Co-reporter:Sakulsuk Unarunotai;Mitchell J. Schultz;Xiaoyu Zhang;Dahl-Young Khang;Changhui Lei;Congjun Wang;Qing Cao;Julio A. N. T. Soares;Scott MacLaren;Ivan Petrov;John A. Rogers
PNAS 2008 Volume 105 (Issue 21 ) pp:7353-7358
Publication Date(Web):2008-05-27
DOI:10.1073/pnas.0710081105
Because of their potential for use in advanced electronic, nanomechanical, and other applications, large two-dimensional,
carbon-rich networks have become an important target to the scientific community. Current methods for the synthesis of these
materials have many limitations including lack of molecular-level control and poor diversity. Here, we present a method for
the synthesis of two-dimensional carbon nanomaterials synthesized by Mo- and Cu-catalyzed cross-linking of alkyne-containing
self-assembled monolayers on SiO2 and Si3N4. When deposited and cross-linked on flat surfaces, spheres, cylinders, or textured substrates, monolayers take the form of
these templates and retain their structure on template removal. These nanomaterials can also be transferred from surface to
surface and suspended over cavities without tearing. This approach to the synthesis of monolayer carbon networks greatly expands
the chemistry, morphology, and size of carbon films accessible for analysis and device applications.
Co-reporter:Jennifer M. Heemstra and Jeffrey S. Moore
Chemical Communications 2004 (Issue 13) pp:1480-1481
Publication Date(Web):25 May 2004
DOI:10.1039/B405980H
Intramolecular cation–π interactions between a methyl pyridinium ion and a phenyl ring stabilize the folded structure of a phenyleneethynylene oligomer.
Co-reporter:Dahui Zhao and Jeffrey S. Moore
Chemical Communications 2003 (Issue 7) pp:807-818
Publication Date(Web):15 Nov 2002
DOI:10.1039/B207442G
This article describes recent developments in the synthesis of macrocycles having rigid, monocyclic skeletons composed of arylene and ethynylene units and the studies on their self-assembling behavior.
Co-reporter:Dahui Zhao and Jeffrey S. Moore
Organic & Biomolecular Chemistry 2003 vol. 1(Issue 20) pp:3471-3491
Publication Date(Web):29 Sep 2003
DOI:10.1039/B308788C
The kinetic and thermodynamic characteristics of polymerizations following a cooperative, nucleation–elongation mechanism are discussed in comparison to those of non-cooperative, isodesmic polymerizations. Nucleation–elongation polymerization is a relatively unexplored avenue of synthetic polymer chemistry and offers some unique and interesting thermodynamic and kinetic attributes not found in the more classical mechanisms of polymer chemistry.
Co-reporter:Aya Tanatani Dr.;Thomas S. Hughes Dr.
Angewandte Chemie International Edition 2002 Volume 41(Issue 2) pp:
Publication Date(Web):18 JAN 2002
DOI:10.1002/1521-3773(20020118)41:2<325::AID-ANIE325>3.0.CO;2-1
A dynamic binding mechanism allows the association of dumbbell-shaped ligand 2 with helical folded oligo(m-phenyleneethynylene)s 1 (see schematic representation). Association constants are oligomer length specific, with a maximum value for the icosamer and docosamer. These oligomers have helical conformations that are highly shape-complementary to 1; shorter and longer oligomers exhibit association constants roughly an order of magnitude smaller.
Co-reporter:Aya Tanatani Dr.;Thomas S. Hughes Dr.
Angewandte Chemie 2002 Volume 114(Issue 2) pp:
Publication Date(Web):17 JAN 2002
DOI:10.1002/1521-3757(20020118)114:2<335::AID-ANGE335>3.0.CO;2-M
Ein dynamischer Bindungsmechanismus ermöglicht die Assoziatbildung des hantelförmigen Liganden 2 mit helicalen gefalteten Oligo(m-phenylenethinylenen) 1 (siehe Schema). Die Assoziationskonstanten hängen von der Länge des Oligomers ab, wobei man die höchsten Werte für das Icosamer und das Docosamer findet. Die Oligomere nehmen helicale Konformationen ein, die formkomplementär zu 1 sind; kürzere und längere Oligomere weisen um etwa eine Größenordnung kleinere Assoziationskonstanten auf.
Co-reporter:Joshua A. Orlicki;Julie L. Thompson;Larry J. Markoski;Kevin N. Sill
Journal of Polymer Science Part A: Polymer Chemistry 2002 Volume 40(Issue 8) pp:936-946
Publication Date(Web):21 FEB 2002
DOI:10.1002/pola.10170
