Co-reporter:Kazuaki Kuwata, Kengo Hanaya, Takeshi Sugai, Mitsuru Shoji
Tetrahedron: Asymmetry 2017 Volume 28, Issue 7(Issue 7) pp:
Publication Date(Web):15 July 2017
DOI:10.1016/j.tetasy.2017.05.007
The chemo-enzymatic synthesis of (R)-5-hydroxymethyl-2-isopropyl-5-methylcyclopent-1-en-1-yl trifluoromethylsulfonate, a potential chiral building block for polycyclic terpenoids containing a five–membered ring having isopropyl and angular methyl substituents, such as erinacin A and dolatriol, was achieved over 11 steps from ethyl 2-oxocyclopentane-1-carboxylate. The key synthetic precursor for this triflate was ethyl (1S,2R)-2-hydroxycyclopentanecarboxylate (>99% ee), which was prepared by a lipase-catalyzed enantioselective hydrolysis of the corresponding racemic acetate. The antipodal (S)-triflate is expected to be the synthetic intermediate for another group of terpenoids involving hamigeran B and stolonidiol. Enantiomerically pure (1R,2S)-hydroxyester (>99% ee) was prepared in high yield using the asymmetric reduction of the oxoester with commercially available carbonyl reductase, “Chiralscreen® OH”-E001.Download high-res image (60KB)Download full-size image
Co-reporter:Ban Fujitani, Kengo Hanaya, Shuhei Higashibayashi, Mitsuru Shoji, Takeshi Sugai
Tetrahedron 2017 Volume 73, Issue 51(Issue 51) pp:
Publication Date(Web):21 December 2017
DOI:10.1016/j.tet.2017.11.008
A unique tricyclic bisacetal, 2,6,9,11-tetraoxatricyclo[6.2.1.03,8]undecane observed in cotylenin A, was constructed in a simplified model compound, based on 6-O-methyl-4-keto-d-glucose as the scaffold to control of the stereochemistry. The key compound was an unprecedented spirolactone formed from the α-(α′-hydroxy)acyloxy dimethyl acetal, accompanied with stereoselective migration of an acyl group. Subsequent intramolecular acetalization between the hydroxy group at the C-3 and the C-1′ hemiacetal position furnished the desired tricyclic bisacetal.Download high-res image (225KB)Download full-size image
Co-reporter:Kazuaki Kuwata, Kengo Hanaya, Shuhei Higashibayashi, Takeshi Sugai, Mitsuru Shoji
Tetrahedron 2017 Volume 73, Issue 41(Issue 41) pp:
Publication Date(Web):12 October 2017
DOI:10.1016/j.tet.2017.08.056
1-Hydroxy-14-isopropyl-3β-methoxymethyl-7β,11β-dimethyl-3α-[((2-trimethylsilyl)ethoxy)methoxy]-1,2-secofusicocca-8,10(14)-dien-2-one, a highly functionalized 1,2-seco fusicoccane diterpene skeleton related to cotylenin A was synthesized in a convergent manner. The A ring segment, i. e., (1′R,2S, 2′E,5S)-2-methoxymethyl-5-[1′-methyl-3’-(trimethylstannyl)prop-2-enyl]-2-[((2″-trimethylsilyl)ethoxy)methoxy]cyclopentanone, was synthesized in 20.1% yield over 18 steps from known (S)-5-isopropenyl-2-methylcyclopent-1-enecarbaldehyde. This was coupled with the C ring segment, i. e., (R)-5-hydroxymethyl-2-isopropyl-5-methylcyclopent-1-en-1-yl trifluoromethylsulfonate, which was prepared according to our previous report. The Stille coupling reaction between alkenylstannane and sterically hindered triflate proceeded successfully in the presence of PEPPSI-SIPr (85%), and the total yield of the target molecule was 17.1% over the longest linear sequences (19 steps) from (S)-5-isopropenyl-2-methylcyclopent-1-enecarbaldehyde.Download high-res image (160KB)Download full-size image
Co-reporter:Hayato Okazaki, Kengo Hanaya, Mitsuru Shoji, Noriyasu Hada, Takeshi Sugai
