Co-reporter:Jakob Danielsson, Diana X. Sun, Xiao-Yang Chen, April L. Risinger, Susan L. Mooberry, and Erik J. Sorensen
Organic Letters September 15, 2017 Volume 19(Issue 18) pp:
Publication Date(Web):August 29, 2017
DOI:10.1021/acs.orglett.7b02349
A robust and scalable route to the taccalonolide skeleton starting from trans-androsterone is presented. The synthesis features a cyclic hydroboration carbonylation reaction, which effectively establishes the trans-hydrindane DE ring junction in a remarkable annulation reaction, as well as a Claisen rearrangement and a catalytic Ullmann-type cyclization. This work is part of a larger effort to uncover new clinical candidates from the taccalonolide class of anticancer agents through advances in chemical synthesis.
Co-reporter:
Israel Journal of Chemistry 2017 Volume 57(Issue 3-4) pp:259-269
Publication Date(Web):2017/04/01
DOI:10.1002/ijch.201600115
AbstractThe alkene is a central functional group in organic synthesis. While myriad reliable methods exist to access this moiety from other functionalities, acceptorless dehydrogenation, or the direct synthesis of alkenes from alkanes with hydrogen gas as the sole byproduct, remains a challenging, albeit highly desirable, transformation. This essay provides an account of our recent efforts toward accessing this difficult reaction class, with particular attention paid to the diverse precedents that informed our explorations. This report highlights the benefits of maintaining a broad range of interests, and we hope that it illustrates the vast connectivity between chemical disciplines.
Co-reporter:Dylan J. Abrams;Julian G. West
Chemical Science (2010-Present) 2017 vol. 8(Issue 3) pp:1954-1959
Publication Date(Web):2017/02/28
DOI:10.1039/C6SC04607J
Dehydroformylation, or the reaction of aldehydes to produce alkenes, hydrogen gas, and carbon monoxide, is a powerful transformation that is underdeveloped despite the high industrial importance of the reverse reaction, hydroformylation. Interestingly, nature routinely performs a related transformation, oxidative dehydroformylation, in the biosynthesis of cholesterol and related sterols under mild conditions using base-metal catalysts. In contrast, chemists have recently developed a non-oxidative dehydroformylation method; however, it requires high temperatures and a precious-metal catalyst. Careful study of both approaches has informed our efforts to design a base-metal catalyzed, mild dehydroformylation method that incorporates benefits from each while avoiding several of their respective disadvantages. Importantly, we show that cooperative base metal catalysis presents a powerful, mechanistically unique approach to reactions which are difficult to achieve using conventional catalyst design.
Co-reporter:Xiao-Yang ChenSeyma Ozturk, Erik J. Sorensen
Organic Letters 2017 Volume 19(Issue 5) pp:
Publication Date(Web):February 22, 2017
DOI:10.1021/acs.orglett.7b00161
The first synthesis of substituted fluorenones directly from benzaldehydes and aryl iodides via a Pd(II)-catalyzed C(sp2)–H functionalization cascade is reported. Featuring anthranilic acid as an inexpensive transient directing group, the process is compatible with a variety of benzaldehydes and aryl iodides. A three-step synthesis of the antiviral drug Tilorone was completed in an excellent overall yield (40%), demonstrating the utility of this method.
Co-reporter:Junjia Liu, T. Aaron Bedell, Julian G. West, Erik J. Sorensen
Tetrahedron 2016 Volume 72(Issue 25) pp:3579-3592
Publication Date(Web):23 June 2016
DOI:10.1016/j.tet.2016.01.044
The discovery and development of new anti-infectives is an important contemporary challenge to modern society. This challenge must be met with matching creativity and enthusiasm by chemists to avoid losing the battle with emerging strains of drug-resistant microbes. A series of case studies from our lab are presented, demonstrating our continued efforts in the areas of synthetic design, total synthesis of natural products, structure revision, and bioactive scaffold diversification. Together, these are used to highlight the power and utility of chemical synthesis to uniquely address challenges in the discovery and development of novel antibiotic compounds, particularly within the context of natural products scaffolds.
Co-reporter:Junjia Liu, Maurice A. Marsini, T. Aaron Bedell, Paul J. Reider, Erik J. Sorensen
Tetrahedron 2016 Volume 72(Issue 26) pp:3713-3717
Publication Date(Web):30 June 2016
DOI:10.1016/j.tet.2016.03.039
The hydrindane (bicyclo[4.3.0]nonane) structural motif (1) and related cis-1-hydrindanone skeleton (2) are common substructures in many natural products. Herein, we describe efficient access to substituted cis-1-hydrindanones enabled by a sequence of Michael reactions. A copper-catalyzed intermolecular Michael addition of a cyclic silyl ketene acetal to a β-substituted-α-alkoxycarbonyl-cyclopentenone enables construction of a quaternary center and is followed, after incorporation of an additional Michael acceptor, by a second, intramolecular addition of a nucleophilic β-ketoester. This strategy affords stereoselective access to substituted bicyclic cis-hydrindanone ring systems containing up to three contiguous stereocenters.
