and moderate ones, two classification models using linear discriminant analysis and support vector machine were further developed. The performance of support vector machine significantly outperforms linear discriminant analysis, with leave-one-out cross-validation and external validation prediction accuracy reaching 0.838 and 0.857, respectively. The resulting models could act as an efficient strategy for estimating the vascular endothelial growth factor receptor-2 inhibiting activity of novel diaryl ureas and provide some insights into the structural features related to the biological activity of these compounds.
Co-reporter:Jin Cai;Wen Zhou;Junqing Chen;Min Sun
Journal of Chemical Crystallography 2009 Volume 39( Issue 2) pp:108-111
Publication Date(Web):2009 February
DOI:10.1007/s10870-008-9435-6
Isosteviol derivative: benzoyloxymethyl (4α,8β,13β)-13-methyl-16-oxo-17-norkauran-18-carbonate ester was synthesized by esterification of isosteviol with chloromethyl benzoate and its crystal structure was determined by X-ray diffraction method. The compound crystallizes in the triclinic space group P1 with unit cell parameters: a = 8.784(3) Å, b = 9.079(3) Å, c = 15.950(6) Å, α = 79.343(6)°, β = 79.061(5)°, γ = 89.849(5)°, Z= 2. The conformation of rings A and B is chair, whereas the conformation of ring C is unsymmetrical twist chair. The carbonyl group at the C20 is coplanar with the benzene ring. The fragment of the ester group occupying the pseudoaxial site of C1 position adopts a zigzag conformation.
Co-reporter:Fang Yang, Aiyuan Gu, Zhongping Chen, Ning Gu, Min Ji
Materials Letters 2008 Volume 62(Issue 1) pp:121-124
Publication Date(Web):15 January 2008
DOI:10.1016/j.matlet.2007.04.111
In order to improve the sensitivity of ultrasound imaging, the contrast agents, a powerful non-invasive and real-time medical imaging technique, are used. However, air or N2 or perfluorocarbon only encapsulated microbubbles which are currently used have lower efficiency and short imaging time. So the novel contrast agents with a higher efficiency are required. To achieve this objective, the strategy that we have explored involves the use of superparamagnetic iron oxide (SPIO) Fe3O4 nanoparticles multilayer emulsion microbubbles. This multilayer structure consists of three layers. The core is poly-d, l-lactide (PLA) encapsulated N2 nanobubble with the SPIO nanoparticles forming oil-in-water (W/O) layer. The outermost is water-in-oil-in-water ((W/O)/W) emulsion layer with PVA solution. Herein we describe the synthesis and characterization of ultrasound imaging microstructure with an overall diameter of around 2μm–8μm. On the one hand, the stable gas encapsulated microstructure can provide a high scattering intensity resulting in high echogenicity, On the other hand, SPIO nanoparticles have shown the potential of high-resolution sonography. So the multiple emulsion microbubbles with SPIO can have double action to enhance the ultrasound imaging. Besides, because SPIO can also serve as magnetic resonance imaging (MRI) contrast agents, such microstructure may be useful for multimodality imaging studies in ultrasound imaging and MRI.
Co-reporter:Jin Cai;Hongtao Wei;Min Sun
Heteroatom Chemistry 2007 Volume 18(Issue 3) pp:236-238
Publication Date(Web):9 APR 2007
DOI:10.1002/hc.20290
Thieno[2,3-b]pyrroles can be synthesized through three steps: Gewald synthesis, alkylation, and Thorpe–Ziegler cyclization. Diethyl 3,6-bis((ethoxycarbonyl)methyl)-4-amino-6H-thieno[2,3-b]pyrrole-2,5-dicarboxylate (13) has been obtained by one-pot method in DMF in good yield and high quality. © 2007 Wiley Periodicals, Inc. Heteroatom Chem 18:236–238, 2007; Published online in Wiley InterScience (www.interscience.wiley.com). DOI 10.1002/hc.20290
Co-reporter:Hai-Feng Liu;Jin Cai
Archiv der Pharmazie 2006 Volume 339(Issue 12) pp:
Publication Date(Web):6 DEC 2006
DOI:10.1002/ardp.200600063
Buxus alkaloids display a wide range of interesting pharmacological activities. Here, we present a short and efficient approach towards one of the alkaloids, buxozine-C. The first synthesis has been achieved with 91% yield starting from cyclovirobuxine-D by forming a tetrahydro-oxazine ring with formaldehyde at room temperature.