Tetrahedron 2013 69(37) pp: 7931-7935
Publication Date(Web):
DOI:10.1016/j.tet.2013.07.018
Co-reporter:Jordi Calveras, Yasuhito Nagai, Israt Sultana, Yuji Ueda, Toshinori Higashi, Mitsuru Shoji, Takeshi Sugai
Tetrahedron 2010 66(24) pp: 4284-4291
Publication Date(Web):
DOI:10.1016/j.tet.2010.04.045
Co-reporter:Manabu Hamada, Yukihiro Niitsu, Chihiro Hiraoka, Ikuko Kozawa, Toshinori Higashi, Mitsuru Shoji, Kazuo Umezawa, Takeshi Sugai
Tetrahedron 2010 66(35) pp: 7083-7087
Publication Date(Web):
DOI:10.1016/j.tet.2010.07.013
Co-reporter:Manabu Hamada, Yoshikazu Inami, Yasuhito Nagai, Toshinori Higashi, Mitsuru Shoji, Seiichiro Ogawa, Kazuo Umezawa, Takeshi Sugai
Tetrahedron: Asymmetry 2009 Volume 20(Issue 18) pp:2105-2111
Publication Date(Web):23 September 2009
DOI:10.1016/j.tetasy.2009.07.049
3,8-Dioxatricyclo[3.2.1.02,4]octane-6-carboxylic acid, whose racemic form is readily available on a large scale, is a versatile starting material for the synthesis of carbasugars and carbocyclic biologically active natural products. In this study, the enzyme-catalyzed kinetic resolution was attempted on a variety of corresponding carboxylic esters. The hydrophobic and hydrophilic properties of ester substituents greatly affected the rate of reaction and the enantioselectivity. Hydrolysis of the corresponding 2′-chloroethyl ester with pig liver esterase worked well in a highly enantioselective manner (E = 116) to give the hydrolyzate (90.6% ee) and unreacted ester recovery (99.4% ee). The hydrolyzate is a precursor for (−)-oseltamivir phosphate, and a route to (3S,4S,5R)-(−)-3-epishikimic acid was developed from the recovered ester.2′-Chloroethyl (1R,2R,4R,5S,6R)-3,8-dioxatricyclo[3.2.1.02.4]octane-6-carboxylateC9H11O4ClEe = 99.4%[α]D23=-5.3 (c 1.02, CHCl3)Source of chirality: pig liver esterase-catalyzed hydrolysisAbsolute configuration: (1R,2S,4R,5S,6R)(1S,2R,4S,5R,6S)-3,8-Dioxatricyclo[3.2.1.02.4]octane-6-carboxylic acidC7H8O4Ee = 90.6%[α]D23=+11.7 (c 1.0, MeOH)Source of chirality: pig liver esterase-catalyzed hydrolysisAbsolute configuration: (1S,2R,4S,5R,6S)2′-Chloroethyl (1R,5R,6S)-5-hydroxy-7-oxabicyclo[4.1.0]hept-2-en-3-carboxylateC9H11O4ClEe = 99.4%[α]D23=+233 (c 1.08, MeOH)Source of chirality: pig liver esterase-catalyzed hydrolysisAbsolute configuration: (1R,5R,6S)(3S,4S,5R)-3,4,5-Trihydroxy-1-cylcohexene-1-carboxylic acidC7H10O5Ee = 99.4%[α]D25=-33.1 (c 0.34, H2O)Source of chirality: pig liver esterase-catalyzed hydrolysisAbsolute configuration: (3S,4S,5R)
Co-reporter:Aya Fujino
Advanced Synthesis & Catalysis 2008 Volume 350( Issue 11-12) pp:1712-1716
Publication Date(Web):
DOI:10.1002/adsc.200800191
Abstract
(R)-3-Hydroxy-3-methyl-5-hexanoic acid p-methoxybenzyl ester 1b was prepared by carbon-chain elongation on both termini of the starting material, (R)-3-benzyloxy-2-methylpropane-1,2-diol 2a, which was prepared by an over-expressed Bacillus subtilis epoxide hydrolase-catalyzed enantioselective hydrolysis of the racemic 1-benzyloxymethyl-1-methyloxirane 3. One of the key steps of the requisite transformation was the Rhodococcus rhodochrous NBRC 15564-mediated hydrolysis of a cyano group to a carboxyl group under neutral conditions, to exclude any racemization of the intermediate and/or product. The enantiomerically pure form of (R)-1b was applied to a new formal total synthesis of taurospongin A.
