Co-reporter:Yajing Peng, Yuqing Ye, Jianyong Liu, Weipeng Lai
Chemical Physics Letters 2016 Volume 647() pp:85-88
Publication Date(Web):March 2016
DOI:10.1016/j.cplett.2016.01.038
Highlights
- •
The two formation mechanisms of 3,4-dinitrofuroxan (DNFO) via nitration of furoxan with two typical nitration reagents BF4NO2 and HNO3 are clarified.
- •
The formation mechanisms are mainly the electrophilic substitutions of nitronium ions from the nitration reagents and the abstractions of protons in the complex intermediates.
- •
BF4− is shown to be a better proton abstracter than HNO3 and H2O due to its no barrier combination with H+.
- •
Chloroform is a feasible solvent and heating properly is necessary for the two reactions.
Co-reporter: Ya-Jing Peng;Yan-Xue Jiang;Dr. Xie Peng; Jian-Yong Liu; Wei-Peng Lai
ChemPhysChem 2016 Volume 17( Issue 4) pp:541-547
Publication Date(Web):
DOI:10.1002/cphc.201500968
Abstract
The reaction pathway of the formation of 3,4-dinitrofuroxan from glyoxime is theoretically investigated under experimental conditions with 25 % nitric acid and dinitrogentetroxide reagents to clarify the mechanism of formation of a furoxan ring by glyoxime. The geometric configurations of minima and transition-state species are optimized at the (U)B3LYP/6-311++G** level. The CCSD(T) and CASSCF(10e,8o)/CASSCF(9e,8o) single-point energy corrections at the same level are performed on top of the optimized geometries. A subsequent dynamic correlation by using NEVPT2/6-311++G**-level single-point energy calculations based on the CASSCF results is also performed to obtain accurate energy values. The formation reaction is analyzed from two processes: glyoxime nitration and 3,4-dinitroglyoxime (nitration product) oxidative cyclization. Calculation results indicate that the electrophilic substitution of nitronium ions from the protonated HNO3 and the abstraction of hydrogen ions by HNO3 molecules are requisites of glyoxime nitration. The formation of a furoxan ring from 3,4-dinitroglyoxime involves two possible mechanisms: 1) oxydehydrogenation by NO2 molecules and the subsequent torsion of NO radical groups to form a ring and 2) the alternation of dehydrogenation and cyclization. The intermediates and transition states in both routes exhibit monoradical and diradical characteristics. Singlet and triplet reactions are considered for the diradical species. Results show that the singlet reaction mechanism is more favorable for cyclization than the triplet reaction. The formation of a furoxan ring from oxime is in accordance with the stepwise intermolecular dehydrogenation and intramolecular torsion to the ring.
Co-reporter:Jinfeng Zhao, Junsheng Chen, Jianyong Liu and Mark R. Hoffmann
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 18) pp:11990-11999
Publication Date(Web):24 Mar 2015
DOI:10.1039/C4CP05651E
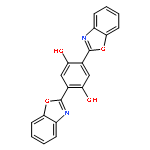
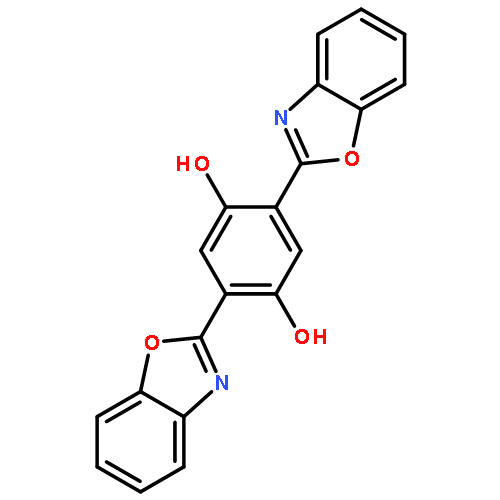
The excited state intramolecular proton transfer (ESIPT) mechanisms of 2-(2-hydroxyphenyl)benzoxazole (HBO), bis-2,5-(2-benzoxazolyl)-hydroquinone (BBHQ) and 2,5-bis(5′-tert-butyl-benzoxazol-2′-yl)hydroquinone (DHBO) have been investigated using time-dependent density functional theory (TDDFT). The calculated vertical excitation energies based on the TDDFT method reproduced the experimental absorption and emission spectra well. Three kinds of stable structures were found on the S1 state potential energy surface (PES). A new ESIPT mechanism that differs from the one proposed previously (Mordzinski et al., Chem. Phys. Lett., 1983, 101, 291. and Lim et al., J. Am. Chem. Soc., 2006, 128, 14542.) is proposed. The new mechanism includes the possibility of simultaneous double proton transfer, or successive single transfers, in