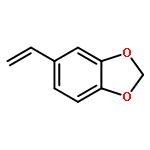
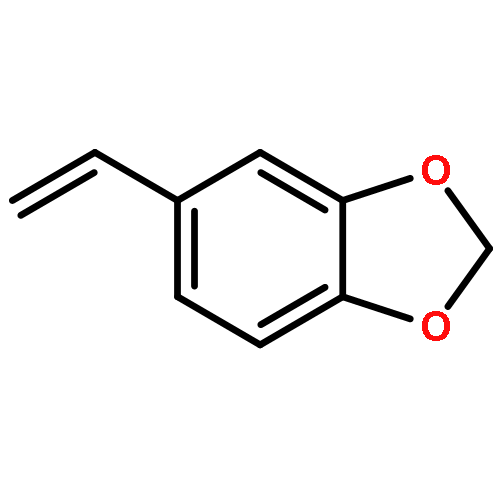
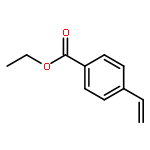
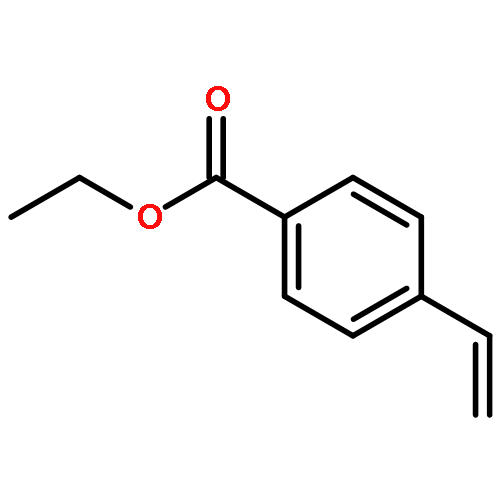
Co-reporter:Sina Rezazadeh, Vijayarajan Devannah, and Donald A. Watson
Journal of the American Chemical Society June 21, 2017 Volume 139(Issue 24) pp:8110-8110
Publication Date(Web):June 8, 2017
DOI:10.1021/jacs.7b04312
Enabled by nickel catalysis, a mild and general catalytic method for C-alkylation of nitroalkanes with unactivated alkyl iodides is described. Compatible with primary, secondary, and tertiary alkyl iodides; and tolerant of a wide range of functional groups, this method allows rapid access to diverse nitroalkanes.
Co-reporter:Andrew P. Cinderella, Bojan Vulovic, and Donald A. Watson
Journal of the American Chemical Society June 14, 2017 Volume 139(Issue 23) pp:7741-7741
Publication Date(Web):June 1, 2017
DOI:10.1021/jacs.7b04364
We report the first example of a silyl-Negishi reaction between secondary zinc organometallics and silicon electrophiles. This palladium-catalyzed process provides direct access to alkyl silanes. The delicate balance of steric and electronic parameters of the employed DrewPhos ligand is paramount to suppressing isomerization and promoting efficient and selective cross-coupling.
Co-reporter:Bojan Vulovic, Andrew P. Cinderella, and Donald A. Watson
ACS Catalysis December 1, 2017 Volume 7(Issue 12) pp:8113-8113
Publication Date(Web):October 30, 2017
DOI:10.1021/acscatal.7b03465
Using a palladium catalyst supported by DrewPhos, the alkylation of monochlorosilanes with primary and secondary alkylmagnesium halides is now possible. Arylation with sterically demanding aromatic magnesium halides is also enabled. This transformation overcomes the high bond strength of the Si–Cl bond (113 kcal/mol) and is a rare example of a transition-metal catalyzed process involving its activation. Because of the availability of both chlorosilanes and organomagnesium halide reagents, this method allows for the preparation of a wide range of alkyl and aryl silanes.Keywords: alkylation; arylation; Grignard reagents; palladium; silane;
Co-reporter:Bojan Vulovic;Donald A. Watson
European Journal of Organic Chemistry 2017 Volume 2017(Issue 34) pp:4996-5009
Publication Date(Web):2017/09/15
DOI:10.1002/ejoc.201700485
Since the discovery of the Heck reaction in the early seventies, the reaction has become a powerful tool in synthetic organic chemistry. By employing heteroatomic instead of traditional carbon electrophiles, the Heck reaction shows an intriguing flexibility. These “hetereoatomic Heck reactions” reinvigorate the area, by offering new routes to highly useful synthetic precursors and structural motifs present in biologically active compounds. This microreview focuses on early developments leading to the heteroatomic-Heck reactions (silyl-Heck, boryl-Heck and intramolecular aza-Heck), current state of the emerging area, as well as that of a few related processes.
Co-reporter:William B. Reid; Jesse J. Spillane; Sarah B. Krause;Donald A. Watson
Journal of the American Chemical Society 2016 Volume 138(Issue 17) pp:5539-5542
Publication Date(Web):April 22, 2016
DOI:10.1021/jacs.6b02914
We report the first example of a boryl-Heck reaction using an electrophilic boron reagent. This palladium-catalyzed process allows for the conversion of terminal alkenes to trans-alkenyl boronic esters using commercially available catecholchloroborane (catBCl). In situ transesterification allows for rapid access to a variety of boronic esters, amides, and other alkenyl boron adducts.
Co-reporter:Scott A. Shuler, Guoyin Yin, Sarah B. Krause, Caroline M. Vesper, and Donald A. Watson
Journal of the American Chemical Society 2016 Volume 138(Issue 42) pp:13830-13833
Publication Date(Web):October 18, 2016
DOI:10.1021/jacs.6b08932
The preparation of unsaturated secondary lactams via the palladium-catalyzed cyclization of O-phenyl hydroxamates onto a pendent alkene is reported. This method provides rapid access to a broad range of lactams that are widely useful building blocks in alkaloid synthesis. Mechanistic studies support an aza-Heck-type pathway.
Co-reporter:Kirk W. Shimkin, Peter G. Gildner, and Donald A. Watson
Organic Letters 2016 Volume 18(Issue 5) pp:988-991
Publication Date(Web):February 11, 2016
DOI:10.1021/acs.orglett.6b00093
Copper catalysis now enables the efficient C-alkylation of nitroalkanes with α-bromonitriles. Using a simple and inexpensive catalyst, this process provides access to β-cyanonitroalkanes. The method is highly tolerant of various functional groups and substitution patterns. These functionally dense products serve as orthogonally masked 1,3-diamines, which can be revealed selectively for access to differentially substituted diamines. These products can also be exploited for the formation of complex cyanoalkenes and 5-aminoisoxazoles.