Co-reporter:Julian G. West;T. Aaron Bedell ; Erik J. Sorensen
Angewandte Chemie 2016 Volume 128( Issue 31) pp:9069-9073
Publication Date(Web):
DOI:10.1002/ange.201603149
Abstract
The fluorination of unactivated C(sp3)−H bonds remains a desirable and challenging transformation for pharmaceutical, agricultural, and materials scientists. Previous methods for this transformation have used bench-stable fluorine atom sources; however, many still rely on the use of UV-active photocatalysts for the requisite high-energy hydrogen atom abstraction event. Uranyl nitrate hexahydrate is described as a convenient, hydrogen atom abstraction catalyst that can mediate fluorinations of certain alkanes upon activation with visible light.
Co-reporter:Julian G. West;T. Aaron Bedell ; Erik J. Sorensen
Angewandte Chemie International Edition 2016 Volume 55( Issue 31) pp:8923-8927
Publication Date(Web):
DOI:10.1002/anie.201603149
Abstract
The fluorination of unactivated C(sp3)−H bonds remains a desirable and challenging transformation for pharmaceutical, agricultural, and materials scientists. Previous methods for this transformation have used bench-stable fluorine atom sources; however, many still rely on the use of UV-active photocatalysts for the requisite high-energy hydrogen atom abstraction event. Uranyl nitrate hexahydrate is described as a convenient, hydrogen atom abstraction catalyst that can mediate fluorinations of certain alkanes upon activation with visible light.
Co-reporter:Jin Choi ; Hoyoon Park ; Hyun Jung Yoo ; Sinae Kim ; Erik J. Sorensen ;Chulbom Lee
Journal of the American Chemical Society 2014 Volume 136(Issue 28) pp:9918-9921
Publication Date(Web):June 25, 2014
DOI:10.1021/ja505721v
A pericyclic approach for the synthesis of six-membered ring structures is described. The method employs 1,3-dienes with a 1-sulfur substituent in a tandem sequence of Diels–Alder and retro-ene reactions. In this pairing of [4 + 2] cycloaddition and 1,5-sigmatropic rearrangement, 1-sulfenyl-1,3-dienes engage in Diels–Alder reactions with electron-deficient dienophiles. Subsequently, the sulfenyl group of the cycloadducts is oxidized and unmasked to form allylic sulfinic acids, which undergo sterospecific reductive transposition via sulfur dioxide extrusion. The sequence can also include an inverse electron demand Diels–Alder reaction by using a 1-sulfonyl-1,3-diene. This combination of two pericyclic events offers novel stereocontrolled access to cyclohexenes that are inaccessible via a direct [4 + 2] cycloaddition route.
Co-reporter:David A. Siler;Jeffrey D. Mighion ; Erik J. Sorensen
Angewandte Chemie International Edition 2014 Volume 53( Issue 21) pp:5332-5335
Publication Date(Web):
DOI:10.1002/anie.201402335
Abstract
A Robinson annulation, van Leusen homologation, and a desymmetrizing CH oxidation enabled an enantiospecific synthesis of the neurotrophic natural product jiadifenolide. From a pulegone-derived building block, a key propellane intermediate was constructed through the use of simple reagents in a highly diastereoselective fashion. A short series of oxidations of this tricylic framework allowed progression to the natural product.
Co-reporter:David A. Siler;Jeffrey D. Mighion ; Erik J. Sorensen
Angewandte Chemie 2014 Volume 126( Issue 21) pp:5436-5439
Publication Date(Web):
DOI:10.1002/ange.201402335
Abstract
A Robinson annulation, van Leusen homologation, and a desymmetrizing CH oxidation enabled an enantiospecific synthesis of the neurotrophic natural product jiadifenolide. From a pulegone-derived building block, a key propellane intermediate was constructed through the use of simple reagents in a highly diastereoselective fashion. A short series of oxidations of this tricylic framework allowed progression to the natural product.
Co-reporter:Keith P. Reber, S. David Tilley, Cheryl A. Carson, and Erik J. Sorensen
The Journal of Organic Chemistry 2013 Volume 78(Issue 19) pp:9584-9607
Publication Date(Web):September 13, 2013
DOI:10.1021/jo401799f
This account describes a strategy for directly forming three of the six rings found in the polyketide natural product hirsutellone B via a novel cyclization cascade. The key step in our approach comprises two transformations: a large-ring-forming, nucleophilic capture of a transient acylketene and an intramolecular Diels–Alder reaction, both of which occur in tandem through thermolyses of appropriately functionalized, polyunsaturated dioxinones. These thermally induced “double cyclization” cascades generate three new bonds, four contiguous stereocenters, and a significant fraction of the polycyclic architecture of hirsutellone B. The advanced macrolactam and macrolactone intermediates that were synthesized by this process possess key features of the hirsutellone framework, including the stereochemically dense decahydrofluorene core and the strained para-cyclophane ring. However, attempts to complete the carbon skeleton of hirsutellone B via transannular carbon–carbon bond formation were undermined by competitive O-alkylation reactions. This account also documents how we adapted to this undesired outcome through an evaluation of several distinct strategies for synthesis, as well as our eventual achievement of a formal total synthesis of hirsutellone B.
Co-reporter:Jimin Kim, John S. Schneekloth and Erik J. Sorensen
Chemical Science 2012 vol. 3(Issue 9) pp:2849-2852
Publication Date(Web):04 Jul 2012
DOI:10.1039/C2SC20669B
A synthesis of 11-methoxy mitragynine pseudoindoxyl, a new member of the mitragynine class of opioid agonists, from a derivative of the Geissman–Waiss lactone is described. An internal attack of an electron-rich aromatic ring on an electrophilic nitrilium ion and a late-stage construction of the functionalized piperidine ring by the method of reductive cyclization are the pivotal transformations; both ring annulations proceed in a highly diastereoselective fashion. The construction of substituted indoxyl frameworks by the interrupted Ugi method offers an attractive alternative to the strategy of oxidatively rearranging indoles.
