Co-reporter:Masaaki Baba, Toshiharu Katori, Megumi Kawabata, Sachi Kunishige, and Takaya Yamanaka
The Journal of Physical Chemistry A 2013 Volume 117(Issue 50) pp:13524-13530
Publication Date(Web):October 1, 2013
DOI:10.1021/jp407327h
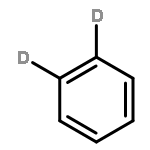
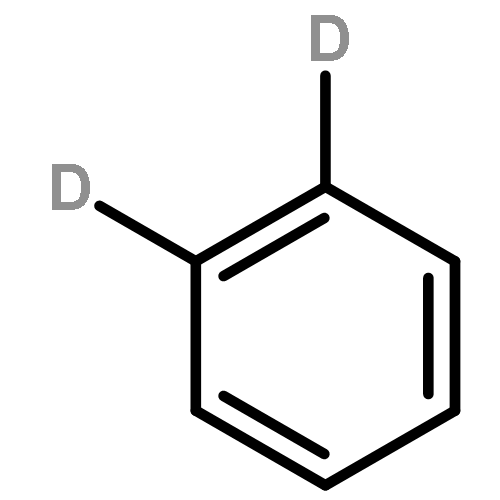
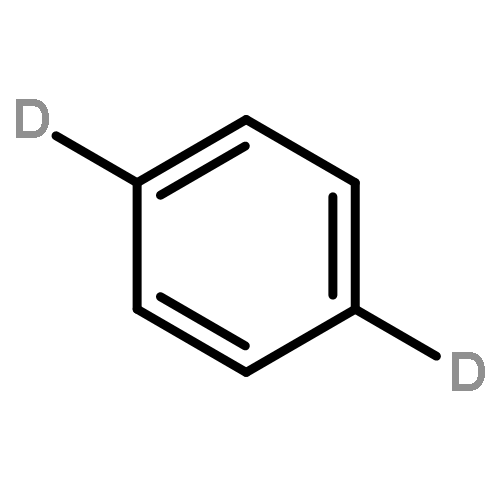
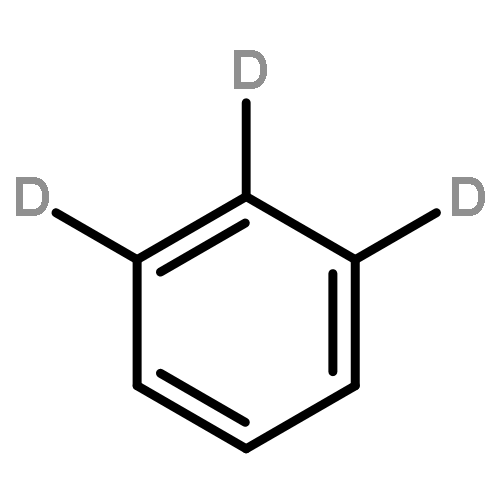
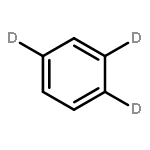
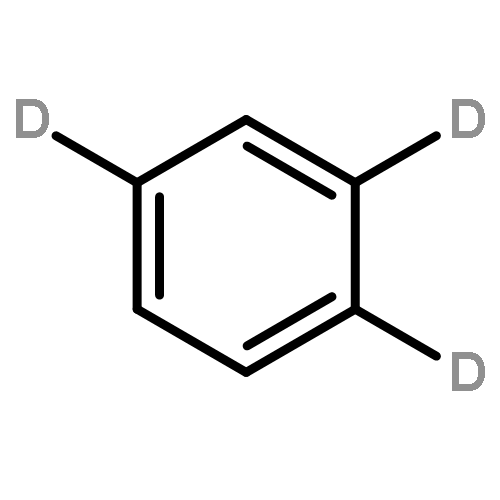
We investigated the S1 and S2 states of linear and zigzag cata-condensed hydrocarbons on the basis of the results of jet spectroscopy and theoretical calculations. The S1 states of anthracene and tetracene are represented by the HOMO → LUMO configuration (Φ(A)), whereas those of phenanthrene and chrysene are represented by HOMO–1 → LUMO and HOMO → LUMO+1 configurations (Φ(B)). We found that the fluorescence lifetime varied with different vibronic levels in the S1 states of linear cata-condensed hydrocarbons due to the mode-selective internal conversion to the S0 state. This selectivity is likely to be seen in the S1 Φ(A) state of the D2h molecule.
