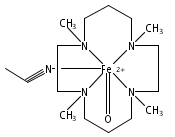
Co-reporter:Tatsuhiko Honda;Tatsuaki Nakanishi;Kei Ohkubo;Shunichi Fukuzumi
The Journal of Physical Chemistry C August 26, 2010 Volume 114(Issue 33) pp:14290-14299
Publication Date(Web):2017-2-22
DOI:10.1021/jp105116y
The reaction of Sn(DPP)(OH)2 (DPP2− = 2,3,5,7,8,10,12,13,15,17,18,20-dodecaphenylporphyrin dianion) with H2F16DPPCOOH (2,3,7,8,12,13,17,18-octakis(3,5-difluorophenyl)-5-(4-carboxyphenyl)-10,15,20-triphenylporphyrin) afforded a porphyrin triad, Sn(DPP)(H2F16DPPCOO)2 (1), in which the Sn(DPP) unit is linked with the two H2F16DPPCOO− units by strong coordination bonds. The H2F16DPPCOO− unit of Sn(DPP)(H2F16DPPCOO)2 was diprotonated by the reaction with trifluoroacetic acid (CF3COOH) to afford a robust electron acceptor−donor−acceptor porphyrin triad, Sn(DPP){(H4F16DPPCOO)(CF3COO)2}2 (2), in which the Sn(DPP) unit and the H4F16DPP2+COO− (H4F16DPPCOO+) unit act as an electron donor and an acceptor, respectively. The photodynamics of 1 was examined by femtosecond laser flash photolysis measurements in PhCN to reveal that the energy transfer occurs from the singlet excited state of the Sn(DPP) unit to the H2F16DPPCOO− unit to generate the singlet excited state of H2F16DPPCOO−. In contrast to the case of 1, the transient absorption spectra of 2 that contains the diprotonated form (H4F16DPPCOO+), observed by femtosecond laser flash photolysis, clearly indicated the occurrence of fast electron transfer from the singlet excited state of the Sn(DPP) unit to the H4F16DPPCOO+ unit. The resulting singlet electron-transfer (ET) state composed of Sn(DPP)•+ and H4F16DPPCOO• decays to the ground state with the rate constant of 1.4 × 1010 s−1 in competition with generation of the triplet ET state, which was also detected by the nanosecond transient absorption spectroscopy. The lifetime of the triplet ET state (50 μs) was much longer than that of the singlet ET state (71 ps) due to the spin-forbidden character of the back electron-transfer process.
Co-reporter:Yuta Saegusa, Tomoya Ishizuka, Yoshihito Shiota, Kazunari Yoshizawa, and Takahiko Kojima
The Journal of Organic Chemistry 2017 Volume 82(Issue 1) pp:322-330
Publication Date(Web):December 14, 2016
DOI:10.1021/acs.joc.6b02419
We report herein unique stepwise protonation at inner imino-nitrogen atoms of a freebase derivative of a quadruply fused porphyrin (H2QFP), which has been newly synthesized. H2QFP has been revealed to have the two inner NH protons on the two nonfused pyrroles by X-ray diffraction analysis and 1H NMR spectroscopy. The first protonation at one of the two imino-nitrogen atoms of the fused pyrroles smoothly proceeds with trifluoroacetic acid (TFA) in CH2Cl2 and the equilibrium constant (K1) of the protonation has been determined to be (1.3 ± 0.1) × 105 M–1. In contrast, the second protonation at the other imino-nitrogen atom is hard to occur unless a large excess amount of TFA is used, as reflected on a much smaller equilibrium constant, K2 = 7.3 ± 0.3 M–1. The stepwise protonation is ascribed to the structural rigidity caused by the ring fusion and the resultant steric repulsion among inner NH atoms of the diprotonated form. Electrochemical studies have revealed that protonation at the pyrrole nitrogen atoms caused positive shifts of the reduction potentials of the QFP derivatives. In addition, the ESR spectrum of the electrochemically one-electron-reduced monoprotonated QFP derivative showed well-resolved hyperfine splitting to represent its unsymmetrical electronic structure due to the monoprotonation.