Co-reporter:Ryohei Kobayashi, Hanghang Huang, Manabu Hamada, Toshinori Higashi, Mitsuru Shoji, Takeshi Sugai
Journal of Molecular Catalysis B: Enzymatic (September 2012) Volume 81() pp:52-57
Publication Date(Web):1 September 2012
DOI:10.1016/j.molcatb.2012.05.006
To date, the enzyme-catalyzed kinetic resolution of the secondary alcohol [Ar-C*H(CH3)OH, Ar = 2′,4′,6′-triisopropylphenyl] has not been available, due to high steric hindrance around the hydroxy group. To achieve resolution, the reaction site was extended by the introduction of two kinds of spacers, [C(O)CH2] and [C(O)C**HCN]. In the first substrate, the recognition of remote chirality [ArC*H(CH3)OC(O)CH2OH] by acylation with Burkholderia cepacia lipase was examined by changing reaction conditions and acyl donors. An E = 22 in the preference of (1′R)-isomer, was recorded with vinyl acetate as an acyl donor at 25 °C. In the second substrate, there was a matched enantiomeric pair [stereoselective ratio at C-1′ = 15, in the preference of (1′R)-isomer] and a mismatched pair [stereoselective ratio at C-1′ = 2.5, in the preference of (1′S)-isomer] based on the relative stereochemistry between the two chiral centers [ArC*H(CH3)OC(O)C**HCNOH].Graphical abstractDownload full-size imageHighlight► Two hydroxyacetate spacers were examined for resolution of hindered alcohols. ► Burkholderia cepacia lipase showed E = 22 on racemic hydroxyacetate derivative. ► Preferred stereoisomer was reversed in cyano(hydroxy)acetate derivative. ► Both enantiomers of “stericol” became available through this study.
Co-reporter:Miyu Furuta, Mitsuru Shoji, Takeshi Sugai
Journal of Molecular Catalysis B: Enzymatic (October 2012) Volume 82() pp:8-11
Publication Date(Web):1 October 2012
DOI:10.1016/j.molcatb.2012.05.017
Among twelve incubated whole-cell yeast strains, two were found to selectively reduce (R)-1,2-(cyclohexylidenedioxy)hept-6-en-3-one, which was derived from d-mannitol. Pichia minuta JCM 3622 and Rhodotorula mucilaginosa NBRC 0889 afforded (2R,3S)-form (97% diastereomeric purity) and (2R,3R)-form (89% diastereomeric purity) of 1,2-(cyclohexylidenedioxy)hept-6-en-3-ols, respectively. As discussed above, the complementary yeast-mediated reduction provided the two diastereomeric glycerol derivatives in high enantiomeric excess.Graphical abstractDownload full-size imageHighlights► Microbial reduction of (R)-1,2-(cyclohexylidenedioxy)hept-6-en-3-one was examined. ► Reduction with Pichia minuta provided (3S)-alcohol. ► Reduction with Rhodotorula mucilaginosa provided (3R)-alcohol. ► Two complementary diastereomeric glycerol derivatives in high ee became available.
