Co-reporter:Robert Otto, Robert Penzis, Friedemann Gaube, Thomas Winckler, Dorothea Appenroth, Christian Fleck, Christian Tränkle, Jochen Lehmann, Christoph Enzensperger
European Journal of Medicinal Chemistry 2014 Volume 87() pp:63-70
Publication Date(Web):24 November 2014
DOI:10.1016/j.ejmech.2014.09.048
•In search for novel anti-Alzheimer drugs we synthesized and tested 7 new compounds.•β- and γ-carbolines with similar residues allow direct comparison.•Cholinesterase inhibition, NMDA antagonism, and effect on M1 receptors.•Compounds antagonize scopolamine-induced cognitive impairment in radial maze test.•γ-Carbolines, esp. compound 6 showed very promising effects in vivo.Nine novel β- and γ-carboline derivatives bearing either methyl-, propargyl- or phenethyl-residues at the indole nitrogen were synthesized and tested as potential anti-Alzheimer drugs. Antagonism of recombinantly expressed NMDA receptors, inhibition of cholinesterases, and radical scavenging properties were determined for all compounds. Some were additionally tested in vivo for their ability to reverse scopolamine-induced cognitive impairment in an 8-arm radial maze experiment with rats. For the most promising candidates, the interaction with muscarinic M1 receptors was also investigated. With this set of compounds assays the influence of the scaffold itself and the substituents can be investigated separately. 5-Methyl-γ-carboline (6) was the most potent (0.25 μmol/100 g b.w.) compound in the in vivo test and might be a good starting point for the development of novel anti-Alzheimer drugs.
Co-reporter:Dina Robaa;Robert Kretschmer;Oliver Siol;Shams ElDin AbulAzm;ElSayeda ElKhawass;Jochen Lehmann
Archiv der Pharmazie 2011 Volume 344( Issue 1) pp:28-36
Publication Date(Web):
DOI:10.1002/ardp.201000121
Abstract
To further investigate SAR in the class of azecine-type dopamine receptor antagonists, we synthesized a series of derivatives, substituted at the indole-NH of the lead compound LE300 by different alkyl chains in addition to phenylpropyl, allyl, propargyl, and acetyl residues. The affinities of the target compounds for all human dopamine receptors (D1–D5) were investigated by radioligand binding assay and their functionality by a calcium assay. Both the affinities and selectivities for the dopamine receptors were found to be affected by the nature of the substituent. The N14-methylated derivative displayed the highest affinities for all D-receptors. In general, the affinities decreased with increasing chain length of the N-alkyl. Different substituents, partly led to altered affinity, and selectivity profile when compared with our lead LE300.
Co-reporter:Dina Robaa;Shams ElDinAbulAzm;Jochen Lehmann
Chemistry & Biodiversity 2011 Volume 8( Issue 3) pp:431-439
Publication Date(Web):
DOI:10.1002/cbdv.201000317
Abstract
Dibenzazecines are a novel class of dopamine receptor antagonists, characterized by their high affinities as well as their tendency for D1 selectivity. Hitherto, the most active dibenzazecines were phenolic in nature; a 3-OH substituent was found to result in the highest affinities. However, the phenolic nature of these compounds mostly renders them unsuitable for in vivo application, due to the poor pharmacokinetic profile, imparted by the phenolic group. A novel dibenzazecine derivative was prepared, with methylenedioxy moiety, connecting C(2) amd C(3), instead of the 3-OH group. The newly synthesized derivative 3 showed high affinities similar to the lead LE404, displaying nanomolar affinities for all dopamine receptor subtypes. Its dibrominated derivative 4, though exhibiting almost a fivefold decrease in affinities, still displayed nanomolar ones for all dopamine receptors, except for D4. In a functional Ca2+ assay, both compounds 3 and 4 were found to possess antagonistic properties towards the dopamine receptors.
Co-reporter:Maria Schulze;Oliver Siol;Werner Meise;Jochen Lehmann
Archiv der Pharmazie 2010 Volume 343( Issue 4) pp:207-214
Publication Date(Web):
DOI:10.1002/ardp.200900267
Abstract
The affinities of tetrahydroprotoberberines for dopamine receptors dramatically decrease after cleaving the central C-N bond to the analogous ten-membered dibenzo[c,g]azecines [1]. In the present work, we also synthesized eleven-membered homologues of these heterocycles and measured the affinities of the resulting dibenzazaundecenes and their underlying homoberberines for human dopamine receptors as well as the cytotoxic effects of all target compounds on human glia cells. The tetracyclic iso-C-homoberberine-derivatives revealed to be D4-selective antagonists, while all other active compounds showed a significant D1/D5 selectivity. Distances in energy-minimized conformations were measured in order to explain our findings.
Co-reporter:Maria Schulze, Franziska K.U. Müller, Jennifer M. Mason, Helmar Görls, Jochen Lehmann, Christoph Enzensperger
Bioorganic & Medicinal Chemistry 2009 Volume 17(Issue 19) pp:6898-6907
Publication Date(Web):1 October 2009
DOI:10.1016/j.bmc.2009.08.028
The moderately flexible 7-methyl-5,6,7,8,9,14-hexahydrodibenz[d,g]azecines are known to be potent dopamine receptor antagonists, whereas the corresponding rigid dibenzo[d,g]quinolizines are inactive. We built the scaffolds of dibenzo[c,g], [c,f] and [d,f]azecines and together with their ring closed, more rigid precursors, evaluated the affinities for the human D1–D5 receptors (radioligand binding) as well as the functionalities (calcium assay) and thus investigated the influence of annelation and conformative flexibility of these compounds on their affinity for human cloned dopamine receptors.