![(2S,2'S)-di-tert-butyl 2,2'-(4,4'-([1,1'-biphenyl]-4,4'-diyl)bis(1H-iMidazole-4,2-diyl))bis(pyrrolidine-1-carboxylate)](http://img.cochemist.com/ccimg/1007900/1007882-23-6.png)
![(2S,2'S)-di-tert-butyl 2,2'-(4,4'-([1,1'-biphenyl]-4,4'-diyl)bis(1H-iMidazole-4,2-diyl))bis(pyrrolidine-1-carboxylate)](http://img.cochemist.com/ccimg/1007900/1007882-23-6_b.png)








![2-Butenoic acid, 3-[(4-chlorophenyl)amino]-, methyl ester, (2Z)-](http://img.cochemist.com/ccimg/920400/920312-68-1.png)
![2-Butenoic acid, 3-[(4-chlorophenyl)amino]-, methyl ester, (2Z)-](http://img.cochemist.com/ccimg/920400/920312-68-1_b.png)
![2-Butenoic acid, 3-[(2-methylphenyl)amino]-, methyl ester, (2Z)-](http://img.cochemist.com/ccimg/920400/920312-56-7.png)
![2-Butenoic acid, 3-[(2-methylphenyl)amino]-, methyl ester, (2Z)-](http://img.cochemist.com/ccimg/920400/920312-56-7_b.png)
![2-Butenoic acid, 3-[(4-methylphenyl)amino]-, methyl ester, (2Z)-](http://img.cochemist.com/ccimg/920400/920312-52-3.png)
![2-Butenoic acid, 3-[(4-methylphenyl)amino]-, methyl ester, (2Z)-](http://img.cochemist.com/ccimg/920400/920312-52-3_b.png)
![2-Butenoic acid, 3-[(1-methylethyl)amino]-, methyl ester, (2Z)-](http://img.cochemist.com/ccimg/920400/920312-29-4.png)
![2-Butenoic acid, 3-[(1-methylethyl)amino]-, methyl ester, (2Z)-](http://img.cochemist.com/ccimg/920400/920312-29-4_b.png)
![2-Butenoic acid, 3-[(2-methylphenyl)amino]-, ethyl ester, (2Z)-](http://img.cochemist.com/ccimg/885500/885469-90-9.png)
![2-Butenoic acid, 3-[(2-methylphenyl)amino]-, ethyl ester, (2Z)-](http://img.cochemist.com/ccimg/885500/885469-90-9_b.png)
![3-PENTEN-2-ONE, 4-[(2-METHYLPHENYL)AMINO]-, (3Z)-](http://img.cochemist.com/ccimg/885500/885469-85-2.png)
![3-PENTEN-2-ONE, 4-[(2-METHYLPHENYL)AMINO]-, (3Z)-](http://img.cochemist.com/ccimg/885500/885469-85-2_b.png)







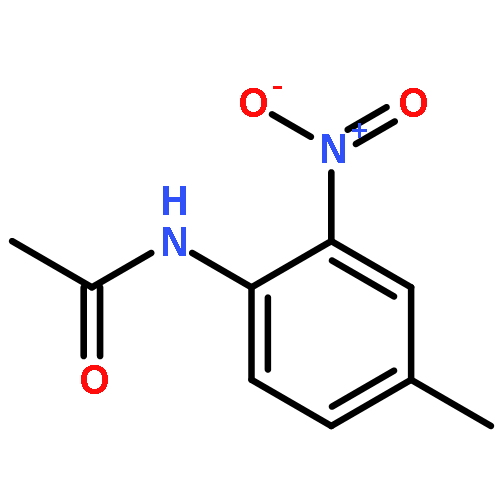
![2-Butenoic acid, 3-[(2-phenylethyl)amino]-, methyl ester, (2Z)-](http://img.cochemist.com/ccimg/848000/847974-57-6.png)
![2-Butenoic acid, 3-[(2-phenylethyl)amino]-, methyl ester, (2Z)-](http://img.cochemist.com/ccimg/848000/847974-57-6_b.png)