Co-reporter:Chika Abe, Takahiro Sugawara, Takuya Machida, Toshinori Higashi, Kengo Hanaya, Mitsuru Shoji, Chen Cao, Takuro Yamamoto, Tomoko Matsuda, Takeshi Sugai
Journal of Molecular Catalysis B: Enzymatic (October 2012) Volume 82() pp:86-91
Publication Date(Web):1 October 2012
DOI:10.1016/j.molcatb.2012.06.010
Whole-cell yeasts and mold-catalyzed reduction of two fluorinated acetophenone derivatives with very bulky substituents on ortho position of aromatic ring, (±)-1′-(2-tert-butyl-2-methyl-1,3-benzodioxol-4-yl)-2′,2′-difluoroethanone and (±)-1′-(2-tert-butyl-2-methyl-1,3-benzodioxol-4-yl)-2′,2′,2′-trifluoroethanone were examined. On the former substrate, Geotrichum candidum NBRC 5767 showed high re-facially selective attack of hydride, while with Pichia angusta JCM 3620, complementary si-facially selective attack proceeded. G. candidum NBRC 5767 was revealed to be potent biocatalyst which provides (1′S)-alcohols from both substrates in a highly facially selective manner. Some unknown reductases were suggested responsible for those reductions, other than so far having been reported acetophenone reductase and trifluoromethyl ketone reductase from G. candidum, comparing the results obtained by applying those enzymes.Graphical abstractDownload full-size imageHighlights► Whole-cell biocatalyst-mediated reduction of two fluorinated acetophenone derivatives with very bulky substituents of aromatic ring was examined. ► Complementary facial selectivity between G. candidum and P. angusta on the attack of hydride to 1′-(2-tert-butyl-2-methyl-1,3-benzodioxol-4-yl)-2′,2′-difluoroethanone was observed. ► So far reported enzyme preparations of acetophenone reductase and trifuoromethyl ketone reductase from G. candidum were applied to the present substrates. ► The above-mentioned two enzymes were not responsible for the formation of major stereoisomers made by whole-cell biocatalyst.
Co-reporter:Manabu Hamada, Toshinori Higashi, Mitsuru Shoji, Kazuo Umezawa, Takeshi Sugai
Journal of Molecular Catalysis B: Enzymatic (November 2010) Volume 67(Issues 1–2) pp:78-84
Publication Date(Web):1 November 2010
DOI:10.1016/j.molcatb.2010.07.009
Candida antarctica lipase B (Novozym 435)-catalyzed transesterification on methyl (±)-3,4-di-O-acetyl-5-O-(tert-butyldimethyl)silyl-3-epi-shikimate worked highly regio- and enantioselective manner. Only (3R,4S,5S)-isomer reacted with an E value over 500, exclusively on C-3 acetate. The regio- and enantioselectivity were greatly affected by the substitution pattern on the hydroxy groups. Towards polyoxygenated carbacycles, the above-mentioned highly selective transformation enabled the subsequent stereoselective inversion and dihydroxylation, to give methyl (3S,4R,5S)-3,4,5-triacetoxy-1-cyclohexenecarboxylate [antipode of naturally occurring methyl (−)-3,4,5-tri-O-acetylshikimate], and methyl (1R,2S,3S,4R,5R)-3,4-diacetoxy-5-(tert-butyldimethyl)silyloxy-1,2-dihydroxy-cyclohexanecarboxylate.Graphical abstractDownload full-size imageResearch highlights▶ C. antarctica lipase B worked on an deriv. of methyl (±)-3-epi-shikimate regioselectively with E > 500.