Co-reporter:Yasuyuki Kowaka, Naofumi Nakayama, Takayoshi Ishimoto, Umpei Nagashima, Takaya Yamanaka, Norifumi Ozawa, Masaaki Baba
Chemical Physics 2012 400() pp: 178-184
Publication Date(Web):25 May 2012
DOI:10.1016/j.chemphys.2012.04.004
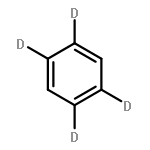
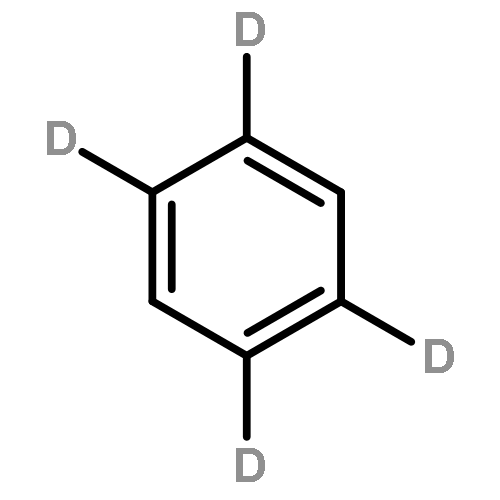
Fluorescence excitation spectra and fluorescence lifetimes at single vibronic levels in the S1S1 state have been observed for jet-cooled pyrene. The fluorescence lifetimes at the zero-vibrational levels of the S11B3uS11B3u states of pyrene-h10h10 and pyrene-d10d10 are 1480 and 1470 ns, respectively, and the relaxation is considered to be dominated by the radiative process. For some vibrational levels, however, the lifetimes are remarkably shorter such as 765 ns at the 221 vibronic level of pyrene-h10h10 (ν22ν22 (b1gb1g); C–H bending and skeletal deforming mode), indicating that nonradiative transition occurs at a specific vibrational level. In this study, we demonstrate that the main process is internal conversion to the S01AgS01Ag state caused by nonadiabatic vibronic interaction via b3ub3u promoting modes.Graphical abstractHighlights► Fluorescence lifetimes were accurately observed for each single vibronic level of pyrene. ► Nonradiative internal conversion in the S1 state is enhanced via specific vibrations. ► The structure of the isolated pyrene molecule was determined by ab initio calculation.
Co-reporter:Masaaki Baba
The Journal of Physical Chemistry A 2011 Volume 115(Issue 34) pp:9514-9519
Publication Date(Web):March 14, 2011
DOI:10.1021/jp111892y
Fast intersystem crossing is observed in the S11nπ* state of N-heterocyclic aromatic hydrocarbons and carbonyl compounds. It is attributed to spin−orbit coupling with the 3ππ* state in the same energy region. The strong singlet−triplet mixing was confirmed by large Zeeman splitting of rotational lines in a high-resolution spectrum. For the S11ππ* state of aromatic hydrocarbons, the observed Zeeman splitting was found to be considerably small, and intersystem crossing was considered to be minor. These facts are in accordance with El-Sayed’s rule, which states spin−orbit coupling is forbidden between the 1ππ* and 3ππ* states. The Zeeman splitting of several derivatives was also observed and the substitution effect on the intersystem crossing rate is discussed.
Co-reporter:Yasuyuki Kowaka, Yoshitake Suganuma, Noritaka Ashizawa, Naofumi Nakayama, Hitoshi Goto, Takayoshi Ishimoto, Umpei Nagashima, Masaaki Baba
Journal of Molecular Spectroscopy 2010 Volume 260(Issue 1) pp:72-76
Publication Date(Web):March 2010
DOI:10.1016/j.jms.2009.11.011
Co-reporter:Masaaki Baba, Koichi Mori, Motohisa Saito, Yasuyuki Kowaka, Yuki Noma, Shunji Kasahara, Takaya Yamanaka, Katsuhiko Okuyama, Takayoshi Ishimoto and Umpei Nagashima
The Journal of Physical Chemistry A 2009 Volume 113(Issue 11) pp:2366-2371
Publication Date(Web):February 20, 2009
DOI:10.1021/jp808550r
Fluorescence excitation spectra and dispersed fluorescence spectra of jet-cooled 9-methylanthracene-h12 and -d12 (9MA-h12 and 9MA-d12) have been observed, and the energy levels of methyl internal rotation (CH3 torsion) in the S0 and S1 states have been analyzed. The molecular symmetry of 9MA is the same as that of toluene (G12). Because of two-fold symmetry in the π system, the potential curve has six-fold barriers to CH3 rotation. In toluene, the barrier height to CH3 rotation V6 is very small, nearly free rotation. As for 9MA-h12, we could fit the level energies by potential curves with the barrier heights of V6(S0) = 118 cm−1 and V6(S1) = 33 cm−1. These barrier heights are remarkably larger than those of toluene and are attributed to hyperconjugation between the π orbitals and methyl group. The dispersed fluorescence spectrum showed broad emission for the excitation of 000 + 386 cm−1 band, indicating that intramolecular vibrational redistribution efficiently occurs, even in the vibronic level of low excess energy of the isolated 9MA molecule.
Co-reporter:Michiru Yamawaki, Yoshio Tatamitani, Atsushi Doi, Shunji Kasahara, Masaaki Baba
Journal of Molecular Spectroscopy 2006 Volume 238(Issue 1) pp:49-55
Publication Date(Web):July 2006
DOI:10.1016/j.jms.2006.04.008
Rotationally resolved ultrahigh-resolution fluorescence excitation spectra of the S1 ← S0 transition of dibenzofuran have been observed using the technique of crossing a collimated molecular beam and the single-mode UV laser beam. 3291 rotational lines of the 000 band and 3047 rotational lines of the 000+443cm−1(5501:b2) band have been assigned. The 000 band has been found to be a b-type transition, in which the transition moment is along the twofold symmetry axis of this molecule, and only the ΔKa = ± 1 transitions were observed. The excited state is identified to be the S11A1(ππ∗) state. In contrast with this, the 000+443cm−1(5501:b2) band has been found to be an a-type transition in which the transition moment is along the long axis in plane. It indicates that the intensity of this vibronic band arises from vibronic coupling with the S21B2(ππ∗) state. We determined the accurate rotational constants and the molecule have been shown to be planar both in the ground and excited states.