Co-reporter:Amber A. S. Gietter-Burch, Roxana E. Mitrut, and Donald A. Watson
Organic Letters 2015 Volume 17(Issue 21) pp:5468-5471
Publication Date(Web):October 26, 2015
DOI:10.1021/acs.orglett.5b02832
We have discovered a highly diastereoselective Michael reaction of α-substituted, β-nitrocarbonyl compounds to deliver highly functionalized stereodiads containing fully substituted nitrogen-bearing centers. Good to excellent yields and diastereoselectivities are observed. This transformation is tolerant of various types of carbonyl groups on the nucleophilic partner, as well as a range of unsaturated electrophiles. Mechanistic investigations are consistent with internal hydrogen bonding in the nitroalkane tautomer as the major factor in the control of diastereoselectivity in these transformations.
Co-reporter:Jesse R. McAtee;Sarah B. Krause ;Donald A. Watson
Advanced Synthesis & Catalysis 2015 Volume 357( Issue 10) pp:2317-2321
Publication Date(Web):
DOI:10.1002/adsc.201500436
Co-reporter:Jesse R. McAtee ; Glenn P. A. Yap ;Donald A. Watson
Journal of the American Chemical Society 2014 Volume 136(Issue 28) pp:10166-10172
Publication Date(Web):July 8, 2014
DOI:10.1021/ja505446y
Using rational ligand design, we have developed of a second-generation ligand, bis(3,5-di-tert-butylphenyl)(tert-butyl)phosphine, for the preparation of allylsilanes using the palladium-catalyzed silyl-Heck reaction. This new ligand provides nearly complete suppression of starting material alkene isomerization that was observed with our first-generation catalyst, providing vastly improved yields of allylsilanes from simple alkene starting materials. The studies quantifying the electronic and steric properties of the new ligand are described. Finally, we report an X-ray crystal structure of a palladium complex resulting from the oxidative addition of Me3SiI using an analogous ligand that provides significant insight into the nature of the catalytic system.
Co-reporter:Amber A. S. Gietter, Peter G. Gildner, Andrew P. Cinderella, and Donald A. Watson
Organic Letters 2014 Volume 16(Issue 11) pp:3166-3169
Publication Date(Web):May 28, 2014
DOI:10.1021/ol5014153
Using a simple copper catalyst, the alkylation of nitroalkanes with α-bromocarbonyls is now possible. This method provides a general, functional group tolerant route to β-nitrocarbonyl compounds, including nitro amides, esters, ketones, and aldehydes. The highly sterically dense, functional group rich products from these reactions can be readily elaborated into a range of complex nitrogen-containing molecules, including highly substituted β-amino acids.
Co-reporter:Jesse R. McAtee, Sara E.S. Martin, Andrew P. Cinderella, William B. Reid, Keywan A. Johnson, Donald A. Watson
Tetrahedron 2014 70(27–28) pp: 4250-4256
Publication Date(Web):
DOI:10.1016/j.tet.2014.03.021
Co-reporter:Sara E. S. Martin ;Donald A. Watson
Journal of the American Chemical Society 2013 Volume 135(Issue 36) pp:13330-13333
Publication Date(Web):August 28, 2013
DOI:10.1021/ja407748z
Vinyl silyl ethers and disiloxanes can now be prepared from aryl-substituted alkenes and related substrates using a silyl-Heck reaction. The reaction employs a commercially available catalyst system and mild conditions. This work represents a highly practical means of accessing diverse classes of vinyl silyl ether substrates in an efficient and direct manner with complete regiomeric and geometric selectivity.
Co-reporter:Phuong T. M. Do, Jesse R. McAtee, Donald A. Watson, and Raul F. Lobo
ACS Catalysis 2013 Volume 3(Issue 1) pp:41
Publication Date(Web):December 12, 2012
DOI:10.1021/cs300673b
The reaction of 2,5-dimethylfuran and ethylene to produce p-xylene represents a potentially important route for the conversion of biomass to high-value organic chemicals. Current preparation methods suffer from low selectivity and produce a number of byproducts. Using modern separation and analytical techniques, the structures of many of the byproducts produced in this reaction when HY zeolite is employed as a catalyst have been identified. From these data, a detailed reaction network is proposed, demonstrating that hydrolysis and electrophilic alkylation reactions compete with the desired Diels–Alder/dehydration sequence. This information will allow the rational identification of more selective catalysts and more selective reaction conditions.Keywords: 2,5-dimethylfuran; 2D-NMR; biomass; ethylene; p-xylene; reaction network; zeolite
Co-reporter:Amber A. S. Gietter, Rachel C. Pupillo, Glenn P. A. Yap, Thomas P. Beebe, Joel Rosenthal and Donald A. Watson
Chemical Science 2013 vol. 4(Issue 1) pp:437-443
Publication Date(Web):23 Oct 2012