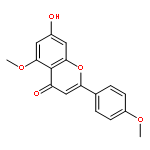
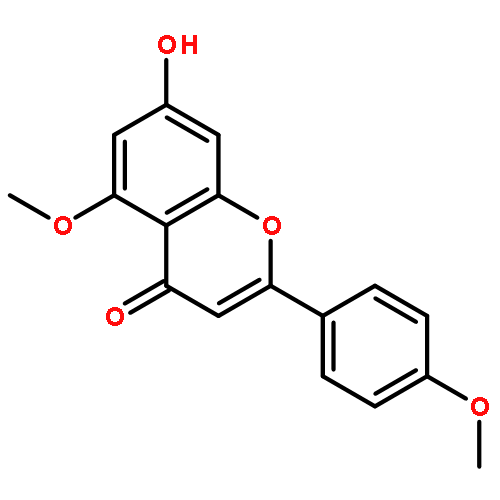
Co-reporter:Shun Hanamura, Kengo Hanaya, Mitsuru Shoji, Takeshi Sugai
Journal of Molecular Catalysis B: Enzymatic (June 2016) Volume 128() pp:19-26
Publication Date(Web):1 June 2016
DOI:10.1016/j.molcatb.2016.03.001
•Lipase-catalyzed transesterification regioselectively proceeded at C-4′ acetate of a peracetylated form of rhoifolin.•Acacetin was synthesized from naringin in 65% overall yield.•Lipase-catalyzed transesterification regioselectively proceeded at C-4′ acetate of a peracetylated form of piceid.•Resveratrol 3,5-diglucoside was synthesized from piceid in 62% overall yield.•The blocking of the proper position with pre-installed sugar side chain was effective for the better regioselectivity.Acacetin and resveratrol 3,5-di-O-β-glucopyranoside were synthesized from naturally abundant naringin and piceid in 65% and 62% overall yield, respectively. The key steps were the regioselective deacetylation of the peracetates of the glycosylated forms with Candida antarctica lipase B (Novozym 435) and Burkholderia cepacia lipase (Amano PS-IM). Deacetylation occurred exclusively at the least hindered position of the aromatic moieties and all acetyl groups in the sugar side chain remained intact. This excellent selectivity enabled regiospecific transformation of the liberated phenolic hydroxy groups, resulting in efficient synthesis of the target molecules.Download high-res image (151KB)Download full-size image
Co-reporter:Maki Sakamoto, Manabu Hamada, Toshinori Higashi, Mitsuru Shoji, Takeshi Sugai
Journal of Molecular Catalysis B: Enzymatic (June 2010) Volume 64(Issues 1–2) pp:96-100
Publication Date(Web):1 June 2010
DOI:10.1016/j.molcatb.2010.02.008
High enantioselectivity (E 94) was observed in Candida antarctica lipase B-catalyzed hydrolysis of the corresponding acetate of racemic title compound. The reaction rate of the slow (S)-isomer was effectively suppressed by lowering the reaction temperature from 25 °C to 5 °C, to allow a five times increase of the enantioselectivity. The ee of the (S)-isomer, reached 97.8% at the reasonable conversion (52%) as the unreacted recovery, and the repetition of the enzymatic reaction provided pure enantiomer. The undesired (R)-isomer was oxidized with IBX and reduced with whole-cell biocatalysis with Candida floricola JCM 9439 to (S)-isomer (63.2% ee), which serves as the enantiomerically enriched substrate for further lipase-catalyzed resolution. The combination of total processes provided over 50% yield of the pure (S)-isomer, exceeding the theoretical limit for the enantiomeric resolution of racemate.
Co-reporter:Kazunori Kitsuda, Jordi Calveras, Yasuhito Nagai, Toshinori Higashi, Takeshi Sugai
Journal of Molecular Catalysis B: Enzymatic (July 2009) Volume 59(Issues 1–3) pp:197-200
Publication Date(Web):1 July 2009
DOI:10.1016/j.molcatb.2009.02.014
A new chemo-enzymatic route to tetra-O-acetyl-l-ribofuranose from d-lyxose is described. Lipase-catalyzed regioselective transesterification of acetate proceeded on C-4 of the d-lyxopyranoside. Subsequently, stereochemistry of liberated secondary alcohol was inverted by way of oxidation and reduction by IBX and NaBH(OAc)3 to give l-ribopyranoside. After deprotection, the furanose–pyranose isomeric mixture was converged to the target molecule, taking advantage of lipase-catalyzed preferential acetylation of primary alcohol on C-5.
