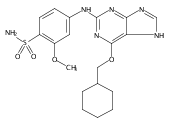
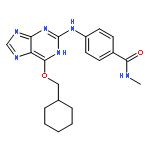
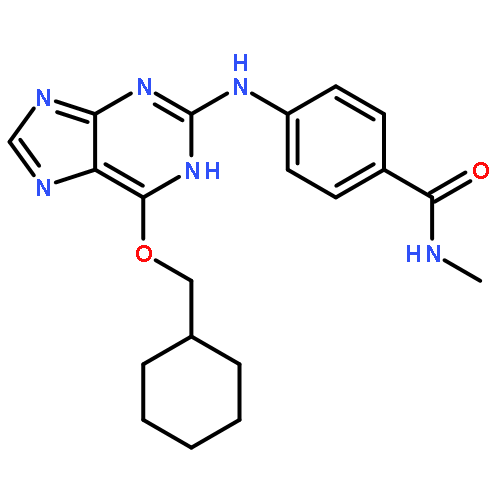
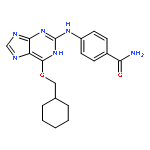
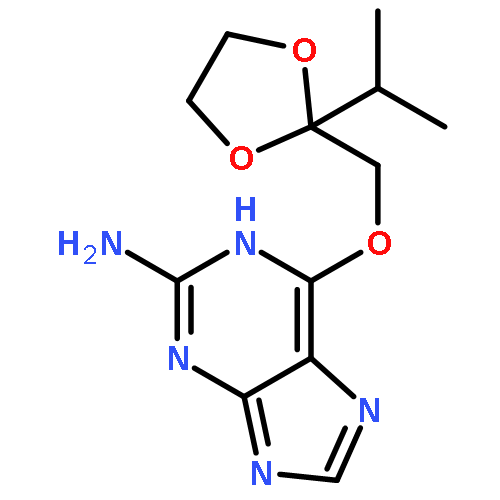
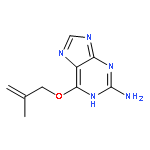
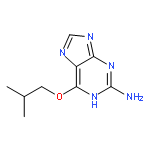
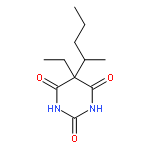
Co-reporter:Dongxia Ma;Congjie Zhang;Zhe-Ning Chen
Physical Chemistry Chemical Physics 2017 vol. 19(Issue 3) pp:2417-2424
Publication Date(Web):2017/01/18
DOI:10.1039/C6CP06215F
In this work, five new palladium(II) complexes have been designed as the model catalysts for methane to methyl trifluoroacetate conversion. All these compounds are analogues of the well-established (bis-NHC)PdBr2 complex (NHC, N-heterocyclic carbenes), derived by complexing the palladium(II) metal ion with the derivatives of bis-2-borabicyclo[1.1.0]but-1(3)-ene (bis-2BB) ligands using the sp2 carbons. Our density functional theory calculation results suggest that the (bis-2BB)PdBr2 catalysts outperform the popular (bis-NHC)PdBr2 complex in the desired catalytic process, and further reveal that the charge-shift bonding in the bis-2BB ligands contributes to the improved catalytic performance. These findings may spark new ideas for experimental design of more efficient organometallic catalysts for C–H bond activation and functionalization.
Co-reporter:Neil Qiang Su and Xin Xu
Journal of Chemical Theory and Computation 2016 Volume 12(Issue 5) pp:2285-2297
Publication Date(Web):March 24, 2016
DOI:10.1021/acs.jctc.6b00197
Recently, we have developed an integration approach for the calculations of ionization potentials (IPs) and electron affinities (EAs) of molecular systems at the level of second-order Møller–Plesset (MP2) (Su, N. Q.; Xu, X. J. Chem. Theory Comput. 11, 4677, 2015), where the full MP2 energy gradient with respect to the orbital occupation numbers was derived but only at integer occupations. The theory is completed here to cover the fractional occupation systems, such that Slater’s transition state concept can be used to have accurate predictions of IPs and EAs. Antisymmetrized Goldstone diagrams have been employed for interpretations and better understanding of the derived equations, where two additional rules were introduced in the present work specifically for hole or particle lines with fractional occupation numbers.
Co-reporter:Jinyang Xi and Xin Xu
Physical Chemistry Chemical Physics 2016 vol. 18(Issue 9) pp:6913-6924
Publication Date(Web):08 Feb 2016
DOI:10.1039/C5CP08065G
Anion–π interaction is a new type of non-covalent interaction. It has attracted growing interest in recent years both theoretically and experimentally. However, the nature of bonding between an anion and an electron-deficient aromatic system has remained elusive. To understand the bonding nature in depth, we have carried out a systematic computational study, using model systems that involve tetraoxacalix[2]arene[2]triazine 1, an electron-deficient macrocyclic host, and four anions, X− (X− = SCN−, NO3−, BF4−, and PF6−), of varied sizes and shapes. The geometries for the 1·X− complexes were optimized using the extended ONIOM (XO) method. The good agreements with the X-ray experimental results provide a validation of our theoretical schemes. The nature of the non-covalent interactions was analyzed with the help of the AIM (atoms in molecules), RDG (reduced density gradient) and LMO-EDA (local molecular orbital-energy decomposition analysis) methods. The results clearly reveal the involvement of anion–π bonding, as well as a weak, yet significant, hydrogen bonding interaction between the benzene C–H on 1 and the anion of NO3− or PF6−. The bonding energies of 1·X− were calculated with the XYG3 functional, and the results were compared with those from MP2, M06-2X and some other functionals with non-covalent interaction corrections (e.g., B3LYP-D3, and ωB97X-D). We conclude that the binding strengths follow the order of 1·NO3− > 1·SCN− > 1·BF4− > 1·PF6−, where the difference between 1·SCN− and 1·BF4− is less significant. The strongest interaction in 1·NO3− comes from: (1) the effective electronic interaction between NO3− and the triazine rings on 1; and (2) the weak hydrogen bonding interaction between the benzene C–H on 1 and nitrate, which cooperates with the anion–π interactions.