![Benzenecarboximidamide, N-[(chloroacetyl)oxy]-4-(trifluoromethyl)-](http://img.cochemist.com/ccimg/844500/844498-78-8.png)
![Benzenecarboximidamide, N-[(chloroacetyl)oxy]-4-(trifluoromethyl)-](http://img.cochemist.com/ccimg/844500/844498-78-8_b.png)
![Benzenecarboximidamide, 2-chloro-N-[(chloroacetyl)oxy]-](http://img.cochemist.com/ccimg/844500/844498-74-4.png)
![Benzenecarboximidamide, 2-chloro-N-[(chloroacetyl)oxy]-](http://img.cochemist.com/ccimg/844500/844498-74-4_b.png)
![ETHANONE, 1-[4-[(4-CHLOROPHENYL)METHOXY]-2-HYDROXYPHENYL]-](http://img.cochemist.com/ccimg/821800/821780-53-4.png)
![ETHANONE, 1-[4-[(4-CHLOROPHENYL)METHOXY]-2-HYDROXYPHENYL]-](http://img.cochemist.com/ccimg/821800/821780-53-4_b.png)
![Benzenecarboximidamide, N-[(chloroacetyl)oxy]-4-fluoro-](http://img.cochemist.com/ccimg/794600/794554-75-9.png)
![Benzenecarboximidamide, N-[(chloroacetyl)oxy]-4-fluoro-](http://img.cochemist.com/ccimg/794600/794554-75-9_b.png)


![Benzenecarboximidamide, N-[(chloroacetyl)oxy]-4-methoxy-](http://img.cochemist.com/ccimg/727400/727363-84-0.png)
![Benzenecarboximidamide, N-[(chloroacetyl)oxy]-4-methoxy-](http://img.cochemist.com/ccimg/727400/727363-84-0_b.png)







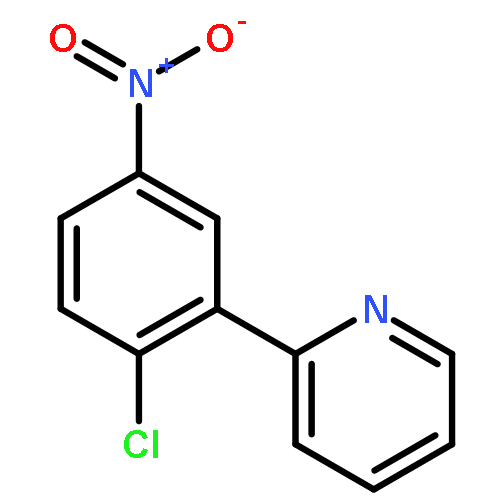
![Ethanone, 1-[2-hydroxy-4-[(4-methoxyphenyl)methoxy]phenyl]-](http://img.cochemist.com/ccimg/503500/503469-24-7.png)
![Ethanone, 1-[2-hydroxy-4-[(4-methoxyphenyl)methoxy]phenyl]-](http://img.cochemist.com/ccimg/503500/503469-24-7_b.png)






![4H-1-Benzopyran-4-one, 3-[(4-fluorophenyl)methylene]-2,3-dihydro-](http://img.cochemist.com/ccimg/193500/193406-85-8.png)
![4H-1-Benzopyran-4-one, 3-[(4-fluorophenyl)methylene]-2,3-dihydro-](http://img.cochemist.com/ccimg/193500/193406-85-8_b.png)


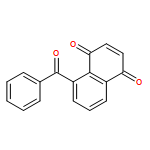


![2-Butenoic acid, 3-[(4-methylphenyl)amino]-, ethyl ester, (2Z)-](http://img.cochemist.com/ccimg/158300/158261-84-8.png)
![2-Butenoic acid, 3-[(4-methylphenyl)amino]-, ethyl ester, (2Z)-](http://img.cochemist.com/ccimg/158300/158261-84-8_b.png)
![Benzoic acid, 2-hydroxy-4-[(4-methoxyphenyl)methoxy]-, methyl ester](http://img.cochemist.com/ccimg/149000/148965-89-3.png)
![Benzoic acid, 2-hydroxy-4-[(4-methoxyphenyl)methoxy]-, methyl ester](http://img.cochemist.com/ccimg/149000/148965-89-3_b.png)