End-group modified hyperbranched polyetherimides were prepared by a one-pot, two-step reaction sequence. General synthetic techniques were developed to prepare both monofunctional terminating segments and the corresponding modified polyetherimide hyperbranched polymers. Monofunctional groups were used to terminate an AB2-type polycondensation reaction, generating capped hyperbranched polymers (HBPs). The composition and constitution of the end groups controlled the solubility and thermal properties of the HBPs. For the same polymer backbone, different end groups were able to shift the glass-transition temperature nearly 100 °C. End-group modification greatly influenced the film-forming ability of the HBPs. © 2002 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem 40: 936–946, 2002
Co-reporter:Keunchan Oh;Kyu-Sung Jeong
Nature 2001 414(6866) pp:889-893
Publication Date(Web):2001-12-20
DOI:10.1038/414889a
The biological function of biomacromolecules such as DNA and enzymes depends on their ability to perform and control mo-lecular association, catalysis, self-replication or other chemical processes. In the case of proteins in particular, the dependence of these functions on the three-dimensional protein conformation is long known1 and has inspired the development of synthetic oligomers and polymers with the capacity to fold in a controlled manner2, 3, 4, 5, 6, 7, but it remains challenging to design these so-called 'foldamers' so that they are capable of inducing or controlling chemical processes and interactions8, 9. Here we show that the stability gained from folding can be used to control the synthesis of oligomers from short chain segments reversibly ligated through an imine metathesis reaction. That is, folding shifts the ligation equilibrium10, 11, 12, 13 in favour of conformationally ordered sequences, so that oligomers having the most stable solution structures form preferentially. Crystallization has previously been used to shift an equilibrium in order to indirectly influence the synthesis of small molecules14, but the present approach to selectively prepare macromolecules with stable conformations directly connects folding and synthesis, emphasizing molecular function rather than structure in polymer synthesis.
Co-reporter:A. Zhu;P. Bharathi;H. G. Drickamer;J. S. Moore
Journal of Polymer Science Part A: Polymer Chemistry 2001 Volume 39(Issue 16) pp:2859-2865
Publication Date(Web):2 JUL 2001
DOI:10.1002/pola.1265
The effect of pressure up to 60 kbar was measured on the luminescence peak location and efficiency for a series of methoxy phenylacetylene dendrimers (MeO). Dendrimers MeO-3, MeO-7, MeO-15, MeO-31, MeO-63, and MeO-127 were studied as neat polymers. MeO-3, MeO-15, MeO-63, and MeO-127 were also investigated in dilute solutions in poly(tert-butyl methacrylate). According to measurements of the dilute solutions, there is a charge-transfer (CT) state that, for the smaller dendrimers, lies well above the π* state; for the larger dendrimers, it is the emitting state at 1 atm. With increasing pressure, the intramolecular CT state is rapidly stabilized, so that at high pressure the emission is from this state for all dendrimers. For the neat polymers, there is an initial redshift that reverses direction at a pressure that is higher for smaller dendrimers. This reversal is attributed to intermolecular CT. There may be changes in the molecular geometry and/or relative orientation of adjacent dendrimers that tend to stabilize the intermolecular CT in the solid state. © 2001 John Wiley & Sons, Inc. J Polym Sci Part A: Polym Chem 39: 2859–2865, 2001
Co-reporter:Jeffrey S. Moore;Peng Zhang
Journal of Polymer Science Part A: Polymer Chemistry 2000 Volume 38(Issue 1) pp:207-219
Publication Date(Web):21 JAN 2000
DOI:10.1002/(SICI)1099-0518(20000101)38:1<207::AID-POLA26>3.0.CO;2-9
Poly(ethylethylene-b-ethylene oxide) (PEE-PEO) diblock copolymers with pyridine-benzoic acid end-groups for heterodimeric hydrogen bonding were designed as a possible means to noncentrosymmetric organizations by spontaneous self-assembly. These end-functionalized polymers were synthesized by anionic living polymerization with protected initiator and terminating reagents. A series of polymeric intermediates with different end-groups was characterized by proton nuclear magnetic resonance, matrix-assisted laser desorption/ionization time-of-flight mass spectrometry, and gel permeation chromatography. Preliminary studies of solid-state organization by differential scanning calorimetry and small-angle X-ray scattering provided evidence for a long-range order that was sensitive to chain length, copolymer composition, and end-group structure. © 2000 John Wiley & Sons, Inc. J Polym Sci A: Polym Chem 38: 207–219, 2000