DOI:10.1039/C2SC21413J
Controlling the molecular topology of electrode–catalyst interfaces is a critical factor in engineering devices with specific electron transport kinetics and catalytic efficiencies. As such, the development of rational methods for the modular construction of tailorable electrode surfaces with robust molecular wires (MWs) exhibiting well-defined molecular topologies, conductivities and morphologies is critical to the evolution and implementation of electrochemical arrays for sensing and catalysis. In response to this need, we have established modular on-surface Sonogashira and Glaser cross-coupling processes to synthetically install arrays of ferrocene-capped MWs onto electrochemically functionalized surfaces. These methods are of comparable convenience and efficiency to more commonly employed Huisgen methods. Furthermore, unlike the Huisgen reaction, this new surface functionalization chemistry generates modified electrodes that do not contain unwanted ancillary metal binding sites, while allowing the bridge between the ferrocenyl moiety and electrode surface to be synthetically tailored. Electrochemical and surface analytical characterization of these platforms demonstrate that the linker topology and connectivity influences the ferrocene redox potential and the kinetics of charge transport at the interface.
Co-reporter:Steven A. Rossi, Kirk W. Shimkin, Qun Xu, Luis M. Mori-Quiroz, and Donald A. Watson
Organic Letters 2013 Volume 15(Issue 9) pp:2314-2317
Publication Date(Web):April 23, 2013
DOI:10.1021/ol401004r
For the first time, a general catalytic procedure for the cross-coupling of primary amides and alkylboronic acids is demonstrated. The key to the success of this reaction was the identification of a mild base (NaOSiMe3) and oxidant (di-tert-butyl peroxide) to promote the copper-catalyzed reaction in high yield. This transformation provides a facile, high-yielding method for the monoalkylation of amides.
Co-reporter:Peter G. Gildner ; Amber A. S. Gietter ; Di Cui ;Donald A. Watson
Journal of the American Chemical Society 2012 Volume 134(Issue 24) pp:9942-9945
Publication Date(Web):June 12, 2012
DOI:10.1021/ja304561c
The C-alkylation of nitroalkanes under mild conditions has been a significant challenge in organic synthesis for more than a century. Herein we report a simple Cu(I) catalyst, generated in situ, that is highly effective for C-benzylation of nitroalkanes using abundant benzyl bromides and related heteroaromatic compounds. This process, which we believe proceeds via a thermal redox mechanism, allows access to a variety of complex nitroalkanes under mild reaction conditions and represents the first step toward the development of a general catalytic system for the alkylation of nitroalkanes.
Co-reporter:Jesse R. McAtee;Sara E. S. Martin;Derek T. Ahneman;Keywan A. Johnson ; Donald A. Watson
Angewandte Chemie 2012 Volume 124( Issue 15) pp:3723-3727
Publication Date(Web):
DOI:10.1002/ange.201200060
Co-reporter:Jesse R. McAtee;Sara E. S. Martin;Derek T. Ahneman;Keywan A. Johnson ; Donald A. Watson
Angewandte Chemie International Edition 2012 Volume 51( Issue 15) pp:3663-3667
Publication Date(Web):
DOI:10.1002/anie.201200060
Co-reporter:Amber A. S. Gietter, Rachel C. Pupillo, Glenn P. A. Yap, Thomas P. Beebe, Joel Rosenthal and Donald A. Watson
Chemical Science (2010-Present) 2013 - vol. 4(Issue 1) pp:NaN443-443
Publication Date(Web):2012/10/23
DOI:10.1039/C2SC21413J
Controlling the molecular topology of electrode–catalyst interfaces is a critical factor in engineering devices with specific electron transport kinetics and catalytic efficiencies. As such, the development of rational methods for the modular construction of tailorable electrode surfaces with robust molecular wires (MWs) exhibiting well-defined molecular topologies, conductivities and morphologies is critical to the evolution and implementation of electrochemical arrays for sensing and catalysis. In response to this need, we have established modular on-surface Sonogashira and Glaser cross-coupling processes to synthetically install arrays of ferrocene-capped MWs onto electrochemically functionalized surfaces. These methods are of comparable convenience and efficiency to more commonly employed Huisgen methods. Furthermore, unlike the Huisgen reaction, this new surface functionalization chemistry generates modified electrodes that do not contain unwanted ancillary metal binding sites, while allowing the bridge between the ferrocenyl moiety and electrode surface to be synthetically tailored. Electrochemical and surface analytical characterization of these platforms demonstrate that the linker topology and connectivity influences the ferrocene redox potential and the kinetics of charge transport at the interface.