![DIBENZ[D,G]AZECINE, 5,6,7,8,9,14-HEXAHYDRO-7-METHYL-](http://img.cochemist.com/ccimg/874700/874656-33-4.png)
![DIBENZ[D,G]AZECINE, 5,6,7,8,9,14-HEXAHYDRO-7-METHYL-](http://img.cochemist.com/ccimg/874700/874656-33-4_b.png)
![Thieno[2,3-f][3]benzazecine, 4,5,6,7,8,13-hexahydro-6-methyl-](http://img.cochemist.com/ccimg/874700/874656-31-2.png)
![Thieno[2,3-f][3]benzazecine, 4,5,6,7,8,13-hexahydro-6-methyl-](http://img.cochemist.com/ccimg/874700/874656-31-2_b.png)
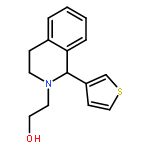
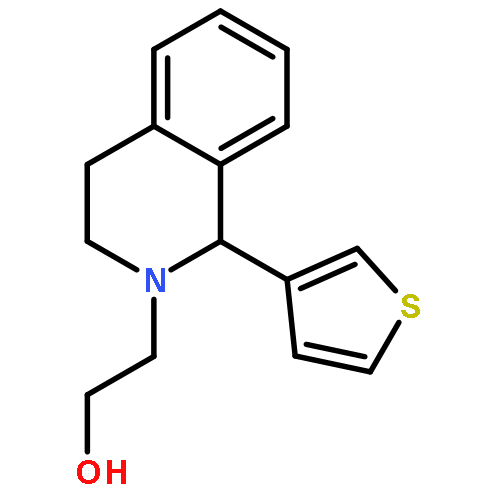
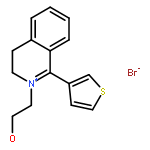
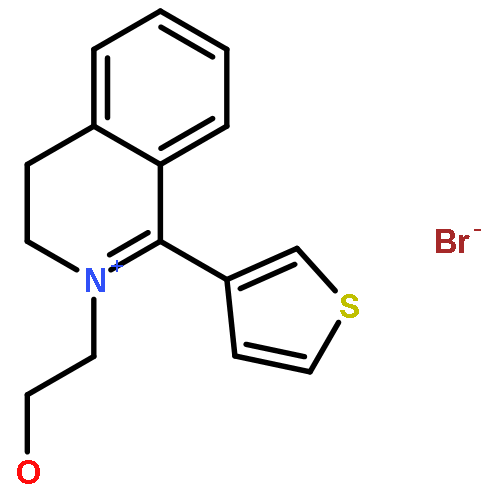


![6H-Benzo[a][1]benzothieno[3,2-h]quinolizine, 5,8,9,14b-tetrahydro-](http://img.cochemist.com/ccimg/107100/107042-09-1.png)
![6H-Benzo[a][1]benzothieno[3,2-h]quinolizine, 5,8,9,14b-tetrahydro-](http://img.cochemist.com/ccimg/107100/107042-09-1_b.png)
![5H-Benzo[a]thieno[3,2-h]quinolizine, 4,7,8,12b-tetrahydro-](http://img.cochemist.com/ccimg/107100/107042-01-3.png)
![5H-Benzo[a]thieno[3,2-h]quinolizine, 4,7,8,12b-tetrahydro-](http://img.cochemist.com/ccimg/107100/107042-01-3_b.png)
![Isoquinolinium, 3,4-dihydro-2-[2-(3-thienyl)ethyl]-, bromide](http://img.cochemist.com/ccimg/107100/107041-99-6.png)
![Isoquinolinium, 3,4-dihydro-2-[2-(3-thienyl)ethyl]-, bromide](http://img.cochemist.com/ccimg/107100/107041-99-6_b.png)
![6H-Dibenzo[a,h]quinolizine, 5,8,9,13b-tetrahydro-](http://img.cochemist.com/ccimg/78000/77929-54-5.png)
![6H-Dibenzo[a,h]quinolizine, 5,8,9,13b-tetrahydro-](http://img.cochemist.com/ccimg/78000/77929-54-5_b.png)
![5H-Pyrido[4,3-b]indole, 5-methyl-](http://img.cochemist.com/ccimg/61500/61406-19-7.png)
![5H-Pyrido[4,3-b]indole, 5-methyl-](http://img.cochemist.com/ccimg/61500/61406-19-7_b.png)
![2-(Benzo[b]thiophen-3-yl)ethanamine](http://img.cochemist.com/ccimg/14600/14585-66-1.png)
![2-(Benzo[b]thiophen-3-yl)ethanamine](http://img.cochemist.com/ccimg/14600/14585-66-1_b.png)



![9-methyl-9H-Pyrido[3,4-b]indole](http://img.cochemist.com/ccimg/2600/2521-07-5.png)
![9-methyl-9H-Pyrido[3,4-b]indole](http://img.cochemist.com/ccimg/2600/2521-07-5_b.png)


![5H-Pyrido[4,3-b]indole](http://img.cochemist.com/ccimg/300/244-69-9.png)
![5H-Pyrido[4,3-b]indole](http://img.cochemist.com/ccimg/300/244-69-9_b.png)