addition to the accepted single proton transfer mechanism. Hydrogen bond strengthening in the excited state was based on primary bond lengths, angles, IR vibrational spectra and hydrogen bond energy. Intramolecular charge transfer based on the frontier molecular orbitals (MOs) also supports the proposed mechanism of the ESIPT reaction. To further elucidate the proposed mechanism, reduced dimensionality PESs of the S0 and S1 states were constructed by keeping the O–H distance fixed at a series of values. The potential barrier heights among the local minima on the S1 surface imply competitive single and double proton transfer branches in the mechanism. Based on the new ESIPT mechanism, the observed fluorescence quenching can be satisfactorily explained.
Co-reporter:Jinfeng Zhao;Junsheng Chen;Peng Song;Jianyong Liu
Journal of Cluster Science 2015 Volume 26( Issue 5) pp:1463-1472
Publication Date(Web):2015 September
DOI:10.1007/s10876-014-0830-1
Sulfoxide is an ideal functional group containing S=O for exploring excited-state hydrogen bond dynamics. Benzene–pyrene–sulfoxide (BPS) molecule, as one of the important fluorescent chemosensors, was selected to complete the hydrogen bond dynamics of sulfoxides functional group connecting to methanol (MeOH). The ground-state and excited-state geometric structures were investigated based on density functional theory and the time-dependent density functional theory methods, respectively. The calculated absorption and emission spectra of BPS chemosensor agreed well with the experimental results, demonstrating the theory we adopted is reasonable and effective. The phenomenon of variable-length S=O and H–O bands in the first excited state (S1) as well as variable-short hydrogen bond S=O···H–O demonstrated that the intermolecular hydrogen bond were strengthened. Bathochromic shift stretching vibrational modes of both S=O and H–O regions in the S1 state manifested hydrogen bond were strengthened authentically. In addition, the frontier molecular orbitals (MOs), depicting the nature of the electronically excited states, supported that the S1 state of BPS–MeOH was a local excited state with a π–π* transition, whereas the second excited state was the charge transfer state.
Co-reporter:Jinfeng Zhao, Hongbin Yao, Jianyong Liu, and Mark R. Hoffmann
The Journal of Physical Chemistry A 2015 Volume 119(Issue 4) pp:681-688
Publication Date(Web):January 2, 2015
DOI:10.1021/jp5120459
The excited state intramolecular proton transfer (ESIPT) mechanisms of 1,8-dihydroxydibenzo[a,h]phenazine (DHBP) in toluene solvent have been investigated based on time-dependent density functional theory (TD-DFT). The results suggest that both a single and double proton transfer mechanisms are relevant, in constrast to the prediction of a single one proposed previously (Piechowska et al. J. Phys. Chem. A 2014, 118, 144–151). The calculated results show that the intramolecular hydrogen bonds were formed in the S0 state, and upon excitation, the intramolecular hydrogen bonds between −OH group and pyridine-type nitrogen atom would be strengthened in the S1 state, which can facilitate the proton transfer process effectively. The calculated vertical excitation energies in the S0 and S1 states reproduce the experimental UV–vis absorption and fluorescence spectra well. The constructed potential energy surfaces of the S0 and S1 states have been used to explain the proton transfer process. Four minima have been found on the S1 state surface, with potential barriers between these excited-state minima of less than 10 kcal/mol, which supports concomitant single and double proton transfer mechanisms. In addition, the fluorescence quenching can be explained reasonably based on the proton transfer process.