![9H-Carbazole, 9-[(1E)-2-(trimethylsilyl)ethenyl]-](http://img.cochemist.com/ccimg/869300/869212-65-7.png)
![9H-Carbazole, 9-[(1E)-2-(trimethylsilyl)ethenyl]-](http://img.cochemist.com/ccimg/869300/869212-65-7_b.png)


![Silane, [(1E)-2-[4-(1,1-dimethylethyl)phenyl]ethenyl]triethyl-](http://img.cochemist.com/ccimg/866400/866363-89-5.png)
![Silane, [(1E)-2-[4-(1,1-dimethylethyl)phenyl]ethenyl]triethyl-](http://img.cochemist.com/ccimg/866400/866363-89-5_b.png)





















![1,3,2-Dioxaborolane, 2-[(1E)-2-cyclohexylethenyl]-4,4,5,5-tetramethyl-](http://img.cochemist.com/ccimg/172600/172512-85-5.png)
![1,3,2-Dioxaborolane, 2-[(1E)-2-cyclohexylethenyl]-4,4,5,5-tetramethyl-](http://img.cochemist.com/ccimg/172600/172512-85-5_b.png)
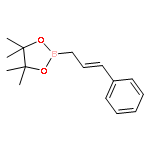
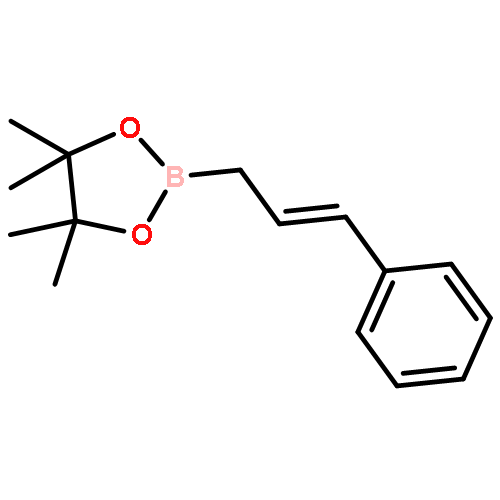
![1,3,2-Dioxaborolane, 4,4,5,5-tetramethyl-2-[(1E)-4-phenyl-1-buten-1-yl]-](/data/chemimg/3759000/172512-84-4.png)
![1,3,2-Dioxaborolane, 4,4,5,5-tetramethyl-2-[(1E)-4-phenyl-1-buten-1-yl]-](/data/chemimg/3759000/172512-84-4_b.png)