Co-reporter:Stephen D. Lotesta, Junjia Liu, Emma V. Yates, Inna Krieger, James C. Sacchettini, Joel S. Freundlich and Erik J. Sorensen
Chemical Science 2011 vol. 2(Issue 7) pp:1258-1261
Publication Date(Web):14 Apr 2011
DOI:10.1039/C1SC00116G
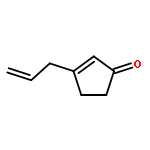
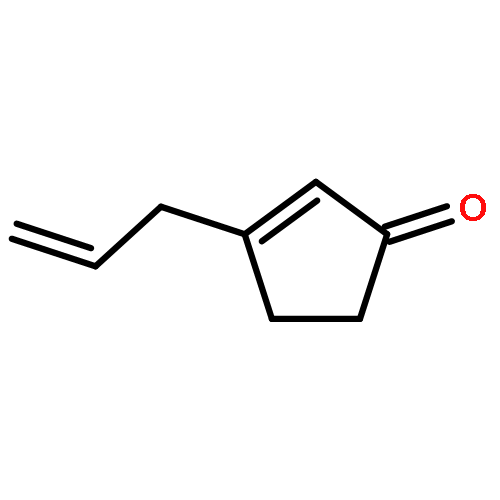
New pleuromutilin-like compounds were synthesized in approximately 11 steps from 3-allylcyclopent-2-enone by a strategy featuring sequential carbonyl addition reactions. Several analogs possessing the C14 tiamulin ester side chain displayed activity in a Mycobacterium tuberculosis mc27000 assay. The results described herein provide a basis for further efforts to expand the structural and stereochemical diversity of the pleuromutilin class of bacterial protein synthesis inhibitors through advances in chemical synthesis.
Co-reporter:Junjia Liu, Stephen D. Lotesta and Erik J. Sorensen
Chemical Communications 2011 vol. 47(Issue 5) pp:1500-1502
Publication Date(Web):16 Nov 2010
DOI:10.1039/C0CC04077K
Two syntheses of the tricyclic carbon skeleton of pleuromutilin are reported. Diastereoselective 1,4-conjugate additions were used to elaborate bicyclic precursors at an early stage of each route, while ring-forming olefin metatheses were executed to complete the pleuromutilin carbon framework. The congeners prepared are appropriately functionalized to enable access to diverse pleuromutilin analogues.
Co-reporter:Carlos A. Guerrero and Erik J. Sorensen
Organic Letters 2011 Volume 13(Issue 19) pp:5164-5167
Publication Date(Web):September 6, 2011
DOI:10.1021/ol2020362
A concise, stereocontrolled synthesis of the citrinadin B core architecture from scalemic, readily available starting materials is disclosed. Highlights include ready access to both cyclic tryptophan tautomer and trans-2,6-disubstituted piperidine fragments, an efficient, stereoretentive mixed Claisen acylation for the coupling of these halves, and further diastereoselective carbonyl addition and oxidative rearrangement for assembly of the core.
Co-reporter:Dr. Hao Xie;Dr. Glenn M. Sammis;Dr. Eric M. Flamme;Christina M. Kraml ;Dr. Erik J. Sorensen
Chemistry - A European Journal 2011 Volume 17( Issue 40) pp:11131-11134
Publication Date(Web):
DOI:10.1002/chem.201102394
Co-reporter:Jillian E. Spangler, Cheryl A. Carson and Erik J. Sorensen
Chemical Science 2010 vol. 1(Issue 2) pp:202-205
Publication Date(Web):11 Jun 2010
DOI:10.1039/C0SC00284D
A stereodivergent synthesis of the [3.3.1] bicyclic core of edaxadiene was completed utilizing a key intramolecular oxidative ketone allylation. Significant discrepancies between the spectroscopic data obtained for the synthetic construct and the natural isolate raised questions about the structural assignment of edaxadiene. A subsequent structural reassignment was validated by completion of a total synthesis of the correct structure of the natural product.
Co-reporter:Brent D. Chandler, Jason T. Roland, Yukai Li and Erik J. Sorensen
Organic Letters 2010 Volume 12(Issue 12) pp:2746-2749
Publication Date(Web):May 19, 2010
DOI:10.1021/ol100845z
When ketones flanked on both sides by nucleophilic atoms react with Seebach’s nitropropenyl pivaloate reagent, direct couplings take place to give two new ring systems and three bonds. Cis-ring fusions are observed in unions leading to 5,5-, 5,6-, and 6,6-bicycles. Densely functionalized and rigid frameworks may be rapidly formed by the chemistry described herein.
Co-reporter:Maurice A. Marsini, Paul J. Reider, and Erik J. Sorensen
The Journal of Organic Chemistry 2010 Volume 75(Issue 21) pp:7479-7482
Publication Date(Web):October 7, 2010
DOI:10.1021/jo1015807
An efficient four-step synthesis of PA-824, a promising antituberculosis drug candidate, has been developed. This concise approach offers significant improvements over the synthetic route currently used for large-scale production.