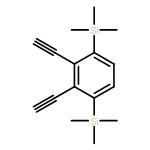



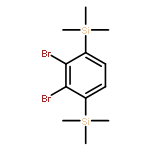
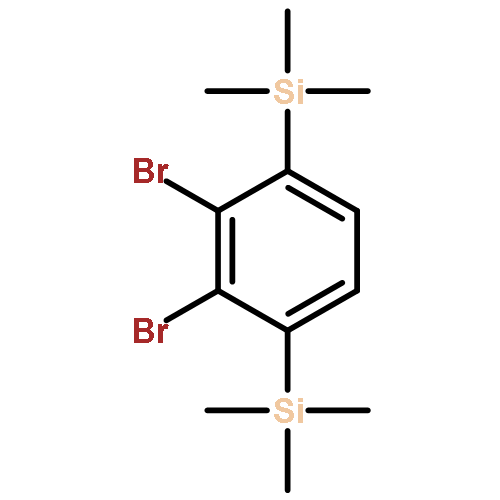






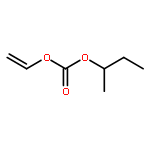




















![Phenol, 4-[(phenylimino)methyl]-, 1-acetate](/data/chemimg/1079300/13031-46-4.png)
![Phenol, 4-[(phenylimino)methyl]-, 1-acetate](/data/chemimg/1079300/13031-46-4_b.png)
![5H-Benzo[a]phenoxazin-5-one,9-(diethylamino)-](http://img.cochemist.com/ccimg/7400/7385-67-3.png)
![5H-Benzo[a]phenoxazin-5-one,9-(diethylamino)-](http://img.cochemist.com/ccimg/7400/7385-67-3_b.png)
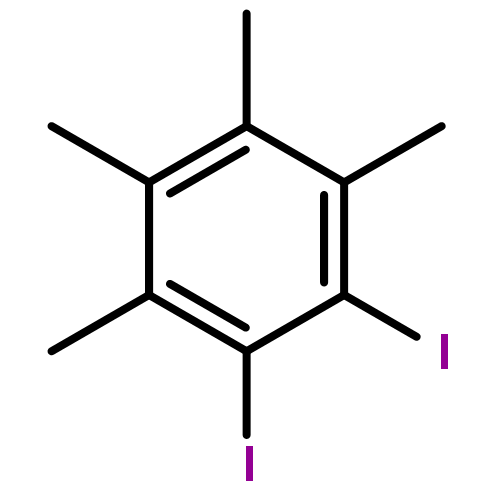
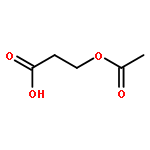

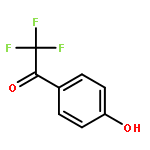



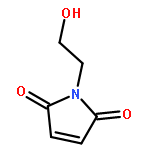
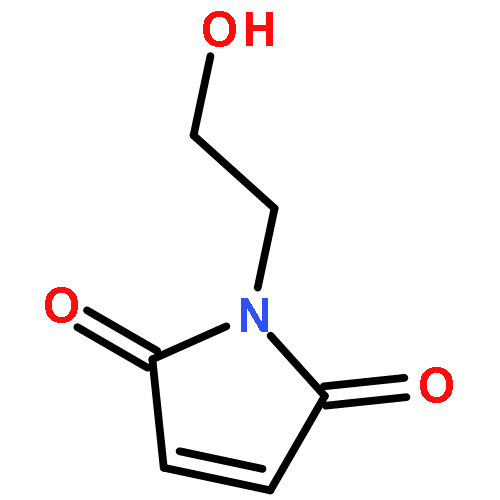