Co-reporter:Neil Qiang Su and Xin Xu
Journal of Chemical Theory and Computation 2015 Volume 11(Issue 10) pp:4677-4688
Publication Date(Web):September 9, 2015
DOI:10.1021/acs.jctc.5b00591
An integration approach is developed to calculate ionization potentials (IPs), and electron affinities (EAs), which is an extension of the D-ΔMBPT(2) method [A. Beste et al., J. Chem. Phys. 2013, 138, 074101]. The latter is an extension of the single-point method of Cohen et al. [A. J. Cohen et al., J. Chem. Theory Comput. 2009, 5, 786] from the perspective of fractional charges. While relaxation effects were included only at the Hartree–Fock (HF) level in the previous methods, such effects are fully taken into account in the present method up to the second-order Møller–Plesset (MP2) level. This is made possible by deriving the full MP2 energy gradient, with respect to the orbital occupation numbers, which is solved through the coupled-perturbed HF (CP-HF) equations.
Co-reporter:Zu-Yong Gong, Sai Duan, Guangjun Tian, Jun Jiang, Xin Xu and Yi Luo
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 19) pp:12698-12707
Publication Date(Web):13 Apr 2015
DOI:10.1039/C5CP01378J
We performed systematic theoretical studies on small anionic water/deuterated water clusters W/D−n=2–6 at both density functional theory (B3LYP) and wavefunction theory (MP2) levels. The focus of the study is to examine the convergence of calculated infrared (IR) spectra with respect to the increasing number of diffuse functions. It is found that at the MP2 level for larger clusters (n = 4–6), only one extra diffuse function is needed to obtain the converged relative IR intensities, while two or three more sets of extra diffuse functions are needed for smaller clusters. Such behaviour is strongly associated with the convergence of the electronic structure of corresponding clusters at the MP2 level. It is striking to observe that at the B3LYP level, the calculated relative IR intensities for all the clusters under investigations are diverse and show no trend of convergence upon increasing the number of diffuse functions. Moreover, the increasing contribution from the extra diffuse functions to the dynamic IR dipole moment indicates that the B3LYP electronic structure also fails to converge. These results manifest that MP2 is a preferential theoretical method, as compared to the widely used B3LYP, for the IR intensity of dipole bounded electron systems.
Co-reporter:Neil Qiang Su and Xin Xu
The Journal of Physical Chemistry A 2015 Volume 119(Issue 9) pp:1590-1599
Publication Date(Web):October 7, 2014
DOI:10.1021/jp507711t
We have systematically analyzed the error accumulations in the adhesive energies for a series of hydrogen molecular chains calculated by various kinds of density functional theory (DFT) methods. In particular, we have focused on some representative doubly hybrid (DH) functionals of either the B2PLYP type (B2PLYP, B2PLYP-D, and B2GP-PLYP) or the XYG3 type (XYG3, XYGJ-OS, and xDH-PBE0). The hydrogen molecular chain models have recently been proposed by Zheng et al. (J. Chem. Phys. 2012, 137, 214106) to identify the delocalization errors (DEs) in thermodynamic properties. From the perspective of DEs, it is shown here that the XYG3 type of DH functionals yield good performance on the calculated adhesive energies due to the minimizing effects of DEs, highlighting the underlying physics for the successes or failures of the approximate functionals. Examination was also extended to HF-DFT, where DFT energies are evaluated with the Hartree–Fock (HF) densities.
Co-reporter:Xian-Yong Yu, Lin Deng, Baishu Zheng, Bi-Rong Zeng, Pinggui Yi and Xin Xu
Dalton Transactions 2014 vol. 43(Issue 4) pp:1524-1533
Publication Date(Web):22 Oct 2013
DOI:10.1039/C3DT51986D
In order to understand the substitution effects of pyrazolylpyridine (pzpy) on the coordination reaction equilibria, the interactions between a series of pzpy-like ligands and biperoxidovanadate ([OV(O2)2(D2O)]−/[OV(O2)2(HOD)]−, abbrv. bpV) have been explored using a combination of multinuclear (1H, 13C, and 51V) magnetic resonance, heteronuclear single quantum coherence (HSQC), and variable temperature NMR in a 0.15 mol L−1 NaCl D2O solution that mimics the physiological conditions. Both the direct NMR data and the equilibrium constants are reported for the first time. A series of new hepta-coordinated peroxidovanadate species [OV(O2)2L]− (L = pzpy-like chelating ligands) are formed due to several competitive coordination interactions. According to the equilibrium constants for products between bpV and the pzpy-like ligands, the relative affinity of the ligands is found to be pzpy > 2-Ester-pzpy ≈ 2-Me-pzpy ≈ 2-Amide-pzpy > 2-Et-pzpy. In the interaction system between bpV and pzpy, a pair of isomers (Isomers A and B) are observed in aqueous solution, which are attributed to different types of coordination modes between the metal center and the ligands, while the crystal structure of NH4[OV(O2)2(pzpy)]·6H2O (CCDC 898554) has the same coordination structure as Isomer A (the main product for pzpy). For the N-substituted ligands, however, Isomer A or B type complexes can also be observed in solution but the molar ratios of the isomer are reversed (i.e., Isomer B type is the main product). These results demonstrate that when the N atom in the pyrazole ring has a substitution group, hydrogen bonding (from the H atom in the pyrazole ring), the steric effect (from alkyl) and the solvation effect (from the ester or amide group) can jointly affect the coordination reaction equilibrium.