Co-reporter:Wataru Suzuki;Hiroaki Kotani;Tomoya Ishizuka;Yoshihito Shiota;Kazunari Yoshizawa
Chemical Communications 2017 vol. 53(Issue 47) pp:6359-6362
Publication Date(Web):2017/06/08
DOI:10.1039/C7CC03635C
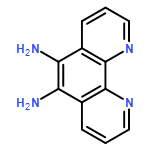
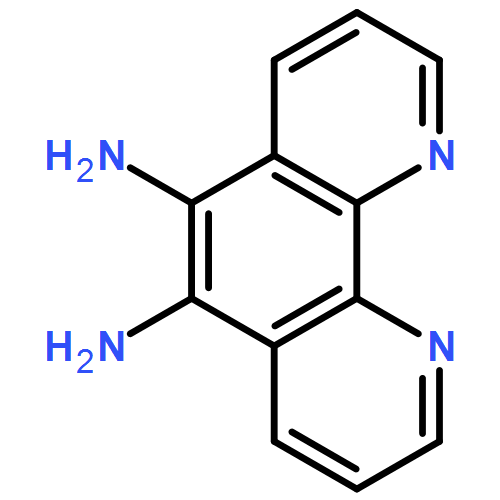
The thermodynamic stability of diprotonated saddle-distorted dodecaphenylporphyrin (H4DPP2+(X−)2) was controlled by the hydrogen-bonding strength of conjugate bases (X−) of strong acids (HX) or acids (R+-COOH) having positively charged moieties. The thermodynamic control of H4DPP2+(X−)2 made it possible to achieve selective formation of supramolecular hetero-triads, H4DPP2+(X−)(Cl−).
Co-reporter:Hiroumi Mitome; Tomoya Ishizuka; Hiroaki Kotani; Yoshihito Shiota; Kazunari Yoshizawa
Journal of the American Chemical Society 2016 Volume 138(Issue 30) pp:9508-9520
Publication Date(Web):July 12, 2016
DOI:10.1021/jacs.6b03785
A ruthenium(II) complex, [Ru(dmdmp)Cl(MeBPA)] (2) (Hdmdmp = N,N-dimethyl-6,7-dimethylpterin, MeBPA = N-methyl-N,N-bis(pyridylmethyl)amine), having a pterin derivative as a proton-accepting ligand, was synthesized and characterized. Complex 2 shows higher basicity than that of a previously reported RuII-pterin complex, [Ru(dmdmp) (TPA)]+ (1) (TPA = tris(2-pyridylmethyl)amine). On the other hand, 1e–-oxidized species of 1 (1OX) exhibits higher electron-acceptability than that of 1e–-oxidized 2 (2OX). Bond dissociation enthalpies (BDE) of the two RuII complexes having Hdmdmp as a ligand in proton-coupled electron transfer (PCET) to generate 1OX and 2OX were calculated to be 85 kcal mol–1 for 1OX and 78 kcal mol–1 for 2OX. The BDE values are large enough to perform H atom transfer from C–H bonds of organic molecules to the 1e–-oxidized complexes through PCET. The second-order rate constants (k) of PCET oxidation reactions were determined for 1OX and 2OX. The logarithms of normalized k values were proportional to the BDE values of C–H bonds of the substrates with slopes of −0.27 for 1OX and −0.44 for 2OX. The difference between 1OX and 2OX in the slopes suggests that the transition states in PCET oxidations of substrates by the two complexes bear different polarization, as reflection of difference in the electron acceptability and basicity of 1OX and 2OX. The more basic 2OX attracts a proton from a C–H bond via a more polarized transition state than that of 1OX; on the contrary, the more electron-deficient 1OX forms less polarized transition states in PCET oxidation reactions of C–H bonds.