Co-reporter:Keith P. Reber, S. David Tilley and Erik J. Sorensen
Chemical Society Reviews 2009 vol. 38(Issue 11) pp:3022-3034
Publication Date(Web):02 Sep 2009
DOI:10.1039/B912599J
The reactive intermediates known as acylketenes exhibit a rich chemistry and have been extensively utilized for many types of inter- and intramolecular bond-forming reactions within the field of organic synthesis. Characteristic reactions of acylketenes include cycloadditions, carbon–carbon bond-forming reactions, and nucleophilic capture with alcohols or amines to give β-keto acid derivatives. In particular, the intramolecular capture of acylketene intermediates with pendant nucleophiles represents a powerful method for forming both medium-sized rings and macrocycles, often in high yield. This tutorial review examines the history, generation, and reactivity of acylketenes with a special focus on their applications in the synthesis of natural products.
Co-reporter:Jessica L. Frie, Christopher S. Jeffrey and Erik J. Sorensen
Organic Letters 2009 Volume 11(Issue 23) pp:5394-5397
Publication Date(Web):October 30, 2009
DOI:10.1021/ol902168g
A stereocontrolled synthesis of a complex pentacycle embodying the molecular architecture of the cortistatin class of natural products was achieved from the (+)-Hajos−Parrish ketone. The cornerstone of our approach is a hypervalent iodine induced tandem intramolecular oxidative dearomatization and nitrile oxide cycloaddition. The manner in which these ring formations were orchestrated has yielded a rather concise strategy for synthesis.
Co-reporter:John S. Schneekloth, Jr., Jimin Kim, Erik J. Sorensen
Tetrahedron 2009 65(16) pp: 3096-3101
Publication Date(Web):
DOI:10.1016/j.tet.2008.08.055
Co-reporter:Jillian E. Spangler, Erik J. Sorensen
Tetrahedron 2009 65(33) pp: 6739-6745
Publication Date(Web):
DOI:10.1016/j.tet.2009.06.063
Co-reporter:Christoph W. Zapf, Bryce A. Harrison, Carmen Drahl,Erik J. Sorensen
Angewandte Chemie International Edition 2005 44(40) pp:6533-6537
Publication Date(Web):
DOI:10.1002/anie.200502119
Co-reporter:Christoph W. Zapf Dr.;Bryce A. Harrison Dr.;Carmen Drahl Dr.
Angewandte Chemie 2005 Volume 117(Issue 40) pp:
Publication Date(Web):15 SEP 2005
DOI:10.1002/ange.200502119
Eine effiziente und hoch diastereoselektive intramolekulare Diels-Alder-Reaktion bildet die Grundlage einer knappen asymmetrischen Synthese des hoch wirksamen antibakteriellen Naturstoffs Abyssomicin C (siehe Formel). Die komplexe Zielstruktur wurde auf drei Fragmente zurückgeführt, die durch zwei Carbonyladditionen miteinander verknüpft wurden.
Co-reporter:Carmen Drahl;Benjamin F. Cravatt
Angewandte Chemie 2005 Volume 117(Issue 36) pp:
Publication Date(Web):8 SEP 2005
DOI:10.1002/ange.200500900
In der Post-Genom-Ära steht man vor der gewaltigen Aufgabe, die Strukturen und Funktionen zehntausender codierter Proteine zuzuweisen. Zur Analyse der Proteinfunktion werden neue Technologien mit großer Bandbreite entwickelt, etwa die aktivitätsbasierte Proteinanalyse (activity-based protein profiling, ABPP). Bei diesem Verfahren sollen auf das aktive Zentrum gerichtete chemische Sonden entwickelt werden, um die Enzyme in ganzen Proteomen zu analysieren. Derartige chemische Proteomtechnologien können von proteinreaktiven Naturstoffen lernen, die Enzyme der unterschiedlichsten Klassen durch ein bemerkenswert vielfältiges Spektrum an Mechanismen kovalent modifizieren und somit eine reiche Quelle an Konzepten für die Entwicklung gegen aktive Zentren gerichteter Proteomsonden bilden. Hier werden wir anhand unterschiedlicher Naturstoffe zeigen, wie ihre Wirkmechanismen zum Fortschritt chemischer Proteomtechnologien wie ABPP beigetragen haben.
Co-reporter:Carmen Drahl;Benjamin F. Cravatt
Angewandte Chemie International Edition 2005 Volume 44(Issue 36) pp:
Publication Date(Web):8 SEP 2005
DOI:10.1002/anie.200500900
Researchers in the post-genome era are confronted with the daunting task of assigning structure and function to tens of thousands of encoded proteins. To realize this goal, new technologies are emerging for the analysis of protein function on a global scale, such as activity-based protein profiling (ABPP), which aims to develop active site-directed chemical probes for enzyme analysis in whole proteomes. For the pursuit of such chemical proteomic technologies, it is helpful to derive inspiration from protein-reactive natural products. Natural products use a remarkably diverse set of mechanisms to covalently modify enzymes from distinct mechanistic classes, thus providing a wellspring of chemical concepts that can be exploited for the design of active-site-directed proteomic probes. Herein, we highlight several examples of protein-reactive natural products and illustrate how their mechanisms of action have influenced and continue to shape the progression of chemical proteomic technologies like ABPP.
Co-reporter:Edward A. Anderson Dr.;Erik J. Alexanian Dr.