Co-reporter:Wenping Guo, Carine Michel, Renate Schwiedernoch, Raphael Wischert, Xin Xu, and Philippe Sautet
Organometallics 2014 Volume 33(Issue 22) pp:6369-6380
Publication Date(Web):October 29, 2014
DOI:10.1021/om5006808
The most challenging step in the production of acrylates from ethylene and CO2 mediated by transition-metal complexes is the release of the acrylate from the metallalactone intermediate formed by coupling of ethylene and CO2. Recently, methyl acrylate formation was achieved from nickelalactones by using methyl iodide (MeI) as the electrophile, and the yield was tuned with different amine and phosphine ligands. Modeling organometallic catalysts with such large ligands accurately is a challenge for computational chemistry. A hybrid approach has been designed here by coupling the double hybrid XYG3 and the hybrid B3LYP exchange correlation functionals, using the extended ONIOM scheme. This approach was then applied to explore the role of the MeI electrophile for the formation of methyl acrylate from the initial nickelalactone complex and to rationalize the effect of the ligands on the yield of methyl acrylate. We show that the choice of ligand has little effect on the main productive pathway. However, it has a significant influence on side reactions, which compete with the productive pathway and are detrimental to methyl acrylate formation. Finally, the need for a very large overstoichiometry of MeI for a good yield of methyl acrylate is explained by the lower polarity of MeI, which avoids the stabilization of nonproductive intermediates. The nature of the limiting intermediates has been validated by comparing calculated and experimental vibrational spectra.
Co-reporter:Neil Qiang Su, Weitao Yang, Paula Mori-Sánchez, and Xin Xu
The Journal of Physical Chemistry A 2014 Volume 118(Issue 39) pp:9201-9211
Publication Date(Web):May 20, 2014
DOI:10.1021/jp5029992
In this work, we examine the fractional charge behaviors of doubly hybrid (DH) functionals. By plotting the ground-state energies E and energy derivatives for atoms and molecules with fractional electron numbers N, we directly quantify the delocalization errors of some representative DH functionals such as B2PLYP, XYG3, and XYGJ-OS. Numerical assessments on ionization potentials (IPs), electron affinities (EAs), and fundamental gaps, from either integer number calculations or energy derivative calculations, are provided. It is shown that the XYG3 type of DH functionals gives good agreement between their energy derivatives and the experimental IPs, EAs, and gaps, as expected from their nearly straight line fractional charge behaviors.
Co-reporter:Igor Ying Zhang;Jun Jiang;Bin Gao;Yi Luo
Science China Chemistry 2014 Volume 57( Issue 10) pp:1399-1404
Publication Date(Web):2014 October
DOI:10.1007/s11426-014-5183-y
Technically, when dealing with a perfect crystal, methods in k-(reciprocal) space that impose periodic boundary conditions (PBC) in conjunction with plane-wave basis sets are widely used. Chemists, however, tend to think of a solid as a giant molecule, which offers a molecular way to describe a solid by using a finite cluster model (FCM). However, FCM may fail to simulate a perfect crystal due to its inevitable boundary effects. We propose an RRS-PBC method that extracts the k-space information of a perfect crystalline solid out of a reduced real space (RRS) of an FCM. We show that the inevitable boundary effects in an FCM are eliminated naturally to achieve converged high-quality band structures.
Co-reporter:Xinlan Wang, Sai Duan, and Xin Xu
The Journal of Physical Chemistry C 2013 Volume 117(Issue 30) pp:15763-15772
Publication Date(Web):July 8, 2013
DOI:10.1021/jp4051879
Density functional theory at the level of (U)B3LYP has been used to investigate the complete oxidation of the Si(111)-7 × 7 surface by water. The results suggest that the initial water dissociation readily occurs across an adjacent adatom–rest atom (Sia–Sir) pair, resulting in either Sia–OH + Sir–H (i.e., the Sia route) or Sir–OH + Sia–H (i.e., the Sir route). Both routes are found to follow the precursor-mediated pathway, while the Sir atom is concluded to be more reactive than the Sia atom toward the initial decomposition of the H2O molecule due to its higher binding affinity to the incident water. With increasing water exposure and reaction temperature, deep oxidation can be accomplished by steady oxygen atom insertion into the Sia–Sis backbonds until the Si4+ oxidation state is eventually developed. Our calculations uncover that the most favorable pathway for deep oxidations begins with the direct water dissociation over the Sia–Sis backbond, followed by H2 liberation. This differs from the recent proposal via the OH insertion. Our deep oxidation mechanism can also be applied to the oxidation of an isolated adatom, which gives an explanation to the experimental observation where more than 50% of Sia are involved in water oxidation. The present work provides the detailed energetics that sheds light on the wet oxidation mechanism of silicon surfaces at the molecular level.