![2-[2-(2-nitrovinyl)phenyl]ethyl benzoate](http://img.cochemist.com/ccimg/139200/139122-16-0.png)
![2-[2-(2-nitrovinyl)phenyl]ethyl benzoate](http://img.cochemist.com/ccimg/139200/139122-16-0_b.png)
![Benzaldehyde, 2-[2-(benzoyloxy)ethyl]-](http://img.cochemist.com/ccimg/139200/139122-15-9.png)
![Benzaldehyde, 2-[2-(benzoyloxy)ethyl]-](http://img.cochemist.com/ccimg/139200/139122-15-9_b.png)
![2-Butenoic acid, 3-[(4-nitrophenyl)amino]-, ethyl ester, (Z)-](http://img.cochemist.com/ccimg/131300/131230-58-5.png)
![2-Butenoic acid, 3-[(4-nitrophenyl)amino]-, ethyl ester, (Z)-](http://img.cochemist.com/ccimg/131300/131230-58-5_b.png)




![Benzenecarboximidamide, N-[(chloroacetyl)oxy]-4-nitro-](http://img.cochemist.com/ccimg/111500/111457-49-9.png)
![Benzenecarboximidamide, N-[(chloroacetyl)oxy]-4-nitro-](http://img.cochemist.com/ccimg/111500/111457-49-9_b.png)








![Benzenecarboximidamide, N-[(chloroacetyl)oxy]-4-methyl-](http://img.cochemist.com/ccimg/93400/93335-40-1.png)
![Benzenecarboximidamide, N-[(chloroacetyl)oxy]-4-methyl-](http://img.cochemist.com/ccimg/93400/93335-40-1_b.png)
![Benzenecarboximidamide, 4-chloro-N-[(chloroacetyl)oxy]-](http://img.cochemist.com/ccimg/93100/93048-83-0.png)
![Benzenecarboximidamide, 4-chloro-N-[(chloroacetyl)oxy]-](http://img.cochemist.com/ccimg/93100/93048-83-0_b.png)
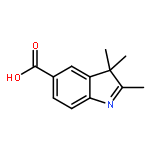
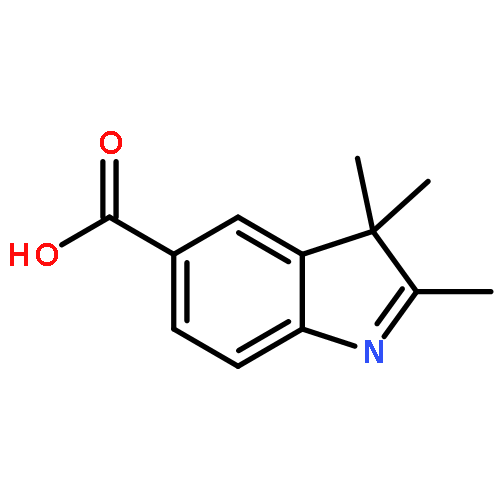

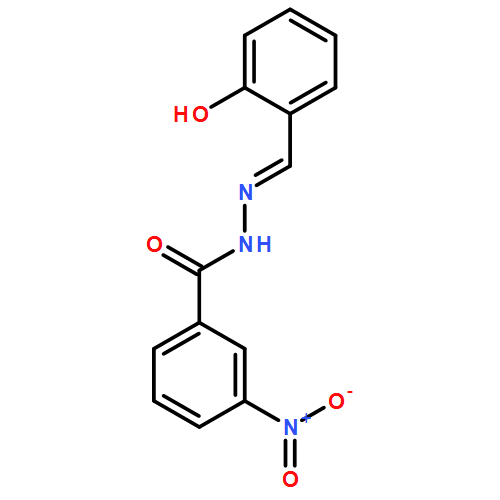
![1(2H)-NAPHTHALENONE, 2-[(2,4-DICHLOROPHENYL)METHYLENE]-3,4-DIHYDRO-](http://img.cochemist.com/ccimg/78400/78364-64-4.png)
![1(2H)-NAPHTHALENONE, 2-[(2,4-DICHLOROPHENYL)METHYLENE]-3,4-DIHYDRO-](http://img.cochemist.com/ccimg/78400/78364-64-4_b.png)








![2-Butenoic acid, 3-[(4-chlorophenyl)amino]-, ethyl ester, (2Z)-](http://img.cochemist.com/ccimg/66600/66558-25-6.png)
![2-Butenoic acid, 3-[(4-chlorophenyl)amino]-, ethyl ester, (2Z)-](http://img.cochemist.com/ccimg/66600/66558-25-6_b.png)
![2-Butenoic acid, 3-[(4-methoxyphenyl)amino]-, ethyl ester, (2Z)-](http://img.cochemist.com/ccimg/66600/66558-24-5.png)
![2-Butenoic acid, 3-[(4-methoxyphenyl)amino]-, ethyl ester, (2Z)-](http://img.cochemist.com/ccimg/66600/66558-24-5_b.png)