Angewandte Chemie 2004 Volume 116(Issue 15) pp:
Publication Date(Web):30 MAR 2004
DOI:10.1002/ange.200353129
Eine rhodiumkatalysierte Alkincyclotrimerisierung, elektrocyclische Dominoreaktionen und eine hydroxygelenkte Dihydroxylierung sind Schlüsselschritte in einer effizienten Synthese des bioaktiven Furanosteroids Viridin (1) aus einem einfachen acyclischen Triin.
Co-reporter:Edward A. Anderson Dr.;Erik J. Alexanian Dr.
Angewandte Chemie International Edition 2004 Volume 43(Issue 15) pp:
Publication Date(Web):30 MAR 2004
DOI:10.1002/anie.200353129
A rhodium-catalyzed alkyne cyclotrimerization, domino electrocyclic reactions, and a hydroxy-directed dihydroxylation are key steps in an efficient synthesis of the bioactive furanosteroid viridin (1) from a simple acyclic triyne.
Co-reporter:Lucy M. Stark;Klaus Pekari
PNAS 2004 101 (33 ) pp:12064-12066
Publication Date(Web):2004-08-17
DOI:10.1073/pnas.0402563101
Substantial structural transformations of much use in complex chemical synthesis can be achieved by channeling the reactivity
of highly unsaturated molecules. This report describes the direct conversion of an acyclic polyunsaturated diketone to the
HIV-1 Rev/Rev-responsive element inhibitor harziphilone under mild reaction conditions.
Co-reporter:Lucy M. Stark;Klaus Pekari
PNAS 2004 101 (33 ) pp:12064-12066
Publication Date(Web):2004-08-17
DOI:10.1073/pnas.0402563101
Substantial structural transformations of much use in complex chemical synthesis can be achieved by channeling the reactivity
of highly unsaturated molecules. This report describes the direct conversion of an acyclic polyunsaturated diketone to the
HIV-1 Rev/Rev-responsive element inhibitor harziphilone under mild reaction conditions.
Co-reporter:Gregory C. Adam;Christopher D. Verwal ;Benjamin F. Cravatt
Angewandte Chemie 2003 Volume 115(Issue 44) pp:
Publication Date(Web):11 NOV 2003
DOI:10.1002/ange.200352576
Durch Aktivitätsprofiling und Reportergruppenmarkierung wurde die Carboxylesterase-1 (CE-1) als spezifisches Target des natürlichen Lactons (−)-FR182877 identifiziert (siehe Schema; tag Reportergruppe). Die gezielte Modifizierung des Naturstoffs kann nun zur Entwicklung aktiverer CE-1-Inhibitoren genutzt werden.
Co-reporter:Gregory C. Adam;Christopher D. Verwal ;Benjamin F. Cravatt
Angewandte Chemie International Edition 2003 Volume 42(Issue 44) pp:
Publication Date(Web):11 NOV 2003
DOI:10.1002/anie.200352576
Activity-based profiling and reporter-group-tagged compounds were used to identify carboxylesterase-1 (CE-1) as a specific target of the natural product (−)-FR182877 (see scheme). Now that a target is known, specific modification of the lactone natural product may lead to even more effective inhibitors of CE-1.
Co-reporter:Dylan J. Abrams, Julian G. West and Erik J. Sorensen
Chemical Science (2010-Present) 2017 - vol. 8(Issue 3) pp:NaN1959-1959
Publication Date(Web):2016/11/09
DOI:10.1039/C6SC04607J
Dehydroformylation, or the reaction of aldehydes to produce alkenes, hydrogen gas, and carbon monoxide, is a powerful transformation that is underdeveloped despite the high industrial importance of the reverse reaction, hydroformylation. Interestingly, nature routinely performs a related transformation, oxidative dehydroformylation, in the biosynthesis of cholesterol and related sterols under mild conditions using base-metal catalysts. In contrast, chemists have recently developed a non-oxidative dehydroformylation method; however, it requires high temperatures and a precious-metal catalyst. Careful study of both approaches has informed our efforts to design a base-metal catalyzed, mild dehydroformylation method that incorporates benefits from each while avoiding several of their respective disadvantages. Importantly, we show that cooperative base metal catalysis presents a powerful, mechanistically unique approach to reactions which are difficult to achieve using conventional catalyst design.
Co-reporter:Keith P. Reber, S. David Tilley and Erik J. Sorensen
Chemical Society Reviews 2009 - vol. 38(Issue 11) pp:NaN3034-3034
Publication Date(Web):2009/09/02
DOI:10.1039/B912599J
The reactive intermediates known as acylketenes exhibit a rich chemistry and have been extensively utilized for many types of inter- and intramolecular bond-forming reactions within the field of organic synthesis. Characteristic reactions of acylketenes include cycloadditions, carbon–carbon bond-forming reactions, and nucleophilic capture with alcohols or amines to give β-keto acid derivatives. In particular, the intramolecular capture of acylketene intermediates with pendant nucleophiles represents a powerful method for forming both medium-sized rings and macrocycles, often in high yield. This tutorial review examines the history, generation, and reactivity of acylketenes with a special focus on their applications in the synthesis of natural products.
Co-reporter:Jillian E. Spangler, Cheryl A. Carson and Erik J. Sorensen
Chemical Science (2010-Present) 2010 - vol. 1(Issue 2) pp:NaN205-205
Publication Date(Web):2010/06/11
DOI:10.1039/C0SC00284D
A stereodivergent synthesis of the [3.3.1] bicyclic core of edaxadiene was completed utilizing a key intramolecular oxidative ketone allylation. Significant discrepancies between the spectroscopic data obtained for the synthetic construct and the natural isolate raised questions about the structural assignment of edaxadiene. A subsequent structural reassignment was validated by completion of a total synthesis of the correct structure of the natural product.