Co-reporter:Igor Ying Zhang and Xin Xu
The Journal of Physical Chemistry Letters 2013 Volume 4(Issue 10) pp:1669-1675
Publication Date(Web):April 30, 2013
DOI:10.1021/jz400695u
An unbiased understanding of complex molecular systems from first-principles critically demands theoretical methods with uniform accuracy for diverse interactions with different natures covering short-, medium-, and long-range correlations. Among the state-of-the-art density functional approximations (DFAs), doubly hybrid (DH) DFAs (e.g., XYG3 in this Letter) provide a remarkable improvement over the conventional DFAs (e.g., B3LYP in this Letter). Even though XYG3 works quite well in many cases of noncovalent bonding interactions (NCIs), it is incomplete in describing the pure long-range dispersive interactions. Here, we address such concerns by adding a scaled long-range contribution from the second-order perturbation theory (PT2). The long-range-corrected XYG3 (lrc-XYG3) is proposed without reparameterizing the three parameters in the original XYG3. Due to its overall excellent performance for all testing sets constructed for various purposes, lrc-XYG3 is the recommended method, which is expected to provide a balanced description of diverse interactions in complex molecular systems.Keywords: density functional theory; dispersion correction; noncovalent bonding interaction; range separation; second-order perturbation theory;
Co-reporter:Xian-Yong Yu, Ping-Gui Yi, Dan-Hong Ji, Bi-Rong Zeng, Xiao-Fang Li and Xin Xu
Dalton Transactions 2012 vol. 41(Issue 13) pp:3684-3694
Publication Date(Web):22 Feb 2012
DOI:10.1039/C2DT12334G
To understand the substitution effects of 4-(pyridin-2-yl)pyrimidine (pprd) on the coordination reaction equilibria, the interactions between a series of the pprd-like ligands and [OV(O2)2(H2O)]− or [OV(O2)2(HOD)]− or [OV(O2)2(D2O)]− (bpV) have been explored by a combination of multinuclear (1H, 13C, and 51V) magnetic resonance, heteronuclear single quantum coherence (HSQC) and variable temperature NMR in a 0.15 mol L−1 NaCl D2O solution that mimics physiological conditions. The direct NMR data are reported for the first time. Competitive coordination interactions result in a series of new hepta-coordinated peroxidovanadate species [OV(O2)2LL′]− (LL′ = pprd-like chelating ligands). The equilibrium constants for the products between bpV and the pprd-like ligands show that the relative affinity of the ligands is pprd ≈ 2-NH2-pprd > 2-Me-pprd > 2-Et-pprd > 4-(6-methylpyridin-2-yl)pyrimidine (abbr. 6′-Me-pprd). When the ligand is pprd, a pair of isomers (Isomer A and B) are observed in aqueous solution, which are attributed to the different types of coordination modes between the metal and the ligands, while the crystal structure of NH4[OV(O2)2(pprd)]·2H2O has the same coordination structure as Isomer A. For substituted pprd ligands, however, only one type of structure (Isomer A or B ) is observed in solution. These results demonstrate that, when the aromatic ring has a substitution group, both the steric effect (from the alkyl) and hydrogen bonding (from the amine) can affect the coordination reaction equilibrium to prevent the appearance of either Isomer B in solution for the ligands 2-Me-pprd, 2-NH2-pprd, 2-Et-pprd, or Isomer A in solution for 6′-Me-pprd.
Co-reporter:Hongping Li, Gang Fu and Xin Xu
Physical Chemistry Chemical Physics 2012 vol. 14(Issue 48) pp:16686-16694
Publication Date(Web):12 Oct 2012
DOI:10.1039/C2CP43176A
In the present work, we have investigated the CO dissociation on corrugated Ru(111) and the stepped Ru(0001) surfaces by means of density functional theory with slab models. Our results show that, while the direct CO dissociation is preferred on the six-fold site of Ru(111), the H-assisted CO dissociation is found to be favored on the B5 site of the stepped Ru(0001) surface. Furthermore, we have studied the effects of co-adsorbed spectator species on the CO dissociation mechanisms. Our results demonstrate that spectators can change the potential energy landscape dramatically, such that different reaction mechanisms can be favored in the presence of different spectators. Neither the H-assisted CO dissociation mechanism nor the CO direct dissociation mechanism should be overlooked at authentic ambient conditions. This work emphasizes a dynamic picture of the reaction mechanisms due to the inherent structural and compositional inhomogeneity on surfaces. Different mechanisms can work together as different active sites will co-exist on a real catalyst surface, and the reaction preferences on an active site can vary as the adsorbate compositions on surfaces are varying during the course of the reactions.
Co-reporter:Igor Ying Zhang and Xin Xu
Physical Chemistry Chemical Physics 2012 vol. 14(Issue 36) pp:12554-12570
Publication Date(Web):23 Apr 2012
DOI:10.1039/C2CP40904F
XYG3 and XYGJ-OS doubly hybrid density functionals (DHDFs) are applied to calculate intermolecular interaction energies of biological relevance. The results are compared with those of the other types of DHDFs (MC3BB and B2PLYP), as well as their dispersion corrected variants (MC3BB-D B2PLYP-D, and B2PLYP-D3), in addition to those obtained from B3LYP, B3LYP-D and B3LYP-D3. The reference data are taken from the S22, S22x5 and JSCH-2005 benchmark sets consisting mainly of DNA base pairs and amino acid pairs. The overall good agreement with the reference values of the extrapolated CCSD(T) complete basis set limit suggests that the XYG3 and XYGJ-OS functionals are valuable tools for applications in biochemistry.
Co-reporter:Zhe-Ning Chen, Gang Fu and Xin Xu
Organic & Biomolecular Chemistry 2012 vol. 10(Issue 47) pp:9491-9500
Publication Date(Web):17 Oct 2012
DOI:10.1039/C2OB26658J
Here we present a systematic theoretical investigation on the mechanisms of Grignard reagent formation (GRF) for CH3Cl reacting with Mg atom, Mg2 and a series of Mg clusters (Mg4–Mg20). Our calculations reveal that the ground state Mg atom is inactive under matrix condition, whereas it is active under metal vapor synthesis (MVS) conditions. On the other hand, the excited state Mg (3P) atom, as produced by laser-ablation, can react with CH3Cl barrierlessly, and hence is active under matrix condition. We predict that the bimagnesium Grignard reagent, though often proposed, can barely be observed experimentally, due to its high reactivity towards additional CH3Cl to produce more stable Grignard reagent dimer, and that the cluster Grignard reagent RMg4X possesses a flat Mg4 unit rather than a tetrahedral geometry. Our calculations further reveal that the radical pathway (T4) is prevalent on Mg, Mg2 and Mgn clusters of small size, while the no-radical pathway (T2), which starts at Mg4, becomes competitive with T4 as the cluster size increases. A structure–reactivity relationship between barrier heights and ionization potentials of Mgn is established. These findings not only resolve controversy in experiment and theory, but also provide insights which can be used in the design of effective synthesis approaches for the preparation of chiral Grignard reagents.