![1-Propanol, 3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-2,2-dimethyl-](http://img.cochemist.com/ccimg/118000/117932-70-4.png)
![1-Propanol, 3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-2,2-dimethyl-](http://img.cochemist.com/ccimg/118000/117932-70-4_b.png)
![Silane, [(2E)-3-(4-methoxyphenyl)-2-propenyl]trimethyl-](http://img.cochemist.com/ccimg/111200/111133-17-6.png)
![Silane, [(2E)-3-(4-methoxyphenyl)-2-propenyl]trimethyl-](http://img.cochemist.com/ccimg/111200/111133-17-6_b.png)
![Silane, [2-(3-methoxyphenyl)ethenyl]trimethyl-, (E)-](/data/chemimg/1049900/105281-53-6.png)
![Silane, [2-(3-methoxyphenyl)ethenyl]trimethyl-, (E)-](/data/chemimg/1049900/105281-53-6_b.png)


![Silane, [(2E)-3-cyclohexyl-2-propenyl]trimethyl-](http://img.cochemist.com/ccimg/91900/91899-33-1.png)
![Silane, [(2E)-3-cyclohexyl-2-propenyl]trimethyl-](http://img.cochemist.com/ccimg/91900/91899-33-1_b.png)








![SILANE, TRIMETHYL[(2E)-4-PHENYL-2-BUTENYL]-](http://img.cochemist.com/ccimg/89600/89567-75-9.png)
![SILANE, TRIMETHYL[(2E)-4-PHENYL-2-BUTENYL]-](http://img.cochemist.com/ccimg/89600/89567-75-9_b.png)