Co-reporter:Songqiu Yang, Jianyong Liu, Panwang Zhou, Junsheng Chen, Keli Han, Guozhong He
Journal of Luminescence 2012 Volume 132(Issue 9) pp:2275-2280
Publication Date(Web):September 2012
DOI:10.1016/j.jlumin.2012.03.062
Subpicosecond fluorescence depletion spectroscopy (FDS) was used to measure the solvation dynamics of coumarin 153 (C153) in methanol. The FDS mechanisms were discussed. A quasi-continuous model was used to describe the solvational relaxation of excited states. The perturbations of the probe pulse on the excited sample system, including up-conversion and stimulated emission, were sufficiently discussed. For a probe molecule used in the FDS experiment, ensuring that the up-conversion perturbation can be negligible is important. FDS was found to be a good technique for measuring the solvation dynamics of C153 in methanol.Highlights► Mechanisms of subpicosecond fluorescence depletion spectroscopy. ► Quasi-continuous model was used to describe the solvational relaxation. ► The solvation dynamics of coumarin 153 in methanol has been measured.
Co-reporter:Yu-Hui Liu, Mohan Singh Mehata, and Jian-Yong Liu
The Journal of Physical Chemistry A 2011 Volume 115(Issue 1) pp:19-24
Publication Date(Web):December 9, 2010
DOI:10.1021/jp1101626
Spectroscopic studies on excited-state proton transfer (ESPT) of hydroxyquinoline (6HQ) have been performed in a previous paper. And a hydrogen-bonded network formed between 6HQ and acetic acid (AcOH) in nonpolar solvents has been characterized. In this work, a time-dependent density functional theory (TDDFT) method at the def-TZVP/B3LYP level was employed to investigate the excited-state proton transfer via hydrogen-bonded AcOH wire for 6HQ. A hydrogen-bonded wire containing three AcOH molecules at least for connecting the phenolic and quinolinic −N− group in 6HQ has been confirmed. The excited-state proton transfer via a hydrogen-bonded wire could result in a keto tautomer of 6HQ and lead to a large Stokes shift in the emission spectra. According to the results of calculated potential energy (PE) curves along different coordinates, a stepwise excited-state proton transfer has been proposed with two steps: first, an anionic hydrogen-bonded wire is generated by the protonation of −N− group in 6HQ upon excitation to the S1 state, which increases the proton-capture ability of the AcOH wire; then, the proton of the phenolic group transfers via the anionic hydrogen-bonded wire, by an overall “concerted” process. Additionally, the formation of the anionic hydrogen-bonded wire as a preliminary step has been confirmed by the hydrogen-bonded parameters analysis of the ESPT process of 6HQ in several protic solvents. Therefore, the formation of anionic hydrogen-bonded wire due to the protonation of the −N− group is essential to strengthen the hydrogen bonding acceptance ability and capture the phenolic proton in the 6HQ chromophore.
Co-reporter:Kun Yang;Xiao-Fang Chen;Wei-Peng Lai
Journal of Molecular Modeling 2011 Volume 17( Issue 5) pp:1017-1027
Publication Date(Web):2011/05/01
DOI:10.1007/s00894-010-0799-0
Nitrosation reactions of malononitrile by three nitrosating agents, HONO, ClNO, and N2O3, have been theoretically investigated at the B3LYP/cc-pVTZ and MP2/cc-pVDZ levels. Two possible competitive paths for nitrosation of malononitrile to give 2-nitroso-malononitrile were proposed: (a) direct C-nitrosation and (b) N-nitrosation and subsequent nitroso transfer from N to C atom. The calculations show that at both B3LYP and MP2 levels, path b is kinetically favored over path a for nitrosations by HONO and N2O3. In the case of ClNO, the B3LYP predicts preference of path b, while the MP2 calculations suggest that both paths have similar rate-determining barriers. The data suggest that N2O3 is the preferred nitrosating agent for the nitrosation of malononitrile in aqueous solution. Transformation of 2-nitroso-malononitrile to form malononitrileoxime via intramolecular proton transfer has also been explored, and it is found that inclusion of an assistant water molecule can drastically accelerate the tautomerization.