Co-reporter:Dr. Igor Ying Zhang; Dr. Xin Xu
ChemPhysChem 2012 Volume 13( Issue 6) pp:1486-1494
Publication Date(Web):
DOI:10.1002/cphc.201100909
Abstract
Density functional theory has become a valuable tool to study surface catalysis. However, due to the scarcity of clean and reliable experimental data on surfaces, the theoretical methods employed to explore heterogeneous catalytic mechanisms are usually less well validated than those for gas-phase reactions. We argue herein that gas-phase reactions and the corresponding surface reactions are related through the Born–Haber cycle and computational catalysis on surfaces will be less meaningful if gas-phase behavior cannot first be suitably determined. In this contribution, we have constructed a set of gas-phase reactions relevant to the Fischer–Tropsch synthesis as a case study. With this set, we have tested the validity of the widely used PBE and B3LYP functionals and found that neither of them are capable of describing all kinds of gas-phase reactions properly, such that some surface reactions may be biased falsely against the others. Significantly, XYG3, which is a double-hybrid functional that includes Hartree–Fock-like exchange and many-body perturbation correlation effects, presents a significant improvement for all of the gas-phase reactions, holding promise for further development for surface catalysis.
Co-reporter:Sai Duan, Xin Xu, Yi Luo and Zhong-Qun Tian
Chemical Communications 2011 vol. 47(Issue 41) pp:11438-11440
Publication Date(Web):21 Sep 2011
DOI:10.1039/C1CC14962H
We present a practical method which demonstrates how the physical and chemical enhancements in SERS for a molecule adsorbed on metal junctions are conceptually coupled through the polarization of the molecule and its surroundings. Calculations with the state-of-the-art density functional reveal that the coupling factor considered in the present work can be as large as 106, such that it is indeed important for certain vibrational modes.
Co-reporter:Gang Liu, Jianming Wu, Igor Ying Zhang, Zhe-Ning Chen, Yong-Wang Li, and Xin Xu
The Journal of Physical Chemistry A 2011 Volume 115(Issue 46) pp:13628-13641
Publication Date(Web):October 17, 2011
DOI:10.1021/jp207641g
Recently, 5-chloromethylfurfural (CMF) was proposed as a central intermediate in the conversion of carbohydrate-based material into useful organic commodities. In the present work, we have calculated the thermochemistry using the highly accurate G4 theory and several state-of-art density functional theory (DFT) methods (e.g., X1, M06-2X, B2PLYP-D, and XYG3) for the conversion from CMF to 5-hydroxymethylfurfural (HMF) and levulinic acid (LA) in water, and that to biofuels 5-ethoxymethylfurfural (EMF) and ethyllevulinate (EL) in alcohol. New reaction mechanisms have been proposed, which complement the well-recognized Horvat mechanisms. The assessment of DFT methods suggested that XYG3 be a viable method for biomass related thermochemistry calculations.
Co-reporter:Zhen-Kun Chu ; Gang Fu ; Wenping Guo
The Journal of Physical Chemistry C 2011 Volume 115(Issue 30) pp:14754-14761
Publication Date(Web):June 29, 2011
DOI:10.1021/jp201454t
There exist 12 crystallographically distinct Al-sites in the ZSM-5 zeolite, associated with which there are various Na-sites. Understanding their locations, while being the key to the understanding of the catalytic properties of this material, remains a great challenge in both experiment and theory. We present here a theoretical survey of the Na+ location along with the Al distribution in ZSM-5 by using hybrid methods, ONIOM (our Own N-layer Integrated molecular Orbital molecular Mechanics) as well as the newly developed extended ONIOM (XO) (Guo, W. P.; Wu, A. A.; Xu, X. Chem. Phys. Lett.2010, 498, 203–208) method. The reliability and efficiency of different methods/models have been systematically tested. Using the T1 Al-site as an example, our calculations demonstrate that the high-level layers of ONIOM models have to include all rings around the [AlO4] tetrahedron to have reliable coordination structures and energetics of different Na-sites, while XO can provide reliable results with 60% savings of computational time as compared to that of ONIOM. Our XO calculations reveal that, in most Al-sites, Na+ preferentially occupies the six-membered-ring sites, and the most favorable Al-sites along with the Na-sites are T8/M6, T10/Z6, and T4/Z6. Conversely, those Al-sites only surrounded by five-membered rings, such as T6 and T3, are predicted to be energetically unfavorable.
Co-reporter:Xin Xu;Igor Ying Zhang;Yousung Jung;William A. Goddard III
PNAS 2011 Volume 108 (Issue 50 ) pp:
Publication Date(Web):2011-12-13
DOI:10.1073/pnas.1115123108
We develop and validate the XYGJ-OS functional, based on the adiabatic connection formalism and Görling-Levy perturbation
theory to second order and using the opposite-spin (OS) ansatz combined with locality of electron correlation. XYGJ-OS with
local implementation scales as N3 with an overall accuracy of 1.28 kcal/mol for thermochemistry, bond dissociation energies, reaction barrier heights, and
nonbonded interactions, comparable to that of 1.06 kcal/mol for the accurate coupled-cluster based G3 method (scales as N7) and much better than many popular density functional theory methods: B3LYP (4.98), PBE0 (4.36), and PBE (12.10).