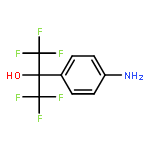
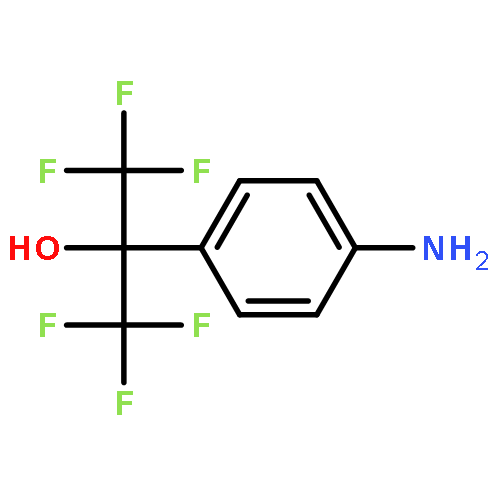


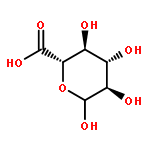














































































































































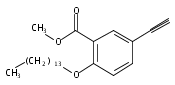























































































































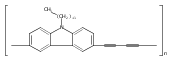



































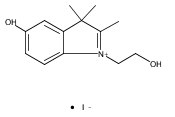








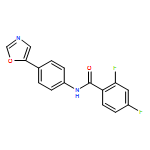

























































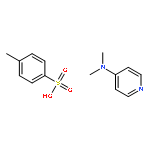
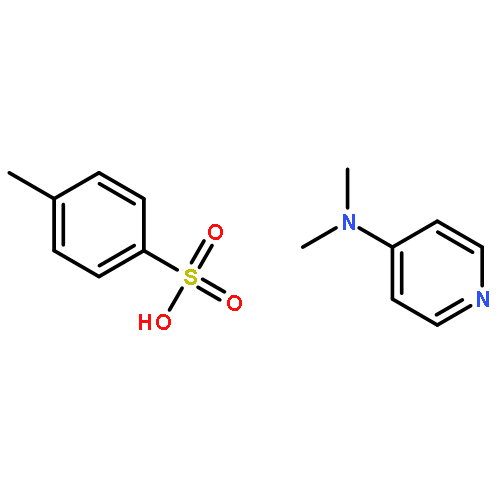


![Poly[[4,5-bis(methoxycarbonyl)-1,3-cyclopentanediyl]-1,2-ethenediyl]](http://img.cochemist.com/ccimg/82500/82441-77-8.png)
![Poly[[4,5-bis(methoxycarbonyl)-1,3-cyclopentanediyl]-1,2-ethenediyl]](http://img.cochemist.com/ccimg/82500/82441-77-8_b.png)
![Acetyl chloride, [2-(2-methoxyethoxy)ethoxy]-](http://img.cochemist.com/ccimg/63900/63881-16-3.png)
![Acetyl chloride, [2-(2-methoxyethoxy)ethoxy]-](http://img.cochemist.com/ccimg/63900/63881-16-3_b.png)
![Ethanol, 2-[2-(2-methoxyethoxy)ethoxy]-, 1-(4-methylbenzenesulfonate)](/data/chemimg/636900/62921-74-8.png)
![Ethanol, 2-[2-(2-methoxyethoxy)ethoxy]-, 1-(4-methylbenzenesulfonate)](/data/chemimg/636900/62921-74-8_b.png)
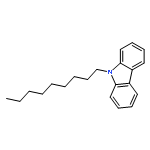