![1H-Indole, 3-ethenyl-1-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/65100/65037-62-9.png)
![1H-Indole, 3-ethenyl-1-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/65100/65037-62-9_b.png)




















![Benzene,[(1E)-3-(trimethylsilyl)-1-propen-1-yl]-](http://img.cochemist.com/ccimg/40600/40595-34-4.png)
![Benzene,[(1E)-3-(trimethylsilyl)-1-propen-1-yl]-](http://img.cochemist.com/ccimg/40600/40595-34-4_b.png)







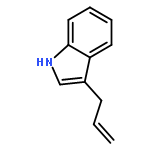
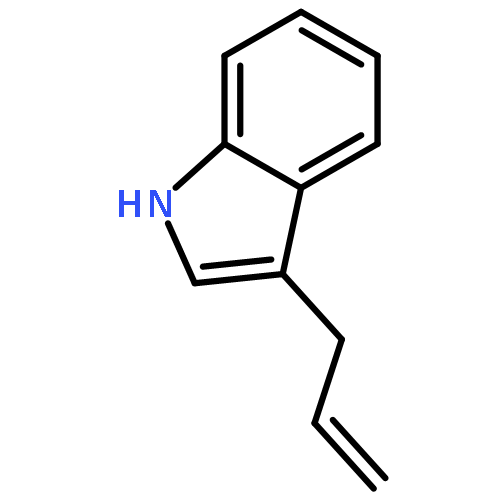




















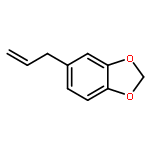
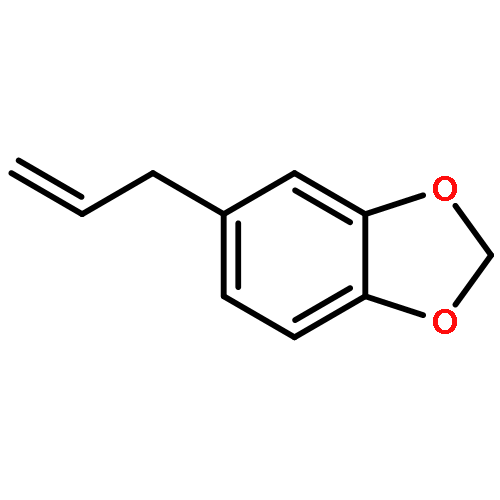


![Silane, [(1E)-2-[4-(1,1-dimethylethyl)phenyl]ethenyl]methyldiphenyl-](http://img.cochemist.com/ccimg/866400/866363-90-8.png)
![Silane, [(1E)-2-[4-(1,1-dimethylethyl)phenyl]ethenyl]methyldiphenyl-](http://img.cochemist.com/ccimg/866400/866363-90-8_b.png)


![Silane, [(1E)-2-(2,4-dimethylphenyl)ethenyl]trimethyl-](http://img.cochemist.com/ccimg/193300/193277-05-3.png)
![Silane, [(1E)-2-(2,4-dimethylphenyl)ethenyl]trimethyl-](http://img.cochemist.com/ccimg/193300/193277-05-3_b.png)




![3-Vinylbicyclo[4.2.0]octa-1,3,5-triene](http://img.cochemist.com/ccimg/99800/99717-87-0.png)
![3-Vinylbicyclo[4.2.0]octa-1,3,5-triene](http://img.cochemist.com/ccimg/99800/99717-87-0_b.png)
![Silane, [(1E)-2-(4-methoxyphenyl)ethenyl]trimethyl-](http://img.cochemist.com/ccimg/76800/76711-42-7.png)
![Silane, [(1E)-2-(4-methoxyphenyl)ethenyl]trimethyl-](http://img.cochemist.com/ccimg/76800/76711-42-7_b.png)


![Benzene,[(1E)-2-(trimethylsilyl)ethenyl]-](http://img.cochemist.com/ccimg/19400/19372-00-0.png)
![Benzene,[(1E)-2-(trimethylsilyl)ethenyl]-](http://img.cochemist.com/ccimg/19400/19372-00-0_b.png)