Co-reporter:Tomoya Ishizuka, Atsuko Watanabe, Hiroaki Kotani, Dachao Hong, Kenta Satonaka, Tohru Wada, Yoshihito Shiota, Kazunari Yoshizawa, Kazuaki Ohara, Kentaro Yamaguchi, Satoshi Kato, Shunichi Fukuzumi, and Takahiko Kojima
Inorganic Chemistry 2016 Volume 55(Issue 3) pp:1154-1164
Publication Date(Web):January 21, 2016
DOI:10.1021/acs.inorgchem.5b02336
A bis-hydroxo-bridged dinuclear CoIII-pyridylmethylamine complex (1) was synthesized and the crystal structure was determined by X-ray crystallography. Complex 1 acts as a homogeneous catalyst for visible-light-driven water oxidation by persulfate (S2O82–) as an oxidant with [RuII(bpy)3]2+ (bpy = 2,2′-bipyridine) as a photosensitizer affording a high quantum yield (44%) with a large turnover number (TON = 742) for O2 formation without forming catalytically active Co-oxide (CoOx) nanoparticles. In the water-oxidation process, complex 1 undergoes proton-coupled electron-transfer (PCET) oxidation as a rate-determining step to form a putative dinuclear bis-μ-oxyl CoIII complex (2), which has been suggested by DFT calculations. Catalytic water oxidation by 1 using [RuIII(bpy)3]3+ as an oxidant in a H216O and H218O mixture was examined to reveal an intramolecular O–O bond formation in the two-electron-oxidized bis-μ-oxyl intermediate, prior to the O2 evolution.
Co-reporter:Hiroaki Kotani; Takumi Sugiyama; Tomoya Ishizuka; Yoshihito Shiota; Kazunari Yoshizawa
Journal of the American Chemical Society 2015 Volume 137(Issue 35) pp:11222-11225
Publication Date(Web):August 24, 2015
DOI:10.1021/jacs.5b06237
Rh(III) complexes having tris(2-pyridylmethyl)amine (TPA) and its derivative as tetradentate ligands showed reversible deprotonation at a methylene moiety of the TPA ligands upon addition of a strong base as confirmed by spectroscopic measurements and X-ray crystallography. Deprotonation selectively occurred at the axial methylene moiety rather than equatorial counterparts because of the thermodynamic stability of corresponding deprotonated complexes. One-electron oxidation of the deprotonated Rh(III)–TPA complex afforded a unique TPA radical bound to the Rh(III) center by a ligand-centered oxidation. This is the first example to demonstrate emergence of the redox-noninnocent character of the TPA ligand.
Co-reporter:Hiroaki Kotani, Suzue Kaida, Tomoya Ishizuka, Miyuki Sakaguchi, Takashi Ogura, Yoshihito Shiota, Kazunari Yoshizawa and Takahiko Kojima
Chemical Science 2015 vol. 6(Issue 2) pp:945-955
Publication Date(Web):17 Oct 2014
DOI:10.1039/C4SC02285H
A mononuclear Cr(V)–oxo complex, [CrV(O)(6-COO−-tpa)](BF4)2 (1; 6-COO−-tpa = N,N-bis(2-pyridylmethyl)-N-(6-carboxylato-2-pyridylmethyl)amine) was prepared through the reaction of a Cr(III) precursor complex with iodosylbenzene as an oxidant. Characterization of 1 was achieved using ESI-MS spectrometry, electron paramagnetic resonance, UV-vis, and resonance Raman spectroscopies. The reduction potential (Ered) of 1 was determined to be 1.23 V vs. SCE in acetonitrile based on analysis of the electron-transfer (ET) equilibrium between 1 and a one-electron donor, [RuII(bpy)3]2+ (bpy = 2,2′-bipyridine). The reorganization energy (λ) of 1 was also determined to be 1.03 eV in ET reactions from phenol derivatives to 1 on the basis of the Marcus theory of ET. The smaller λ value in comparison with that of an Fe(IV)–oxo complex (2.37 eV) is caused by the small structural change during ET due to the dπ character of the electron-accepting LUMO of 1. When benzyl alcohol derivatives (R-BA) with different oxidation potentials were employed as substrates, corresponding aldehydes were obtained as the 2e−-oxidized products in moderate yields as determined from 1H NMR and GC-MS measurements. One-step UV-vis spectral changes were observed in the course of the oxidation reactions of BA derivatives by 1 and a kinetic isotope effect (KIE) was observed in the oxidation reactions for deuterated BA derivatives at the benzylic position as substrates. These results indicate that the rate-limiting step is a concerted proton-coupled electron transfer (PCET) from substrate to 1. In sharp contrast, in the oxidation of trimethoxy-BA (Eox = 1.22 V) by 1, trimethoxy-BA radical cation was observed by UV-vis spectroscopy. Thus, it was revealed that the mechanism of the oxidation reaction changed from one-step PCET to stepwise ET–proton transfer (ET/PT), depending on the redox potentials of R-BA.
