Co-reporter:Yao-Yue Fan, Li-She Gan, Hong-Chun Liu, Heng Li, Cheng-Hui Xu, Jian-Ping Zuo, Jian Ding, and Jian-Min Yue
Organic Letters September 1, 2017 Volume 19(Issue 17) pp:
Publication Date(Web):August 23, 2017
DOI:10.1021/acs.orglett.7b02181
Phainanolide A (1), a highly modified triterpenoid incorporating an unprecedented 6/9/6 heterotricyclic system and a highly oxygenated 5,5-spirocyclic ketal lactone, along with three new triterpenoids 2–4 were isolated from Phyllanthus hainanensis. Their structures were completely elucidated by a combination of diverse methods including 2D NMR, quantum chemical NMR and ECD calculations, and NMR data analogy with model compounds. Compounds 1–4 exhibited both remarkable cytotoxic and immunosuppressive activities.
Co-reporter:Lingfeng Chen, Weitao Fu, Chen Feng, Rong Qu, Linjiang Tong, Lulu Zheng, Bo Fang, Yinda Qiu, Jie Hu, Yuepiao Cai, Jianpeng Feng, Hua Xie, Jian Ding, Zhiguo Liu, Guang Liang
European Journal of Medicinal Chemistry 2017 Volume 140(Volume 140) pp:
Publication Date(Web):10 November 2017
DOI:10.1016/j.ejmech.2017.08.061
•Three-step SAR explorations were performed based on the binding mode of lead compound.•Active compounds showed good pharmacokinetics properties.•19e and 19h exhibited great potency and selectivity against EGFR L858R/T790M kinase.•19e and 19h significantly inhibited tumor growth in H1975 xenograft mouse model.Mutated epidermal growth factor receptor (EGFR) is a major driver of non-small cell lung cancer (NSCLC). The EGFRT790M secondary mutation has become a leading cause of clinically-acquired resistance to gefitinib and erlotinib. Herein, we present a structure-based design approach to increase the potency and selectivity of the previously reported reversible EGFR inhibitor 7, at the kinase and cellular levels. Three-step structure-activity relationship exploration led to promising compounds 19e and 19h with unique chemical structure and binding mode from the other third-generation tyrosine kinase inhibitors. In a human NSCLC xenograft model, 19e and 19h exhibited dose-dependent tumor growth suppression without toxicity. These selective inhibitors are promising drug candidates for EGFRT790M-driven NSCLC.Download high-res image (206KB)Download full-size image
Co-reporter:Danni Sun, Hongchun Liu, Xiaoyang Dai, Xingling Zheng, Juan Yan, Rongrui Wei, Xuhong Fu, Min Huang, Aijun Shen, Xun Huang, Jian Ding, Meiyu Geng
Cancer Letters 2017 Volume 406(Volume 406) pp:
Publication Date(Web):10 October 2017
DOI:10.1016/j.canlet.2017.06.029
•Aspirin inhibits mTORC1 signaling in an AMPK-independent manner.•Aspirin disrupts the mTOR-raptor interaction.•mTORC1 inhibition contributed to the synergistic effect of aspirin combining with sorafenib.Aspirin is associated with a reduced risk of cancer and delayed progression of malignant disease. Adenosine 5‘-monophosphate (AMP)-activated protein kinase (AMPK)-mTOR signaling is believed to partially contribute to these anticancer effects, although the mechanism is unclear. In this study, we revealed the mechanism underlying the effects of aspirin on AMPK-mTOR signaling, and described a mechanism-based rationale for the use of aspirin in cancer therapy. We found that aspirin inhibited mTORC1 signaling through AMPK-dependent and -independent manners. Aspirin inhibited the AMPK-TSC pathway, thus resulting in the suppression of mTORC1 activity. In parallel, it directly disrupted the mTOR-raptor interaction. Additionally, the combination of aspirin and sorafenib showed synergetic effects via inhibiting mTORC1 signaling and the PI3K/AKT, MAPK/ERK pathways. Aspirin and sorafenib showed synergetic anticancer efficacy in the SMMC-7721 model. Our study provides mechanistic insights and a mechanism-based rationale for the roles of aspirin in cancer treatment.
Co-reporter:Guo-Rui Gao, Meng-Yuan Li, Lin-Jiang Tong, Li-Xin Wei, Jian Ding, Hua Xie, Wen-Hu Duan