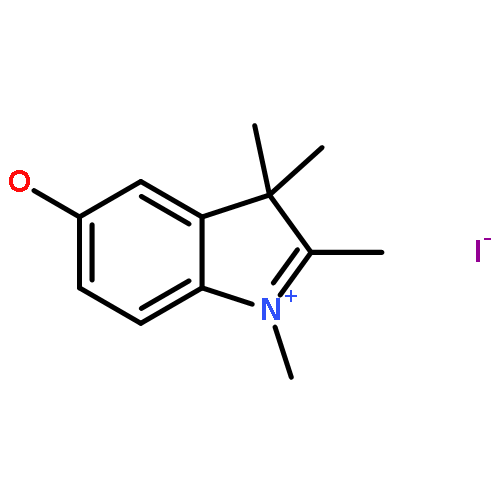







![3-[4-(HYDROXYMETHYL)PHENYL]-1-PROPANOL](http://img.cochemist.com/ccimg/36400/36308-36-8.png)
![3-[4-(HYDROXYMETHYL)PHENYL]-1-PROPANOL](http://img.cochemist.com/ccimg/36400/36308-36-8_b.png)


![1-[2-(BENZYLOXY)-6-HYDROXYPHENYL]ETHANONE](/data/chemimg/409600/25038-78-2.png)
![1-[2-(BENZYLOXY)-6-HYDROXYPHENYL]ETHANONE](/data/chemimg/409600/25038-78-2_b.png)
![Poly[(5,7-dihydro-1,3,5,7-tetraoxobenzo[1,2-c:4,5-c']dipyrrole-2,6(1H,3H)-diyl)-1,4-phenyleneoxy-1,4-phenylene]](http://img.cochemist.com/ccimg/25100/25036-53-7.png)
![Poly[(5,7-dihydro-1,3,5,7-tetraoxobenzo[1,2-c:4,5-c']dipyrrole-2,6(1H,3H)-diyl)-1,4-phenyleneoxy-1,4-phenylene]](http://img.cochemist.com/ccimg/25100/25036-53-7_b.png)





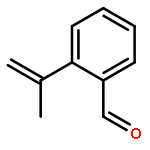


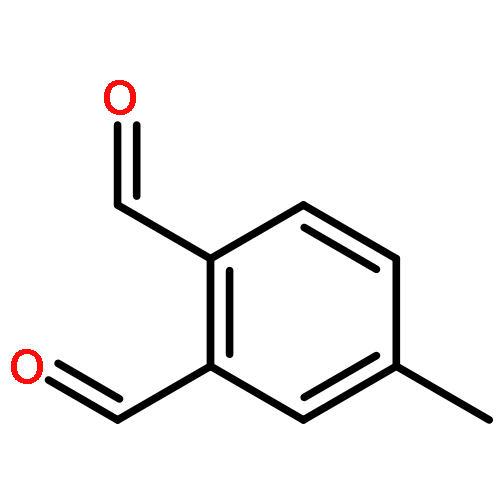


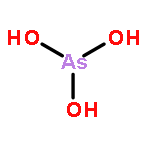

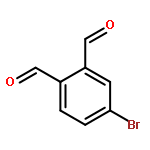



![3-Oxabicyclo[3.2.0]heptane-2,4-dione](http://img.cochemist.com/ccimg/4500/4462-96-8.png)
![3-Oxabicyclo[3.2.0]heptane-2,4-dione](http://img.cochemist.com/ccimg/4500/4462-96-8_b.png)















![Spiro[2H-1-benzopyran-2,2'-[2H]indole]-1'(3'H)-ethanol, 5'-hydroxy-3',3'-dimethyl-6-nitro-](/data/chemimg/171100/1253909-23-7.png)
![Spiro[2H-1-benzopyran-2,2'-[2H]indole]-1'(3'H)-ethanol, 5'-hydroxy-3',3'-dimethyl-6-nitro-](/data/chemimg/171100/1253909-23-7_b.png)
![Spiro[2H-1-benzopyran-2,2'-[2H]indole]-5',8-diol, 1',3'-dihydro-1',3',3'-trimethyl-6-nitro-](/data/chemimg/171100/1253909-21-5.png)
![Spiro[2H-1-benzopyran-2,2'-[2H]indole]-5',8-diol, 1',3'-dihydro-1',3',3'-trimethyl-6-nitro-](/data/chemimg/171100/1253909-21-5_b.png)