Co-reporter:Stephen D. Lotesta, Junjia Liu, Emma V. Yates, Inna Krieger, James C. Sacchettini, Joel S. Freundlich and Erik J. Sorensen
Chemical Science (2010-Present) 2011 - vol. 2(Issue 7) pp:NaN1261-1261
Publication Date(Web):2011/04/14
DOI:10.1039/C1SC00116G
New pleuromutilin-like compounds were synthesized in approximately 11 steps from 3-allylcyclopent-2-enone by a strategy featuring sequential carbonyl addition reactions. Several analogs possessing the C14 tiamulin ester side chain displayed activity in a Mycobacterium tuberculosis mc27000 assay. The results described herein provide a basis for further efforts to expand the structural and stereochemical diversity of the pleuromutilin class of bacterial protein synthesis inhibitors through advances in chemical synthesis.
Co-reporter:Jimin Kim, John S. Schneekloth and Erik J. Sorensen
Chemical Science (2010-Present) 2012 - vol. 3(Issue 9) pp:NaN2852-2852
Publication Date(Web):2012/07/04
DOI:10.1039/C2SC20669B
A synthesis of 11-methoxy mitragynine pseudoindoxyl, a new member of the mitragynine class of opioid agonists, from a derivative of the Geissman–Waiss lactone is described. An internal attack of an electron-rich aromatic ring on an electrophilic nitrilium ion and a late-stage construction of the functionalized piperidine ring by the method of reductive cyclization are the pivotal transformations; both ring annulations proceed in a highly diastereoselective fashion. The construction of substituted indoxyl frameworks by the interrupted Ugi method offers an attractive alternative to the strategy of oxidatively rearranging indoles.
Co-reporter:Junjia Liu, Stephen D. Lotesta and Erik J. Sorensen
Chemical Communications 2011 - vol. 47(Issue 5) pp:NaN1502-1502
Publication Date(Web):2010/11/16
DOI:10.1039/C0CC04077K
Two syntheses of the tricyclic carbon skeleton of pleuromutilin are reported. Diastereoselective 1,4-conjugate additions were used to elaborate bicyclic precursors at an early stage of each route, while ring-forming olefin metatheses were executed to complete the pleuromutilin carbon framework. The congeners prepared are appropriately functionalized to enable access to diverse pleuromutilin analogues.

![Rhodium, tetrakis[m-[(aR)-1,3-dihydro-1,3-dioxo-a-tricyclo[3.3.1.13,7]dec-1-yl-2H-isoindole-2-acetato-kO2:kO2']]di-, (Rh-Rh)](http://img.cochemist.com/ccimg/909400/909393-65-3.png)
![Rhodium, tetrakis[m-[(aR)-1,3-dihydro-1,3-dioxo-a-tricyclo[3.3.1.13,7]dec-1-yl-2H-isoindole-2-acetato-kO2:kO2']]di-, (Rh-Rh)](http://img.cochemist.com/ccimg/909400/909393-65-3_b.png)
![Rhodium, tetrakis[m-[(aS)-1,3-dihydro-1,3-dioxo-a-tricyclo[3.3.1.13,7]dec-1-yl-2H-isoindole-2-acetato-kO2:kO2']]di-, (Rh-Rh)](http://img.cochemist.com/ccimg/909400/909389-99-7.png)
![Rhodium, tetrakis[m-[(aS)-1,3-dihydro-1,3-dioxo-a-tricyclo[3.3.1.13,7]dec-1-yl-2H-isoindole-2-acetato-kO2:kO2']]di-, (Rh-Rh)](http://img.cochemist.com/ccimg/909400/909389-99-7_b.png)
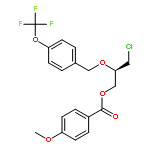
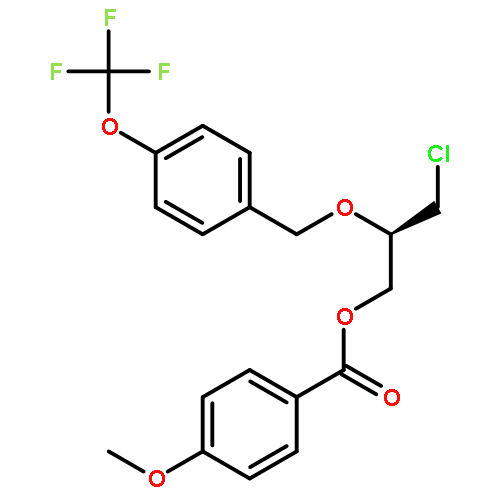
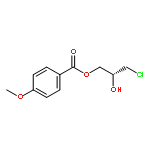
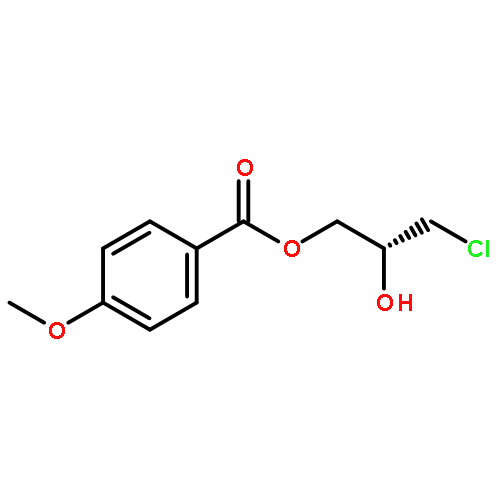