Chinese Chemical Letters 2015 Volume 26(Issue 9) pp:1165-1168
Publication Date(Web):September 2015
DOI:10.1016/j.cclet.2015.07.016
Inhibition of VEGFR-2 signaling pathway has already become one of the most promising approaches for the treatment of cancer. In this study, we describe the design, synthesis, and biological evaluation of a series of O-linked indoles as potent inhibitors of VEGFR-2. Among these compounds, 18 showed significant anti-angiogenesis activities via VEGFR-2 in enzymatic proliferation assays, with IC50 value of 3.8 nmol/L. Kinase selectivity profiling revealed that 18 was a multitargeted inhibitor, and it also exhibited good potency against VEGFR-1, PDGFR-α and β.In an effort to discover potent VEGFR-2 inhibitors, a series of 2,4 or 4,6-disubstituted O-linked indoles derivatives were designed and synthesized. The structural activity relationships led to identification of a potential VEGFR-2 inhibitor compound 18.
Co-reporter:Xiang Wang;Jian Ding;Ling-hua Meng
Acta Pharmacologica Sinica 2015 36(10) pp:1170-1176
Publication Date(Web):2015-09-14
DOI:10.1038/aps.2015.71
The pivotal roles of phosphatidylinositol 3-kinases (PI3Ks) in human cancers have inspired active development of small molecules to inhibit these lipid kinases. However, the first-generation pan-PI3K and dual-PI3K/mTOR inhibitors have encountered problems in clinical trials, with limited efficacies as a monotherapeutic agent as well as a relatively high rate of side effects. It is increasingly recognized that different PI3K isoforms play non-redundant roles in particular tumor types, which has prompted the development of isoform-selective inhibitors for pre-selected patients with the aim for improving efficacy while decreasing undesirable side effects. The success of PI3K isoform-selective inhibitors is represented by CAL101 (Idelalisib), a first-in-class PI3Kδ-selective small-molecule inhibitor that has been approved by the FDA for the treatment of chronic lymphocytic leukemia, indolent B-cell non-Hodgkin's lymphoma and relapsed small lymphocytic lymphoma. Inhibitors targeting other PI3K isoforms are also being extensively developed. This review focuses on the recent progress in development of PI3K isoform-selective inhibitors for cancer therapy. A deeper understanding of the action modes of novel PI3K isoform-selective inhibitors will provide valuable information to further validate the concept of targeting specific PI3K isoforms, while the identification of biomarkers to stratify patients who are likely to benefit from the therapy will be essential for the success of these agents.
Co-reporter:Min Huang, Aijun Shen, Jian Ding, Meiyu Geng
Trends in Pharmacological Sciences (January 2014) Volume 35(Issue 1) pp:41-50
Publication Date(Web):1 January 2014
DOI:10.1016/j.tips.2013.11.004
•Tumor heterogeneity is the key factor underlying limited response rate to targeted therapy.•Personalized medicines depend on biomarkers for selecting patients and directing therapy.•Co-development of predictive and response biomarkers is required for drug development.•PDX model-based trials mimicking human patients may improve biomarker discovery.The tremendous advances achieved in the understanding of cancer biology have delivered unprecedented progress in molecularly targeted cancer therapy in the past decade. The fast growing category of targeted anticancer agents available for clinical use is accompanied by a conceptual revolution in anticancer drug development. Nevertheless, molecularly targeted cancer therapy remains challenged by a high failure rate and an extremely small proportion of patients that can benefit. It is pivotal to take lessons from the past and seek new solutions. This review discusses conceptual progress and remaining challenges in molecularly targeted cancer therapy, and proposes feasible alternatives to increase chances of clinical success in the future.