![1(2H)-Naphthalenone, 2-[(4-fluorophenyl)methylene]-3,4-dihydro-](http://img.cochemist.com/ccimg/66100/66045-89-4.png)
![1(2H)-Naphthalenone, 2-[(4-fluorophenyl)methylene]-3,4-dihydro-](http://img.cochemist.com/ccimg/66100/66045-89-4_b.png)
![1(2H)-Naphthalenone, 2-[(2-chlorophenyl)methylene]-3,4-dihydro-](http://img.cochemist.com/ccimg/66100/66045-83-8.png)
![1(2H)-Naphthalenone, 2-[(2-chlorophenyl)methylene]-3,4-dihydro-](http://img.cochemist.com/ccimg/66100/66045-83-8_b.png)
![5-(Bromomethyl)benzo[c][1,2,5]thiadiazole](http://img.cochemist.com/ccimg/65900/65858-50-6.png)
![5-(Bromomethyl)benzo[c][1,2,5]thiadiazole](http://img.cochemist.com/ccimg/65900/65858-50-6_b.png)




![2-Thiophenecarboximidamide, N-[(chloroacetyl)oxy]-](http://img.cochemist.com/ccimg/63500/63417-80-1.png)
![2-Thiophenecarboximidamide, N-[(chloroacetyl)oxy]-](http://img.cochemist.com/ccimg/63500/63417-80-1_b.png)
![4H-1-Benzopyran-4-one, 3-[(4-chlorophenyl)methylene]-2,3-dihydro-](http://img.cochemist.com/ccimg/61700/61661-20-9.png)
![4H-1-Benzopyran-4-one, 3-[(4-chlorophenyl)methylene]-2,3-dihydro-](http://img.cochemist.com/ccimg/61700/61661-20-9_b.png)
![1(2H)-Naphthalenone, 2-[(3-chlorophenyl)methylene]-3,4-dihydro-](http://img.cochemist.com/ccimg/61700/61661-18-5.png)
![1(2H)-Naphthalenone, 2-[(3-chlorophenyl)methylene]-3,4-dihydro-](http://img.cochemist.com/ccimg/61700/61661-18-5_b.png)
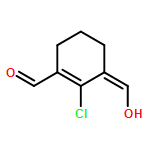
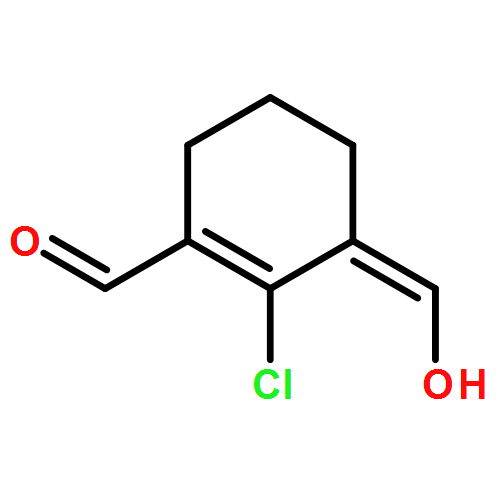