Co-reporter:Shohei Taketomi, Masayoshi Asano, Toshinori Higashi, Mitsuru Shoji, Takeshi Sugai
Journal of Molecular Catalysis B: Enzymatic (December 2012) Volume 84() pp:83-88
Publication Date(Web):1 December 2012
DOI:10.1016/j.molcatb.2012.01.020
To synthesize (R)-terbutaline hydrochloride, a potent β2-adrenoceptor-stimulating agent, asymmetric reduction of a substituted α-chloroacetophenone derivative with cultured whole-cell biocatalyst of the yeast Williopsis californica JCM 3600 was developed as the key reaction. The reduction proceeded by a si-facial attack of hydride in a highly enantioselective manner. Co-factor generation was enhanced by applying glycerol as the carbon source.Graphical abstractDownload full-size imageHighlights► Towards (R)-terbutalin, asymmetric reduction of a chloroketone was examined. ► Reduction with Williopsis californica proceeded by a si-facial hydride attack. ► In the reduction, co-factor generation was enhanced by glycerol as the carbon source. ► Substrate was easily provided by lipase-catalyzed deprotection of an intermediate.
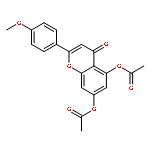
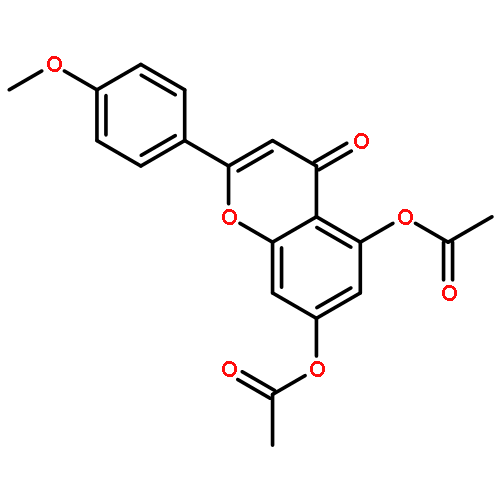
Co-reporter:Ryohei Kobayashi, Takasi Itou, Kengo Hanaya, Mitsuru Shoji, Noriyasu Hada, Takeshi Sugai
Journal of Molecular Catalysis B: Enzymatic (August 2013) Volume 92() pp:14-18
Publication Date(Web):1 August 2013
DOI:10.1016/j.molcatb.2013.03.002
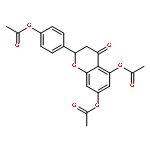
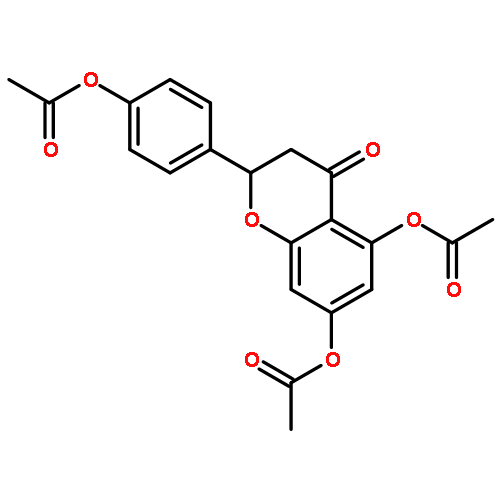
•Luteolin was synthesized in 8 steps and 36% overall yield from naringin.•Lipase-catalyzed transesterification proceeded regioselectively on C-4′.•Oxidation provided desired C-3′and C-4′ catechol functionality.•Diglycoside side chain worked as the protective group on C-7 hydroxy group.Luteolin [3′,4′,5,7-tetrahydroxyflavone], having multiple biological effects such as anti-inflammation, anti-allergy and anti-cancer, was prepared by chemo-enzymatic synthesis from naringin, a naturally abundant flavonoid glycoside. On the occasion of Candida antarctica lipase B (Novozym 435)-catalyzed transesterification on peracetylated form of naringin, an acetate on C-4′ was exclusively deprotected to give the key intermediate. The oxidation with 2-iodoxybenzoic acid (IBX) followed by the reductive workup provided regioselectively C-3′and C-4′ catechol functionality. After protection of the above-mentioned diol with methoxymethyl (MOM) groups and subsequent hydrolysis of all acetyl groups, a dehydrogenative introduction of double bond between C-2 and C-3 was done by the treatment with I2. Acid-catalyzed simultaneous removal of MOM groups and glycoside provided luteolin in total 8 steps and 36% overall yield from the starting material. Throughout the synthesis, diglycoside side chain effectively worked as the protective group on C-7 hydroxy group.Download full-size image