Co-reporter:Bing Jin, Jian-Yong Liu
Chemical Physics Letters 2010 Volume 491(1–3) pp:33-38
Publication Date(Web):7 May 2010
DOI:10.1016/j.cplett.2010.03.065
Co-reporter:Ya-Hui Guo, Hai-Xiang He, Jian-Yong Liu, Guo-Zhong He
Journal of Molecular Structure: THEOCHEM 2010 Volume 947(1–3) pp:119-122
Publication Date(Web):15 May 2010
DOI:10.1016/j.theochem.2010.02.004
The harmonic generations of molecular ion H2+ and D2+ exposed to an intense laser field have been studied with the collinear reduced-dimensionality model. The numerical solution of the time-dependent Schrödinger equation for H2+ and its isotopic variant D2+ shows that high-order harmonic generation is sensitive to the initial vibrational states of the molecular ion. By comparing the spectra of H2+ with D2+, it is shown more intense harmonics are generated in lighter isotopes.
Co-reporter:Li-Chuan Zhou, Jian-Yong Liu, Guang-Jiu Zhao, Ying Shi, Xiao-Jun Peng, Ke-Li Han
Chemical Physics 2007 Volume 333(2–3) pp:179-185
Publication Date(Web):30 March 2007
DOI:10.1016/j.chemphys.2007.01.019
The ultrafast dynamics of a synthesized near-infrared heptamethine cyanine dye has been studied in the typical alcoholic and aprotic solvents using the femtosecond time-resolved stimulated emission pumping fluorescence depletion (FS TR SEP FD) technique. The faster decay on the hundreds of the femtosecond time scale and the slower decay on the order of picosecond are found. The intramolecular vibrational redistribution (IVR) and the ultrafast solvent inertial relaxation should account for the faster decay, while the slower decay is attributed to the diffusive solvent relaxation. The time constants of the slower decay increase with the hydrogen-bonding energy in alcoholic solvent, and the time constants of the slower decay decrease with the dipole moments of the aprotic solvent molecules due to the interactions between the aprotic molecules and the excited dye molecule.
Co-reporter:Jindou Huang, Shuhao Wen, Jianyong Liu, Guozhong He
Journal of Natural Gas Chemistry (May 2012) Volume 21(Issue 3) pp:302-307
Publication Date(Web):1 May 2012
DOI:10.1016/S1003-9953(11)60368-X
In this study, we have performed first-principles screened exchanged hybrid density function theory with the HSE06 function calculations of the C-Mo, C-W, N-Nb and N-Ta codoped anatase TiO2 systems to investigate the effect of codoping on the electronic structure of TiO2. The calculated results demonstrate that (W(s)+C(s)) codoped TiO2 narrows the band gap significantly, and have little influence on the position of conduction band edges, therefore, enhances the efficiency of the photocatalytic hydrogen generation from water and the photodegradation of organic pollutants. Moreover, the proper oxygen pressure and temperature are two key factors during synthesis which should be carefully under control so that the desired (W(s)+C(s)) codoped TiO2 can be obtained.