![2-Buten-1-one, 3-[(4-methoxyphenyl)amino]-1-phenyl-, (Z)-](http://img.cochemist.com/ccimg/56600/56570-41-3.png)
![2-Buten-1-one, 3-[(4-methoxyphenyl)amino]-1-phenyl-, (Z)-](http://img.cochemist.com/ccimg/56600/56570-41-3_b.png)
![2-BUTEN-1-ONE, 3-[(4-METHYLPHENYL)AMINO]-1-PHENYL-, (2Z)-](http://img.cochemist.com/ccimg/56600/56570-40-2.png)
![2-BUTEN-1-ONE, 3-[(4-METHYLPHENYL)AMINO]-1-PHENYL-, (2Z)-](http://img.cochemist.com/ccimg/56600/56570-40-2_b.png)
![2-Buten-1-one, 1-phenyl-3-[(phenylmethyl)amino]-, (2Z)-](http://img.cochemist.com/ccimg/56600/56570-39-9.png)
![2-Buten-1-one, 1-phenyl-3-[(phenylmethyl)amino]-, (2Z)-](http://img.cochemist.com/ccimg/56600/56570-39-9_b.png)
![3-Penten-2-one, 4-[(4-nitrophenyl)amino]-, (3Z)-](http://img.cochemist.com/ccimg/56600/56570-37-7.png)
![3-Penten-2-one, 4-[(4-nitrophenyl)amino]-, (3Z)-](http://img.cochemist.com/ccimg/56600/56570-37-7_b.png)
![3-Penten-2-one, 4-[(4-chlorophenyl)amino]-, (Z)-](http://img.cochemist.com/ccimg/56600/56570-36-6.png)
![3-Penten-2-one, 4-[(4-chlorophenyl)amino]-, (Z)-](http://img.cochemist.com/ccimg/56600/56570-36-6_b.png)
![3-Penten-2-one, 4-[(4-methoxyphenyl)amino]-, (Z)-](http://img.cochemist.com/ccimg/56600/56570-35-5.png)
![3-Penten-2-one, 4-[(4-methoxyphenyl)amino]-, (Z)-](http://img.cochemist.com/ccimg/56600/56570-35-5_b.png)
![3-Penten-2-one, 4-[(4-methylphenyl)amino]-, (Z)-](http://img.cochemist.com/ccimg/56600/56570-34-4.png)
![3-Penten-2-one, 4-[(4-methylphenyl)amino]-, (Z)-](http://img.cochemist.com/ccimg/56600/56570-34-4_b.png)
![2-[(2-METHYLPHENYL)METHYLIDENE]-3,4-DIHYDRONAPHTHALEN-1-ONE](http://img.cochemist.com/ccimg/54800/54752-32-8.png)
![2-[(2-METHYLPHENYL)METHYLIDENE]-3,4-DIHYDRONAPHTHALEN-1-ONE](http://img.cochemist.com/ccimg/54800/54752-32-8_b.png)
![2-[(3-METHYLPHENYL)METHYLIDENE]-3,4-DIHYDRONAPHTHALEN-1-ONE](http://img.cochemist.com/ccimg/54800/54752-31-7.png)
![2-[(3-METHYLPHENYL)METHYLIDENE]-3,4-DIHYDRONAPHTHALEN-1-ONE](http://img.cochemist.com/ccimg/54800/54752-31-7_b.png)
![2-[(4-METHYLPHENYL)METHYLIDENE]-3,4-DIHYDRONAPHTHALEN-1-ONE](http://img.cochemist.com/ccimg/54800/54752-30-6.png)
![2-[(4-METHYLPHENYL)METHYLIDENE]-3,4-DIHYDRONAPHTHALEN-1-ONE](http://img.cochemist.com/ccimg/54800/54752-30-6_b.png)
![Benzoic acid, 4-nitro-, [(2-hydroxyphenyl)methylene]hydrazide](http://img.cochemist.com/ccimg/50400/50366-20-6.png)
![Benzoic acid, 4-nitro-, [(2-hydroxyphenyl)methylene]hydrazide](http://img.cochemist.com/ccimg/50400/50366-20-6_b.png)
![1(2H)-Naphthalenone, 3,4-dihydro-2-[(4-methoxyphenyl)methylene]-](http://img.cochemist.com/ccimg/49700/49629-37-0.png)
![1(2H)-Naphthalenone, 3,4-dihydro-2-[(4-methoxyphenyl)methylene]-](http://img.cochemist.com/ccimg/49700/49629-37-0_b.png)
![(2e)-2-[(4-chlorophenyl)methylidene]-3,4-dihydronaphthalen-1-one](http://img.cochemist.com/ccimg/49600/49545-70-2.png)
![(2e)-2-[(4-chlorophenyl)methylidene]-3,4-dihydronaphthalen-1-one](http://img.cochemist.com/ccimg/49600/49545-70-2_b.png)


![Benzoic acid, [(5-bromo-2-hydroxyphenyl)methylene]hydrazide](http://img.cochemist.com/ccimg/41400/41377-36-0.png)
![Benzoic acid, [(5-bromo-2-hydroxyphenyl)methylene]hydrazide](http://img.cochemist.com/ccimg/41400/41377-36-0_b.png)