![2-Oxazolidinone, 4-[(acetyloxy)methyl]-3-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/855500/855422-09-2.png)
![2-Oxazolidinone, 4-[(acetyloxy)methyl]-3-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/855500/855422-09-2_b.png)
![2-PYRROLIDINONE, 5-[(ACETYLOXY)METHYL]-1-[(4-METHYLPHENYL)SULFONYL]-](http://img.cochemist.com/ccimg/855500/855422-08-1.png)
![2-PYRROLIDINONE, 5-[(ACETYLOXY)METHYL]-1-[(4-METHYLPHENYL)SULFONYL]-](http://img.cochemist.com/ccimg/855500/855422-08-1_b.png)


![Rhodium, bis[m-[a,a,a',a'-tetramethyl-1,3-benzenedipropanoato(2-)-kO1,kO'3:kO3,kO'1]]di-, (Rh-Rh)](http://img.cochemist.com/ccimg/819100/819050-89-0.png)
![Rhodium, bis[m-[a,a,a',a'-tetramethyl-1,3-benzenedipropanoato(2-)-kO1,kO'3:kO3,kO'1]]di-, (Rh-Rh)](http://img.cochemist.com/ccimg/819100/819050-89-0_b.png)


![(1R,3s,5S)-3-(3-Isopropyl-5-methyl-4H-1,2,4-triazol-4-yl)-8-azabicyclo[3.2.1]octane](http://img.cochemist.com/ccimg/423200/423165-07-5.png)
![(1R,3s,5S)-3-(3-Isopropyl-5-methyl-4H-1,2,4-triazol-4-yl)-8-azabicyclo[3.2.1]octane](http://img.cochemist.com/ccimg/423200/423165-07-5_b.png)
![4-Pentenamide, N-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/413200/413188-48-4.png)
![4-Pentenamide, N-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/413200/413188-48-4_b.png)











![2-Oxazolidinone, 3-[(2E)-1-oxo-2-pentenyl]-](http://img.cochemist.com/ccimg/192400/192379-68-3.png)
![2-Oxazolidinone, 3-[(2E)-1-oxo-2-pentenyl]-](http://img.cochemist.com/ccimg/192400/192379-68-3_b.png)


![5H-Imidazo[2,1-b][1,3]oxazine, 6,7-dihydro-2-nitro-6-[[4-(trifluoromethoxy)phenyl]methoxy]-, (6S)-](http://img.cochemist.com/ccimg/187300/187235-37-6.png)
![5H-Imidazo[2,1-b][1,3]oxazine, 6,7-dihydro-2-nitro-6-[[4-(trifluoromethoxy)phenyl]methoxy]-, (6S)-](http://img.cochemist.com/ccimg/187300/187235-37-6_b.png)





![Carbamic acid, [(4-methylphenyl)sulfonyl]-, 3-butenyl ester](http://img.cochemist.com/ccimg/140000/139951-63-6.png)
![Carbamic acid, [(4-methylphenyl)sulfonyl]-, 3-butenyl ester](http://img.cochemist.com/ccimg/140000/139951-63-6_b.png)




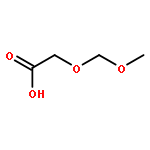
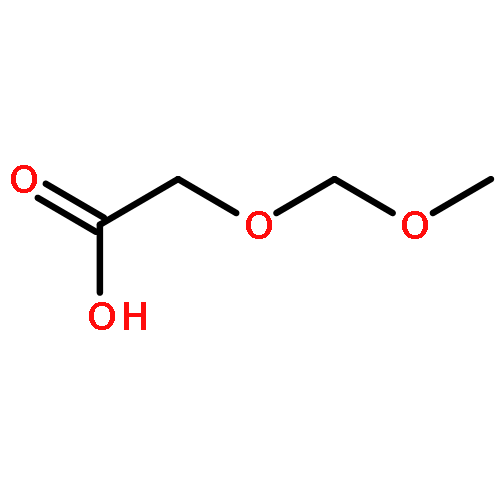
![Silane, [(2,2-dimethyl-4-methylene-4H-1,3-dioxin-6-yl)oxy]trimethyl-](http://img.cochemist.com/ccimg/130600/130573-43-2.png)
![Silane, [(2,2-dimethyl-4-methylene-4H-1,3-dioxin-6-yl)oxy]trimethyl-](http://img.cochemist.com/ccimg/130600/130573-43-2_b.png)
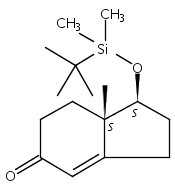

![2-Butenal, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, (2E)-](http://img.cochemist.com/ccimg/119200/119125-30-3.png)
![2-Butenal, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, (2E)-](http://img.cochemist.com/ccimg/119200/119125-30-3_b.png)
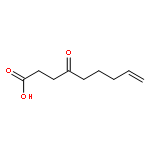


![Phenol, 4-[[[(1,1-dimethylethyl)diphenylsilyl]oxy]methyl]-](http://img.cochemist.com/ccimg/113100/113068-75-0.png)
![Phenol, 4-[[[(1,1-dimethylethyl)diphenylsilyl]oxy]methyl]-](http://img.cochemist.com/ccimg/113100/113068-75-0_b.png)




![2-Oxazolidinone, 3-[(2E)-1-oxo-3-phenyl-2-propenyl]-](http://img.cochemist.com/ccimg/109300/109299-93-6.png)
![2-Oxazolidinone, 3-[(2E)-1-oxo-3-phenyl-2-propenyl]-](http://img.cochemist.com/ccimg/109300/109299-93-6_b.png)