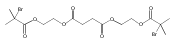






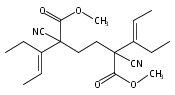










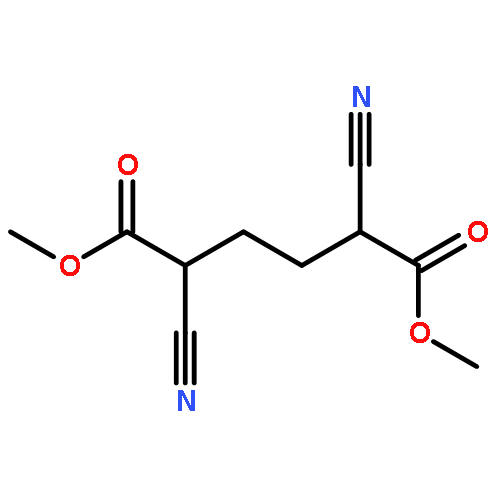
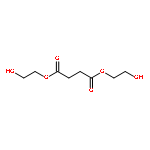










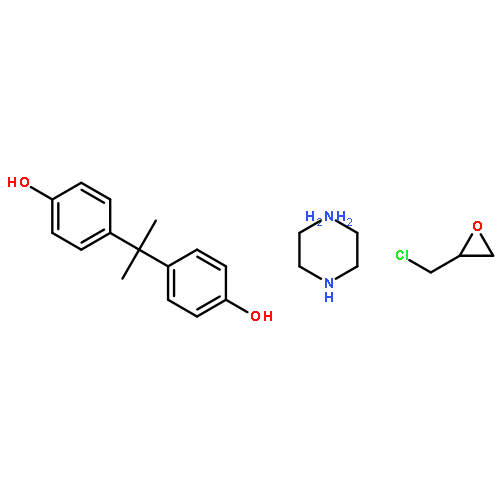
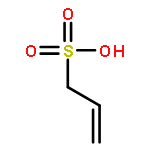
![Poly[(1,3-dihydro-1,3-isobenzofurandiyl)oxy]](http://img.cochemist.com/ccimg/27000/26966-00-7.png)
![Poly[(1,3-dihydro-1,3-isobenzofurandiyl)oxy]](http://img.cochemist.com/ccimg/27000/26966-00-7_b.png)
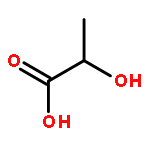
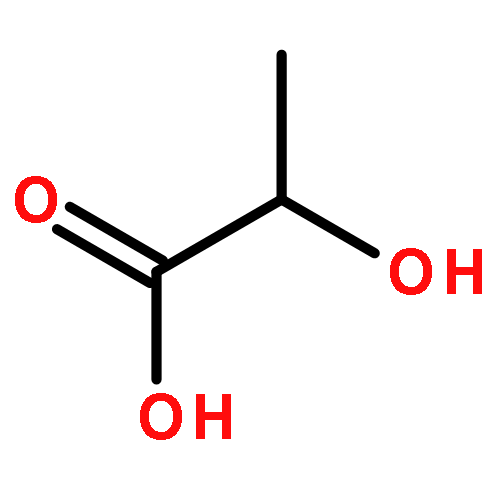
![Propanoic acid, 2-bromo-2-methyl-, 1,1'-[(2S)-1',3'-dihydro-1',3',3'-trimethyl-6-nitrospiro[2H-1-benzopyran-2,2'-[2H]indole]-5',8-diyl] ester](/data/chemimg/112000/960011-28-3.png)
![Propanoic acid, 2-bromo-2-methyl-, 1,1'-[(2S)-1',3'-dihydro-1',3',3'-trimethyl-6-nitrospiro[2H-1-benzopyran-2,2'-[2H]indole]-5',8-diyl] ester](/data/chemimg/112000/960011-28-3_b.png)