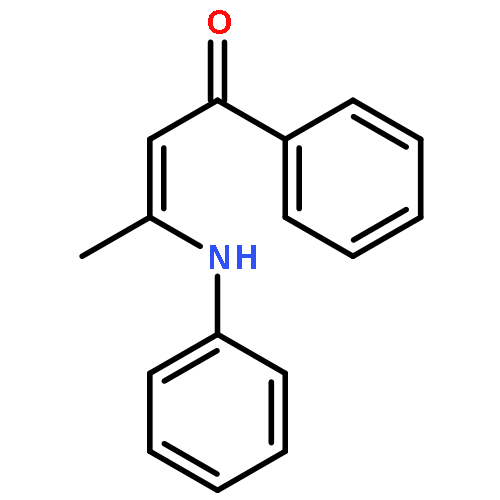
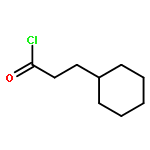
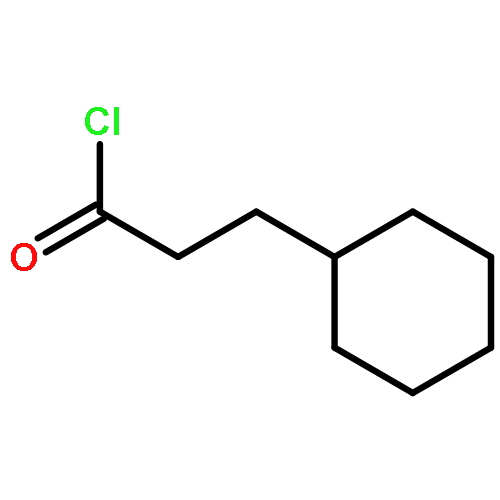
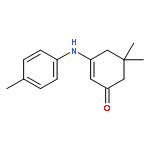
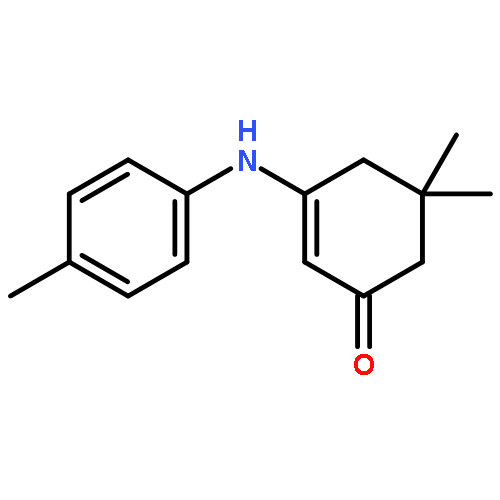
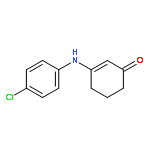
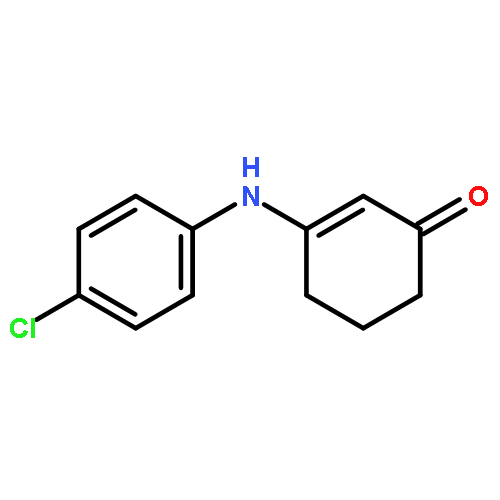
![2-Cyclohexen-1-one, 3-[(4-methylphenyl)amino]-](http://img.cochemist.com/ccimg/36700/36646-74-9.png)
![2-Cyclohexen-1-one, 3-[(4-methylphenyl)amino]-](http://img.cochemist.com/ccimg/36700/36646-74-9_b.png)


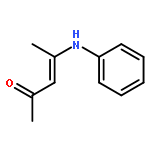
![3-[(4-METHOXYPHENYL)METHYLIDENE]CHROMEN-4-ONE](http://img.cochemist.com/ccimg/30800/30779-94-3.png)
![3-[(4-METHOXYPHENYL)METHYLIDENE]CHROMEN-4-ONE](http://img.cochemist.com/ccimg/30800/30779-94-3_b.png)
![4H-1-Benzopyran-4-one, 2,3-dihydro-3-[(4-methylphenyl)methylene]-](http://img.cochemist.com/ccimg/30800/30779-92-1.png)
![4H-1-Benzopyran-4-one, 2,3-dihydro-3-[(4-methylphenyl)methylene]-](http://img.cochemist.com/ccimg/30800/30779-92-1_b.png)