Co-reporter:Li Rao ; Qiang Cui
Journal of the American Chemical Society 2010 Volume 132(Issue 51) pp:18092-18102
Publication Date(Web):December 3, 2010
DOI:10.1021/ja103742k
We carry out a theoretical analysis of factors that dictate the binding affinity and selectivity of the copper efflux regulator (CueR) toward different metal ions (Cu+, Ag+, Au+, Zn2+, and Hg2+). In addition to a simplified active-site model, we have established a computational framework based on quantum mechanical/molecular mechanical (QM/MM) and Poisson−Boltzmann approaches that allows us, for the first time, to systematically analyze the protein contribution to transition metal binding affinity and selectivity. We find that the QM/MM model leads to relative binding affinities that are consistent with observations from transcription induction experiments, while an active-site model does not, which highlights the importance of explicitly considering the protein environment for a thorough understanding of metal binding properties of metalloproteins. Regarding the trends in binding affinity, our analysis highlights both intrinsic properties of a metal ion and protein contributions. Specifically, the softness and desolvation penalty of a metal ion make large contributions to the binding affinity; for example, we find that the large desolvation penalty for Zn2+ rather than any stereoelectronic factor (e.g., linear vs tetrahedron coordination) is the key reason that Zn2+ binds much more weakly than Hg2+ to CueR. Moreover, our results explicitly demonstrate that the electrostatic environment of CueR is well-tuned to favor the binding of coinage metal ions over divalent ions. Finally, our analyses highlight the importance of considering the proper solution reference (i.e., the metal ion bound to buffer ligands vs water molecules) when discussing the binding affinity of metal ions to proteins.
Co-reporter:Igor Ying Zhang and Xin Xu
Physical Chemistry Chemical Physics 2012 - vol. 14(Issue 36) pp:NaN12570-12570
Publication Date(Web):2012/04/23
DOI:10.1039/C2CP40904F
XYG3 and XYGJ-OS doubly hybrid density functionals (DHDFs) are applied to calculate intermolecular interaction energies of biological relevance. The results are compared with those of the other types of DHDFs (MC3BB and B2PLYP), as well as their dispersion corrected variants (MC3BB-D B2PLYP-D, and B2PLYP-D3), in addition to those obtained from B3LYP, B3LYP-D and B3LYP-D3. The reference data are taken from the S22, S22x5 and JSCH-2005 benchmark sets consisting mainly of DNA base pairs and amino acid pairs. The overall good agreement with the reference values of the extrapolated CCSD(T) complete basis set limit suggests that the XYG3 and XYGJ-OS functionals are valuable tools for applications in biochemistry.
Co-reporter:Zu-Yong Gong, Sai Duan, Guangjun Tian, Jun Jiang, Xin Xu and Yi Luo
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 19) pp:NaN12707-12707
Publication Date(Web):2015/04/13
DOI:10.1039/C5CP01378J
We performed systematic theoretical studies on small anionic water/deuterated water clusters W/D−n=2–6 at both density functional theory (B3LYP) and wavefunction theory (MP2) levels. The focus of the study is to examine the convergence of calculated infrared (IR) spectra with respect to the increasing number of diffuse functions. It is found that at the MP2 level for larger clusters (n = 4–6), only one extra diffuse function is needed to obtain the converged relative IR intensities, while two or three more sets of extra diffuse functions are needed for smaller clusters. Such behaviour is strongly associated with the convergence of the electronic structure of corresponding clusters at the MP2 level. It is striking to observe that at the B3LYP level, the calculated relative IR intensities for all the clusters under investigations are diverse and show no trend of convergence upon increasing the number of diffuse functions. Moreover, the increasing contribution from the extra diffuse functions to the dynamic IR dipole moment indicates that the B3LYP electronic structure also fails to converge. These results manifest that MP2 is a preferential theoretical method, as compared to the widely used B3LYP, for the IR intensity of dipole bounded electron systems.
Co-reporter:Hongping Li, Gang Fu and Xin Xu
Physical Chemistry Chemical Physics 2012 - vol. 14(Issue 48) pp:NaN16694-16694
Publication Date(Web):2012/10/12
DOI:10.1039/C2CP43176A
In the present work, we have investigated the CO dissociation on corrugated Ru(111) and the stepped Ru(0001) surfaces by means of density functional theory with slab models. Our results show that, while the direct CO dissociation is preferred on the six-fold site of Ru(111), the H-assisted CO dissociation is found to be favored on the B5 site of the stepped Ru(0001) surface. Furthermore, we have studied the effects of co-adsorbed spectator species on the CO dissociation mechanisms. Our results demonstrate that spectators can change the potential energy landscape dramatically, such that different reaction mechanisms can be favored in the presence of different spectators. Neither the H-assisted CO dissociation mechanism nor the CO direct dissociation mechanism should be overlooked at authentic ambient conditions. This work emphasizes a dynamic picture of the reaction mechanisms due to the inherent structural and compositional inhomogeneity on surfaces. Different mechanisms can work together as different active sites will co-exist on a real catalyst surface, and the reaction preferences on an active site can vary as the adsorbate compositions on surfaces are varying during the course of the reactions.