Co-reporter:Daisuke Tokoshima, Kengo Hanaya, Mitsuru Shoji, Takeshi Sugai
Journal of Molecular Catalysis B: Enzymatic (15 December 2013) Volume 97() pp:95-99
Publication Date(Web):15 December 2013
DOI:10.1016/j.molcatb.2013.07.021
•P. minuta reduced 2-chloro-1-(3-nitrophenyl)ethanone to give (R)-alcohol.•Substrate is easily prepared by nitration of 2-chlorophenylethanone.•Amberlite XAD-7 effectively worked for a reservoir of the substrate.•The product was transferred to (R)-phenylephrine, an α1-adrenergic receptor agonist.•Five-step transformation proceeded without loss of the ee (98.0%).The incubated whole-cell biocatalyst of Pichia minuta JCM 3622 reduced 2-chloro-1-(3-nitrophenyl)ethanone to provide (R)-2-chloro-1-(3-nitrophenyl)ethanol with 99.2% ee in 87% isolated yield in the presence of Amberlite XAD-7 as a reservoir for the hydrophobic, crystalline and toxic substrate. The product was transformed to (R)-1-(3-hydroxyphenyl)-2-methylaminoethanol (phenylephrine, 1a), a selective α1-adrenergic receptor agonist, in 98.0% ee over five steps.Download full-size image
Co-reporter:Kento Asami, Takuya Machida, Sonna Jung, Kengo Hanaya, Mitsuru Shoji, Takeshi Sugai
Journal of Molecular Catalysis B: Enzymatic (15 December 2013) Volume 97() pp:106-109
Publication Date(Web):15 December 2013
DOI:10.1016/j.molcatb.2013.08.003
•W. californica reduced 1-[3,5-bis(dimethylcarbamoyloxy)phenyl]-2-chloroethanone to give (R)-alcohol.•Substrate for the reduction was easily prepared by chemo-enzymatic procedure from commercially available 1-(3,5-diacetoxyphenyl)ethanone.•Addition of glycerol (10%, v/v) assisted the co-factor regeneration as well as the dissolution of the crystalline substrate.•The product was transferred to enantiomerically pure (R)-bambuterol, a prodrug of (R)-terbutalin.•Seven-step synthesis of (R)-bambuterol was achieved from commercially available material in total 30% yield.To achieve the synthesis of (R)-bambuterol, a prodrug of (R)-terbutaline, asymmetric reduction of 1-[3,5-bis(dimethylcarbamoyloxy)phenyl]-2-chloroethanone with whole cells of Williopsis californica JCM 3600 pre-incubated on glycerol as a carbon source was examined. Initially, the insolubility of this crystalline substrate (mp 126–127 °C) in the incubation broth was an obstacle that needed to be overcome. To solve the problem, the concentration of glycerol was increased to 10% during reduction. Glycerol worked well for recycling oxido-reduction cofactors and for enhancing the water solubility of the substrate. The reduction proceeded smoothly to give enantiomerically pure (R)-alcohol in 81% isolated yield.Download full-size image