![Benzenamine, 4-[2-(dimethylamino)ethoxy]-2-methoxy-](http://img.cochemist.com/ccimg/927700/927672-74-0.png)
![Benzenamine, 4-[2-(dimethylamino)ethoxy]-2-methoxy-](http://img.cochemist.com/ccimg/927700/927672-74-0_b.png)

![1H-Benz[de]isoquinoline-1,3(2H)-dione, 6-bromo-2-octyl-](/data/chemimg/110700/865443-57-8.png)
![1H-Benz[de]isoquinoline-1,3(2H)-dione, 6-bromo-2-octyl-](/data/chemimg/110700/865443-57-8_b.png)
![6-Bromoimidazo[1,2-a]pyrimidine-3-carbaldehyde](http://img.cochemist.com/ccimg/865200/865156-67-8.png)
![6-Bromoimidazo[1,2-a]pyrimidine-3-carbaldehyde](http://img.cochemist.com/ccimg/865200/865156-67-8_b.png)
![IMIDAZO[1,5-A]PYRIDINE-1-CARBOXALDEHYDE, 7-BROMO-](http://img.cochemist.com/ccimg/865200/865156-47-4.png)
![IMIDAZO[1,5-A]PYRIDINE-1-CARBOXALDEHYDE, 7-BROMO-](http://img.cochemist.com/ccimg/865200/865156-47-4_b.png)
![Benzenamine, 4-[(2-chloro-4-pyrimidinyl)oxy]-](http://img.cochemist.com/ccimg/853300/853299-33-9.png)
![Benzenamine, 4-[(2-chloro-4-pyrimidinyl)oxy]-](http://img.cochemist.com/ccimg/853300/853299-33-9_b.png)
![2-METHOXY-4-[4-(4-METHYLPIPERAZIN-1-YL)PIPERIDIN-1-YL]ANILINE](http://img.cochemist.com/ccimg/761500/761440-75-9.png)
![2-METHOXY-4-[4-(4-METHYLPIPERAZIN-1-YL)PIPERIDIN-1-YL]ANILINE](http://img.cochemist.com/ccimg/761500/761440-75-9_b.png)
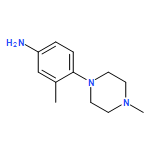
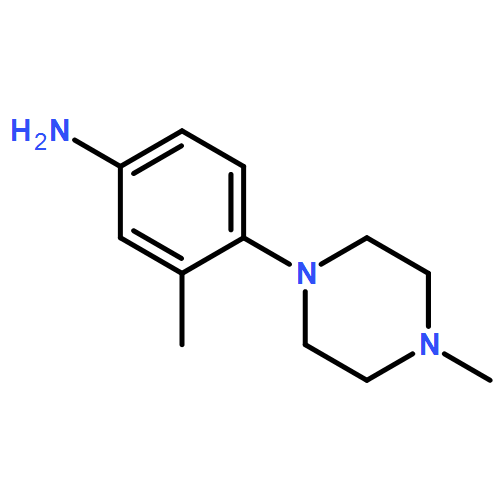
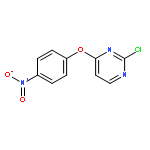
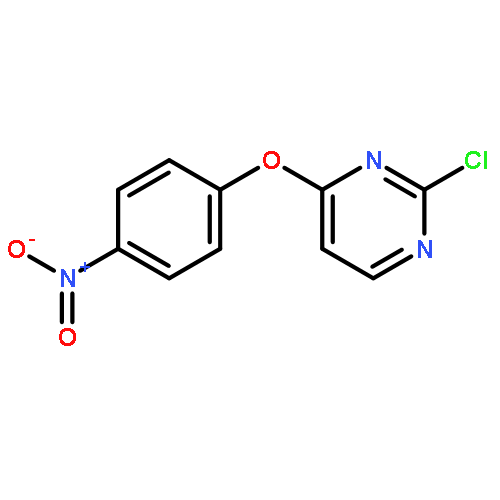
































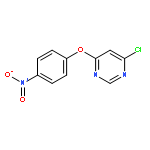
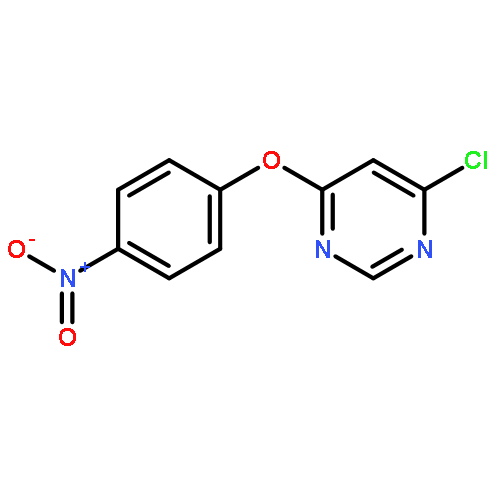
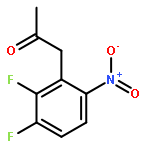


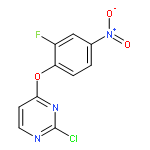
![Benzenamine, 4-[(6-chloro-4-pyrimidinyl)oxy]-](http://img.cochemist.com/ccimg/105200/105130-28-7.png)
![Benzenamine, 4-[(6-chloro-4-pyrimidinyl)oxy]-](http://img.cochemist.com/ccimg/105200/105130-28-7_b.png)


![6H-2,8b-Epoxyoxireno[6,7]azuleno[5,4-e]-1,3-benzodioxol-6-one, 10-(acetyloxy)-3a,3b,3c,4a,5,5a,8a,9,10,10a-decahydro-5,5a-dihydroxy-4a-](/data/chemimg/1952200/73061-92-4.png)
![6H-2,8b-Epoxyoxireno[6,7]azuleno[5,4-e]-1,3-benzodioxol-6-one, 10-(acetyloxy)-3a,3b,3c,4a,5,5a,8a,9,10,10a-decahydro-5,5a-dihydroxy-4a-](/data/chemimg/1952200/73061-92-4_b.png)