Co-reporter:Jinyang Xi and Xin Xu
Physical Chemistry Chemical Physics 2016 - vol. 18(Issue 9) pp:NaN6924-6924
Publication Date(Web):2016/02/08
DOI:10.1039/C5CP08065G
Anion–π interaction is a new type of non-covalent interaction. It has attracted growing interest in recent years both theoretically and experimentally. However, the nature of bonding between an anion and an electron-deficient aromatic system has remained elusive. To understand the bonding nature in depth, we have carried out a systematic computational study, using model systems that involve tetraoxacalix[2]arene[2]triazine 1, an electron-deficient macrocyclic host, and four anions, X− (X− = SCN−, NO3−, BF4−, and PF6−), of varied sizes and shapes. The geometries for the 1·X− complexes were optimized using the extended ONIOM (XO) method. The good agreements with the X-ray experimental results provide a validation of our theoretical schemes. The nature of the non-covalent interactions was analyzed with the help of the AIM (atoms in molecules), RDG (reduced density gradient) and LMO-EDA (local molecular orbital-energy decomposition analysis) methods. The results clearly reveal the involvement of anion–π bonding, as well as a weak, yet significant, hydrogen bonding interaction between the benzene C–H on 1 and the anion of NO3− or PF6−. The bonding energies of 1·X− were calculated with the XYG3 functional, and the results were compared with those from MP2, M06-2X and some other functionals with non-covalent interaction corrections (e.g., B3LYP-D3, and ωB97X-D). We conclude that the binding strengths follow the order of 1·NO3− > 1·SCN− > 1·BF4− > 1·PF6−, where the difference between 1·SCN− and 1·BF4− is less significant. The strongest interaction in 1·NO3− comes from: (1) the effective electronic interaction between NO3− and the triazine rings on 1; and (2) the weak hydrogen bonding interaction between the benzene C–H on 1 and nitrate, which cooperates with the anion–π interactions.
Co-reporter:Dongxia Ma, Congjie Zhang, Zhe-Ning Chen and Xin Xu
Physical Chemistry Chemical Physics 2017 - vol. 19(Issue 3) pp:NaN2424-2424
Publication Date(Web):2016/12/08
DOI:10.1039/C6CP06215F
In this work, five new palladium(II) complexes have been designed as the model catalysts for methane to methyl trifluoroacetate conversion. All these compounds are analogues of the well-established (bis-NHC)PdBr2 complex (NHC, N-heterocyclic carbenes), derived by complexing the palladium(II) metal ion with the derivatives of bis-2-borabicyclo[1.1.0]but-1(3)-ene (bis-2BB) ligands using the sp2 carbons. Our density functional theory calculation results suggest that the (bis-2BB)PdBr2 catalysts outperform the popular (bis-NHC)PdBr2 complex in the desired catalytic process, and further reveal that the charge-shift bonding in the bis-2BB ligands contributes to the improved catalytic performance. These findings may spark new ideas for experimental design of more efficient organometallic catalysts for C–H bond activation and functionalization.
Co-reporter:Sai Duan, Xin Xu, Yi Luo and Zhong-Qun Tian
Chemical Communications 2011 - vol. 47(Issue 41) pp:NaN11440-11440
Publication Date(Web):2011/09/21
DOI:10.1039/C1CC14962H
We present a practical method which demonstrates how the physical and chemical enhancements in SERS for a molecule adsorbed on metal junctions are conceptually coupled through the polarization of the molecule and its surroundings. Calculations with the state-of-the-art density functional reveal that the coupling factor considered in the present work can be as large as 106, such that it is indeed important for certain vibrational modes.
Co-reporter:Xian-Yong Yu, Lin Deng, Baishu Zheng, Bi-Rong Zeng, Pinggui Yi and Xin Xu
Dalton Transactions 2014 - vol. 43(Issue 4) pp:NaN1533-1533
Publication Date(Web):2013/10/22
DOI:10.1039/C3DT51986D
In order to understand the substitution effects of pyrazolylpyridine (pzpy) on the coordination reaction equilibria, the interactions between a series of pzpy-like ligands and biperoxidovanadate ([OV(O2)2(D2O)]−/[OV(O2)2(HOD)]−, abbrv. bpV) have been explored using a combination of multinuclear (1H, 13C, and 51V) magnetic resonance, heteronuclear single quantum coherence (HSQC), and variable temperature NMR in a 0.15 mol L−1 NaCl D2O solution that mimics the physiological conditions. Both the direct NMR data and the equilibrium constants are reported for the first time. A series of new hepta-coordinated peroxidovanadate species [OV(O2)2L]− (L = pzpy-like chelating ligands) are formed due to several competitive coordination interactions. According to the equilibrium constants for products between bpV and the pzpy-like ligands, the relative affinity of the ligands is found to be pzpy > 2-Ester-pzpy ≈ 2-Me-pzpy ≈ 2-Amide-pzpy > 2-Et-pzpy. In the interaction system between bpV and pzpy, a pair of isomers (Isomers A and B) are observed in aqueous solution, which are attributed to different types of coordination modes between the metal center and the ligands, while the crystal structure of NH4[OV(O2)2(pzpy)]·6H2O (CCDC 898554) has the same coordination structure as Isomer A (the main product for pzpy). For the N-substituted ligands, however, Isomer A or B type complexes can also be observed in solution but the molar ratios of the isomer are reversed (i.e., Isomer B type is the main product). These results demonstrate that when the N atom in the pyrazole ring has a substitution group, hydrogen bonding (from the H atom in the pyrazole ring), the steric effect (from alkyl) and the solvation effect (from the ester or amide group) can jointly affect the coordination reaction equilibrium.
Co-reporter:Zhe-Ning Chen, Gang Fu and Xin Xu
Organic & Biomolecular Chemistry 2012 - vol. 10(Issue 47) pp:NaN9500-9500
Publication Date(Web):2012/10/17
DOI:10.1039/C2OB26658J
Here we present a systematic theoretical investigation on the mechanisms of Grignard reagent formation (GRF) for CH3Cl reacting with Mg atom, Mg2 and a series of Mg clusters (Mg4–Mg20). Our calculations reveal that the ground state Mg atom is inactive under matrix condition, whereas it is active under metal vapor synthesis (MVS) conditions. On the other hand, the excited state Mg (3P) atom, as produced by laser-ablation, can react with CH3Cl barrierlessly, and hence is active under matrix condition. We predict that the bimagnesium Grignard reagent, though often proposed, can barely be observed experimentally, due to its high reactivity towards additional CH3Cl to produce more stable Grignard reagent dimer, and that the cluster Grignard reagent RMg4X possesses a flat Mg4 unit rather than a tetrahedral geometry. Our calculations further reveal that the radical pathway (T4) is prevalent on Mg, Mg2 and Mgn clusters of small size, while the no-radical pathway (T2), which starts at Mg4, becomes competitive with T4 as the cluster size increases. A structure–reactivity relationship between barrier heights and ionization potentials of Mgn is established. These findings not only resolve controversy in experiment and theory, but also provide insights which can be used in the design of effective synthesis approaches for the preparation of chiral Grignard reagents.