![7-Oxabicyclo[4.1.0]hept-2-ene-3-carboxylic acid, 5-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, methyl ester, (1R,5R,6R)-](/data/chemimg/3739700/1486515-86-9.png)
![7-Oxabicyclo[4.1.0]hept-2-ene-3-carboxylic acid, 5-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, methyl ester, (1R,5R,6R)-](/data/chemimg/3739700/1486515-86-9_b.png)
![2-Propenoic acid, 3-[4-(acetyloxy)-3,5-bis(3-methyl-2-butenyl)phenyl]-, methyl ester, (E)-](http://img.cochemist.com/ccimg/107400/107390-44-3.png)
![2-Propenoic acid, 3-[4-(acetyloxy)-3,5-bis(3-methyl-2-butenyl)phenyl]-, methyl ester, (E)-](http://img.cochemist.com/ccimg/107400/107390-44-3_b.png)
![1H-Pyrazole,5-(4-methylphenyl)-1-[4-(methylsulfonyl)phenyl]-3-(trifluoromethyl)-](http://img.cochemist.com/ccimg/675700/675605-72-8.png)
![1H-Pyrazole,5-(4-methylphenyl)-1-[4-(methylsulfonyl)phenyl]-3-(trifluoromethyl)-](http://img.cochemist.com/ccimg/675700/675605-72-8_b.png)


![1,4-Dioxaspiro[4.5]dec-7-ene, 7-methyl-](http://img.cochemist.com/ccimg/83400/83313-55-7.png)
![1,4-Dioxaspiro[4.5]dec-7-ene, 7-methyl-](http://img.cochemist.com/ccimg/83400/83313-55-7_b.png)
![N-ETHYL-N-(2-HYDROXYCYCLOHEXYL)-4-[(1-METHYL-1H-TETRAZOL-5-YL)SUL<WBR />FANYL]BUTANAMIDE](http://img.cochemist.com/ccimg/78900/78844-86-7.png)
![N-ETHYL-N-(2-HYDROXYCYCLOHEXYL)-4-[(1-METHYL-1H-TETRAZOL-5-YL)SUL<WBR />FANYL]BUTANAMIDE](http://img.cochemist.com/ccimg/78900/78844-86-7_b.png)
![2-[(1,1-dimethylpiperidinium-3-yl)oxy]-2-oxo-1,1-diphenylethanolate](http://img.cochemist.com/ccimg/26000/25990-43-6.png)
![2-[(1,1-dimethylpiperidinium-3-yl)oxy]-2-oxo-1,1-diphenylethanolate](http://img.cochemist.com/ccimg/26000/25990-43-6_b.png)
![5-hydroxy-2-(4-methoxyphenyl)-7-[3,4,5-trihydroxy-6-[(3,4,5-trihydroxy-6-methyloxan-2-yl)oxymethyl]oxan-2-yl]oxychromen-4-one](http://img.cochemist.com/ccimg/20700/20633-93-6.png)
![5-hydroxy-2-(4-methoxyphenyl)-7-[3,4,5-trihydroxy-6-[(3,4,5-trihydroxy-6-methyloxan-2-yl)oxymethyl]oxan-2-yl]oxychromen-4-one](http://img.cochemist.com/ccimg/20700/20633-93-6_b.png)


![4H-1-Benzopyran-4-one,7-[[2-O-(6-deoxy-伪-L-mannopyranosyl)-尾-D-glucopyranosyl]oxy]-5-hydroxy-2-(4-hydroxyphenyl)-](http://img.cochemist.com/ccimg/17400/17306-46-6.png)
![4H-1-Benzopyran-4-one,7-[[2-O-(6-deoxy-伪-L-mannopyranosyl)-尾-D-glucopyranosyl]oxy]-5-hydroxy-2-(4-hydroxyphenyl)-](http://img.cochemist.com/ccimg/17400/17306-46-6_b.png)