![6H-2,8b-Epoxyoxireno[6,7]azuleno[5,4-e]-1,3-benzodioxol-6-one,3a,3b,3c,4a,5,5a,8a,9,10,10a-decahydro-5,5a-dihydroxy-4a-(hydroxymethyl)-7,9-dimethyl-10a-(1-methylethenyl)-2-(1E,3E)-1,3-tridecadien-1-yl-,(2S,3aR,3bS,3cS,4aR,5S,5aS,8aR,8bR,9R,10aR)-](http://img.cochemist.com/ccimg/33500/33465-16-6.png)
![6H-2,8b-Epoxyoxireno[6,7]azuleno[5,4-e]-1,3-benzodioxol-6-one,3a,3b,3c,4a,5,5a,8a,9,10,10a-decahydro-5,5a-dihydroxy-4a-(hydroxymethyl)-7,9-dimethyl-10a-(1-methylethenyl)-2-(1E,3E)-1,3-tridecadien-1-yl-,(2S,3aR,3bS,3cS,4aR,5S,5aS,8aR,8bR,9R,10aR)-](http://img.cochemist.com/ccimg/33500/33465-16-6_b.png)
![1H-Benz[de]isoquinoline-1,3(2H)-dione,6-bromo-2-methyl-](/data/chemimg/60600/4116-90-9.png)
![1H-Benz[de]isoquinoline-1,3(2H)-dione,6-bromo-2-methyl-](/data/chemimg/60600/4116-90-9_b.png)
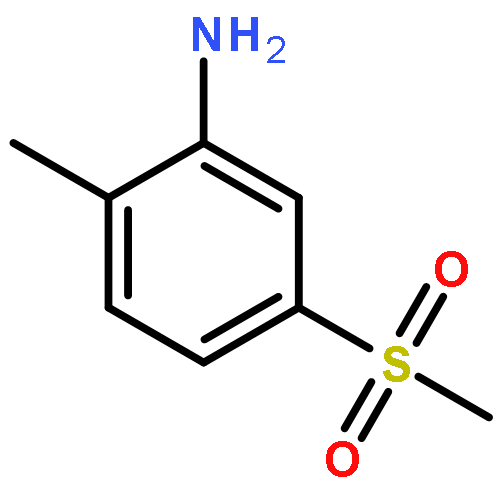
![6H-2,8b-Epoxyoxireno[6,7]azuleno[5,4-e]-1,3-benzodioxol-6-one,3a,3b,3c,4a,5,5a,8a,9,10,10a-decahydro-5,5a-dihydroxy-4a-(hydroxymethyl)-7,9-dimethyl-10a-(1-methylethenyl)-2-nonyl-,(2S,3aR,3bS,3cS,4aR,5S,5aS,8aR,8bR,9R,10aR)-](http://img.cochemist.com/ccimg/1500/1404-62-2.png)
![6H-2,8b-Epoxyoxireno[6,7]azuleno[5,4-e]-1,3-benzodioxol-6-one,3a,3b,3c,4a,5,5a,8a,9,10,10a-decahydro-5,5a-dihydroxy-4a-(hydroxymethyl)-7,9-dimethyl-10a-(1-methylethenyl)-2-nonyl-,(2S,3aR,3bS,3cS,4aR,5S,5aS,8aR,8bR,9R,10aR)-](http://img.cochemist.com/ccimg/1500/1404-62-2_b.png)
![Benzenamine, 3-[(methylsulfonyl)methyl]-](http://img.cochemist.com/ccimg/262000/261925-02-4.png)
![Benzenamine, 3-[(methylsulfonyl)methyl]-](http://img.cochemist.com/ccimg/262000/261925-02-4_b.png)


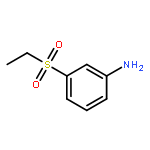
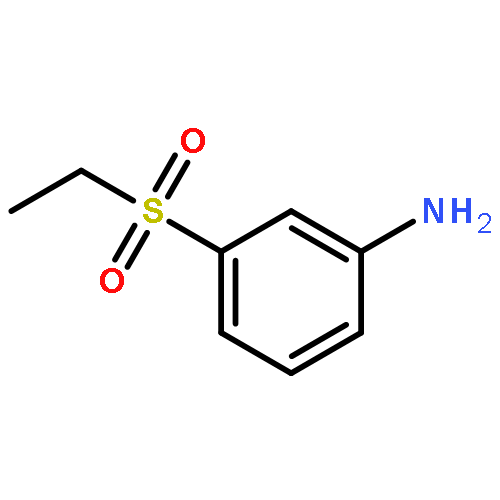
![{4-[(Methylsulfonyl)methyl]phenyl}amine hydrochloride](http://img.cochemist.com/ccimg/24200/24176-70-3.png)
![{4-[(Methylsulfonyl)methyl]phenyl}amine hydrochloride](http://img.cochemist.com/ccimg/24200/24176-70-3_b.png)