![3-[(4-Methoxyphenyl)amino]-5,5-dimethylcyclohex-2-en-1-one](http://img.cochemist.com/ccimg/24800/24706-48-7.png)
![3-[(4-Methoxyphenyl)amino]-5,5-dimethylcyclohex-2-en-1-one](http://img.cochemist.com/ccimg/24800/24706-48-7_b.png)
![2-Cyclohexen-1-one, 3-[(4-chlorophenyl)amino]-5,5-dimethyl-](http://img.cochemist.com/ccimg/24800/24706-47-6.png)
![2-Cyclohexen-1-one, 3-[(4-chlorophenyl)amino]-5,5-dimethyl-](http://img.cochemist.com/ccimg/24800/24706-47-6_b.png)
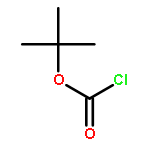

![2-Butenoic acid, 3-[(phenylmethyl)amino]-, ethyl ester, (2Z)-](http://img.cochemist.com/ccimg/21800/21759-74-0.png)
![2-Butenoic acid, 3-[(phenylmethyl)amino]-, ethyl ester, (2Z)-](http://img.cochemist.com/ccimg/21800/21759-74-0_b.png)
![Benzenecarboximidamide, N-[(chloroacetyl)oxy]-](http://img.cochemist.com/ccimg/21300/21251-95-6.png)
![Benzenecarboximidamide, N-[(chloroacetyl)oxy]-](http://img.cochemist.com/ccimg/21300/21251-95-6_b.png)






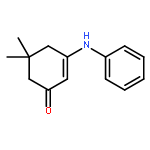
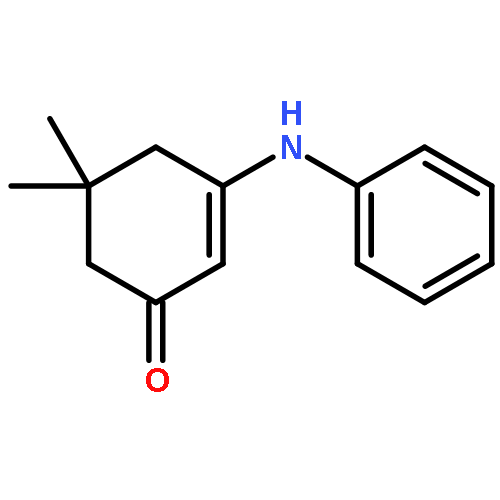

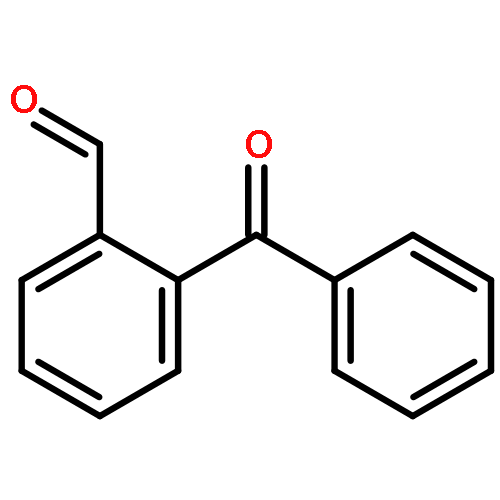
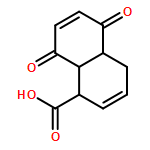










![Benzoic acid,2-[(2-hydroxyphenyl)methylene]hydrazide](http://img.cochemist.com/ccimg/3300/3232-37-9.png)
![Benzoic acid,2-[(2-hydroxyphenyl)methylene]hydrazide](http://img.cochemist.com/ccimg/3300/3232-37-9_b.png)
![2-Butenoic acid, 3-[(1-methylethyl)amino]-, ethyl ester, (2Z)-](http://img.cochemist.com/ccimg/1000/926-84-1.png)
![2-Butenoic acid, 3-[(1-methylethyl)amino]-, ethyl ester, (2Z)-](http://img.cochemist.com/ccimg/1000/926-84-1_b.png)