Co-reporter:Guo-Hong Fan, Xing Li, Jian-Yong Liu, Guo-Zhong He
Computational and Theoretical Chemistry (15 February 2014) Volume 1030() pp:
Publication Date(Web):15 February 2014
DOI:10.1016/j.comptc.2013.12.010
•Excited properties of a covalent organic framework are studied.•Calculated spectra compare well with experiment result.•Adjacent layers are more stable if layers are horizontally offset.•Eclipsed AA stacking structure promotes the carrier mobility.•Low-symmetry offset structure is more conducive to luminescence.Excited-state properties of a blue luminescent two-dimensional (2D) layered covalent organic framework (COF) have been studied by the time-dependent density-functional tight-binding (TD-DFTB) method and compared with available experimental data. For single layer fragment, signatures in absorption and emission spectra are revealed, which show the typical π–π∗ transitions and intramolecular charge transfer (ICT) character. The influence of interlayer stacking has also been considered. Our calculations on the interlayer spacing of the COF suggest that adjacent layers are considerably more stable if their stacking arrangement is horizontally offset by ≈1.4 Å compared with the eclipsed AA stacking structure. Excite-states calculations of high-symmetry eclipsed AA stacking and low-symmetry offset stacking are also considered. The low-symmetry offset stacking shows higher luminescence intensity but lower carrier transport ability. These calculations will be helpful for understanding interlayer excitation and carrier mobility, and the balance of the two processes is essential to the designing of the COF-based optoelectronic devices.
Co-reporter:Jinfeng Zhao, Junsheng Chen, Jianyong Liu and Mark R. Hoffmann
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 18) pp:NaN11999-11999
Publication Date(Web):2015/03/24
DOI:10.1039/C4CP05651E
The excited state intramolecular proton transfer (ESIPT) mechanisms of 2-(2-hydroxyphenyl)benzoxazole (HBO), bis-2,5-(2-benzoxazolyl)-hydroquinone (BBHQ) and 2,5-bis(5′-tert-butyl-benzoxazol-2′-yl)hydroquinone (DHBO) have been investigated using time-dependent density functional theory (TDDFT). The calculated vertical excitation energies based on the TDDFT method reproduced the experimental absorption and emission spectra well. Three kinds of stable structures were found on the S1 state potential energy surface (PES). A new ESIPT mechanism that differs from the one proposed previously (Mordzinski et al., Chem. Phys. Lett., 1983, 101, 291. and Lim et al., J. Am. Chem. Soc., 2006, 128, 14542.) is proposed. The new mechanism includes the possibility of simultaneous double proton transfer, or successive single transfers, in addition to the accepted single proton transfer mechanism. Hydrogen bond strengthening in the excited state was based on primary bond lengths, angles, IR vibrational spectra and hydrogen bond energy. Intramolecular charge transfer based on the frontier molecular orbitals (MOs) also supports the proposed mechanism of the ESIPT reaction. To further elucidate the proposed mechanism, reduced dimensionality PESs of the S0 and S1 states were constructed by keeping the O–H distance fixed at a series of values. The potential barrier heights among the local minima on the S1 surface imply competitive single and double proton transfer branches in the mechanism. Based on the new ESIPT mechanism, the observed fluorescence quenching can be satisfactorily explained.
.jpg)
![1H-Imidazole, 1-methyl-2-[(4-methylphenyl)azo]-](http://img.cochemist.com/ccimg/198400/198347-67-0.png)
![1H-Imidazole, 1-methyl-2-[(4-methylphenyl)azo]-](http://img.cochemist.com/ccimg/198400/198347-67-0_b.png)
![1,4-Benzenediol, 2,5-bis[5-(1,1-dimethylethyl)-2-benzoxazolyl]-](http://img.cochemist.com/ccimg/152200/152158-88-8.png)
![1,4-Benzenediol, 2,5-bis[5-(1,1-dimethylethyl)-2-benzoxazolyl]-](http://img.cochemist.com/ccimg/152200/152158-88-8_b.png)


![dibenzo[a,h]phenazine-1,8-diol](http://img.cochemist.com/ccimg/26900/26846-41-3.png)
![dibenzo[a,h]phenazine-1,8-diol](http://img.cochemist.com/ccimg/26900/26846-41-3_b.png)
![Phenol, 2-[(methylimino)methyl]-](http://img.cochemist.com/ccimg/3200/3117-65-5.png)
![Phenol, 2-[(methylimino)methyl]-](http://img.cochemist.com/ccimg/3200/3117-65-5_b.png)