![3-[(e)-2-butenoyl]-1,3-oxazolidin-2-one](http://img.cochemist.com/ccimg/109300/109299-92-5.png)
![3-[(e)-2-butenoyl]-1,3-oxazolidin-2-one](http://img.cochemist.com/ccimg/109300/109299-92-5_b.png)
![Magnesium, bromo[4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]phenyl]-](/data/chemimg/1664800/107539-52-6.png)
![Magnesium, bromo[4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]phenyl]-](/data/chemimg/1664800/107539-52-6_b.png)
![1H-Cyclopenta[c]furan-5(3H)-one, 3a,4-dihydro-3a-methyl-](http://img.cochemist.com/ccimg/104800/104728-34-9.png)
![1H-Cyclopenta[c]furan-5(3H)-one, 3a,4-dihydro-3a-methyl-](http://img.cochemist.com/ccimg/104800/104728-34-9_b.png)
![11H-Benzo[b]fluoren-11-one, 2-methoxy-](http://img.cochemist.com/ccimg/100600/100516-15-2.png)
![11H-Benzo[b]fluoren-11-one, 2-methoxy-](http://img.cochemist.com/ccimg/100600/100516-15-2_b.png)



![Silane,(1,1-dimethylethyl)[(3-methoxy-1-methylene-2-propenyl)oxy]dimethyl-](http://img.cochemist.com/ccimg/71800/71735-01-8.png)
![Silane,(1,1-dimethylethyl)[(3-methoxy-1-methylene-2-propenyl)oxy]dimethyl-](http://img.cochemist.com/ccimg/71800/71735-01-8_b.png)



![Benzenesulfonamide, 4-methyl-N-[2-(2-propenyl)phenyl]-](http://img.cochemist.com/ccimg/51400/51315-69-6.png)
![Benzenesulfonamide, 4-methyl-N-[2-(2-propenyl)phenyl]-](http://img.cochemist.com/ccimg/51400/51315-69-6_b.png)
![2-[3-(1-CYANOETHYL)-5-(1H-1,2,4-TRIAZOL-1-YLMETHYL)PHENYL]-2-METH<WBR />YLPROPANENITRILE](http://img.cochemist.com/ccimg/37500/37428-58-3.png)
![2-[3-(1-CYANOETHYL)-5-(1H-1,2,4-TRIAZOL-1-YLMETHYL)PHENYL]-2-METH<WBR />YLPROPANENITRILE](http://img.cochemist.com/ccimg/37500/37428-58-3_b.png)
![Bicyclo[2.2.1]hept-5-ene-2-carboxaldehyde, 2-methyl-, (1R,2R,4R)-rel-](/data/chemimg/1204300/34618-31-0.png)
![Bicyclo[2.2.1]hept-5-ene-2-carboxaldehyde, 2-methyl-, (1R,2R,4R)-rel-](/data/chemimg/1204300/34618-31-0_b.png)
![Bicyclo[2.2.1]hept-5-ene-2-carboxaldehyde, 2-methyl-, (1R,2S,4R)-rel-](/data/chemimg/1200600/34618-30-9.png)
![Bicyclo[2.2.1]hept-5-ene-2-carboxaldehyde, 2-methyl-, (1R,2S,4R)-rel-](/data/chemimg/1200600/34618-30-9_b.png)
]chloro(pyridine)-, (OC-6-42)-](http://img.cochemist.com/ccimg/23300/23295-32-1.png)
]chloro(pyridine)-, (OC-6-42)-](http://img.cochemist.com/ccimg/23300/23295-32-1_b.png)
![Benzene,[(1S)-1-isocyanoethyl]-](http://img.cochemist.com/ccimg/21900/21872-32-2.png)
![Benzene,[(1S)-1-isocyanoethyl]-](http://img.cochemist.com/ccimg/21900/21872-32-2_b.png)
![Carbamic acid, [(4-methylphenyl)sulfonyl]-, 2-propenyl ester](http://img.cochemist.com/ccimg/18400/18303-03-2.png)
![Carbamic acid, [(4-methylphenyl)sulfonyl]-, 2-propenyl ester](http://img.cochemist.com/ccimg/18400/18303-03-2_b.png)


























![Bicyclo[2.2.1]hept-2-ene,5-methylene-](http://img.cochemist.com/ccimg/700/694-91-7.png)
![Bicyclo[2.2.1]hept-2-ene,5-methylene-](http://img.cochemist.com/ccimg/700/694-91-7_b.png)
![Magnesium, bromo[3-(1,3-dioxolan-2-yl)propyl]-](http://img.cochemist.com/ccimg/91100/91083-05-5.png)
![Magnesium, bromo[3-(1,3-dioxolan-2-yl)propyl]-](http://img.cochemist.com/ccimg/91100/91083-05-5_b.png)