Co-reporter:Xian-Yong Yu, Ping-Gui Yi, Dan-Hong Ji, Bi-Rong Zeng, Xiao-Fang Li and Xin Xu
Dalton Transactions 2012 - vol. 41(Issue 13) pp:NaN3694-3694
Publication Date(Web):2012/02/22
DOI:10.1039/C2DT12334G
To understand the substitution effects of 4-(pyridin-2-yl)pyrimidine (pprd) on the coordination reaction equilibria, the interactions between a series of the pprd-like ligands and [OV(O2)2(H2O)]− or [OV(O2)2(HOD)]− or [OV(O2)2(D2O)]− (bpV) have been explored by a combination of multinuclear (1H, 13C, and 51V) magnetic resonance, heteronuclear single quantum coherence (HSQC) and variable temperature NMR in a 0.15 mol L−1 NaCl D2O solution that mimics physiological conditions. The direct NMR data are reported for the first time. Competitive coordination interactions result in a series of new hepta-coordinated peroxidovanadate species [OV(O2)2LL′]− (LL′ = pprd-like chelating ligands). The equilibrium constants for the products between bpV and the pprd-like ligands show that the relative affinity of the ligands is pprd ≈ 2-NH2-pprd > 2-Me-pprd > 2-Et-pprd > 4-(6-methylpyridin-2-yl)pyrimidine (abbr. 6′-Me-pprd). When the ligand is pprd, a pair of isomers (Isomer A and B) are observed in aqueous solution, which are attributed to the different types of coordination modes between the metal and the ligands, while the crystal structure of NH4[OV(O2)2(pprd)]·2H2O has the same coordination structure as Isomer A. For substituted pprd ligands, however, only one type of structure (Isomer A or B ) is observed in solution. These results demonstrate that, when the aromatic ring has a substitution group, both the steric effect (from the alkyl) and hydrogen bonding (from the amine) can affect the coordination reaction equilibrium to prevent the appearance of either Isomer B in solution for the ligands 2-Me-pprd, 2-NH2-pprd, 2-Et-pprd, or Isomer A in solution for 6′-Me-pprd.
Co-reporter:Neil Qiang Su and Xin Xu
Chemical Communications 2016 - vol. 52(Issue 96) pp:NaN13860-13860
Publication Date(Web):2016/09/08
DOI:10.1039/C6CC04886B
The exchange–correlation functional holds the key to the success or failure of density functional theory calculations. When evaluating the functional performances, the current literature has focused much more on the ability of the method to reproduce exact electronic energies than on exact geometries. As all calculations have to start from the right geometries, the present feature article explores the functional performances on geometry predictions involving covalently and non-covalently bonded systems, as well as transition state structures with emphasis on the XYG3 type of doubly hybrid (xDH) functionals. The results are also presented for some challenging cases, demonstrating the usefulness of the xDH functionals in general.

![Thieno[2',3':4,5]thieno[3,2-b]thieno[2',3':4,5]thieno[2,3-d]thiophene](http://img.cochemist.com/ccimg/124800/124796-79-8.png)
![Thieno[2',3':4,5]thieno[3,2-b]thieno[2',3':4,5]thieno[2,3-d]thiophene](http://img.cochemist.com/ccimg/124800/124796-79-8_b.png)


































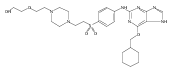
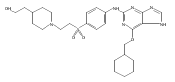

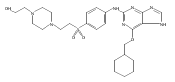



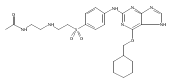








![Benzenesulfonamide, 3-[[6-(cyclohexylmethoxy)-1H-purin-2-yl]amino]-](http://img.cochemist.com/ccimg/722000/721925-82-2.png)
![Benzenesulfonamide, 3-[[6-(cyclohexylmethoxy)-1H-purin-2-yl]amino]-](http://img.cochemist.com/ccimg/722000/721925-82-2_b.png)
![Benzenesulfonamide,4-[2-[6-(cyclohexylmethoxy)-1H-purin-2-yl]hydrazino]-](http://img.cochemist.com/ccimg/722000/721925-72-0.png)
![Benzenesulfonamide,4-[2-[6-(cyclohexylmethoxy)-1H-purin-2-yl]hydrazino]-](http://img.cochemist.com/ccimg/722000/721925-72-0_b.png)
![2-Thiophenesulfonamide,5-[[[6-(cyclohexylmethoxy)-1H-purin-2-yl]amino]methyl]-](http://img.cochemist.com/ccimg/722000/721925-70-8.png)
![2-Thiophenesulfonamide,5-[[[6-(cyclohexylmethoxy)-1H-purin-2-yl]amino]methyl]-](http://img.cochemist.com/ccimg/722000/721925-70-8_b.png)
![Ethanol, 2-[[6-(cyclohexylmethoxy)-1H-purin-2-yl]amino]-](http://img.cochemist.com/ccimg/722000/721925-62-8.png)
![Ethanol, 2-[[6-(cyclohexylmethoxy)-1H-purin-2-yl]amino]-](http://img.cochemist.com/ccimg/722000/721925-62-8_b.png)