Co-reporter:Hiroaki Kotani, Tomomi Yagi, Tomoya Ishizuka and Takahiko Kojima
Chemical Communications 2015 vol. 51(Issue 69) pp:13385-13388
Publication Date(Web):13 Jul 2015
DOI:10.1039/C5CC03012A
We have synthesised a novel copper(II) complex with a pyridine pendant as a proton relay port for electrocatalytic 4e− reduction of O2 in water. The enhancement of the electrocatalytic O2 reduction via protonation of the pyridine pendant is demonstrated in comparison with a copper(II) complex without the pyridine pendant.
Co-reporter:Yuta Saegusa, Tomoya Ishizuka, Keiyu Komamura, Soji Shimizu, Hiroaki Kotani, Nagao Kobayashi and Takahiko Kojima
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 22) pp:15001-15011
Publication Date(Web):13 May 2015
DOI:10.1039/C5CP01420D
Here, we report the effects of ring fusion, which causes expansion of the π-conjugation circuits of the porphyrin derivatives to the fused meso-aryl groups, on the aromaticity and the magnetic properties of porphyrin derivatives. These studies revealed the facts that the ring fusion with five-membered rings causes not only the remarkable red shifts of the absorption bands and narrowed HOMO–LUMO gaps, but also the contribution of anti-aromatic resonance forms to the magnetic properties as observed in the 1H NMR spectra. The optical absorption and magnetic circular dichroism (MCD) spectroscopies indicate that the increase in the number of the fused rings causes stabilization of the LUMO level of the porphyrin derivatives and as a result induces the loosening of the LUMO degeneracy that is generally observed for porphyrins. The electronic structure of a quadruply fused porphyrin derivative was experimentally clarified by the ESR studies on the 1e−-oxidized and 1e−-reduced species in THF. Furthermore, we revealed the substituent effects of the fused meso-aryl groups of quadruply fused porphyrins (QFPs) on the crystal structures, absorption spectra and redox potentials; the sensitiveness of the substituent effects shows that the π-conjugation circuits extended to the fused meso-aryl groups. Additionally, the elongation of the bond lengths between the pyrrolic nitrogen and the central metal ions in QFP–metal complexes causes a remarkable increase of the Lewis acidity of the central metal ions.
Co-reporter:Hiroumi Mitome, Tomoya Ishizuka, Yoshihito Shiota, Kazunari Yoshizawa and Takahiko Kojima
Dalton Transactions 2015 vol. 44(Issue 7) pp:3151-3158
Publication Date(Web):22 Dec 2014
DOI:10.1039/C4DT03358B
Ruthenium(II) complexes of PQQTME, a trimethyl ester derivative of redox-active PQQ (pyrroloquinolinequinone), were prepared using a tridentate ligand, 2,2′:6′,2′′-terpyridine (terpy) as an auxiliary ligand. The characterization of the complexes was performed by spectroscopic methods, X-ray crystallography, and electrochemical measurements. In one complex, the pyridine site of PQQTME binds to the [RuII(terpy)] unit as a tridentate ligand, and a silver(I) ion is coordinated by the quinone moiety in a bidentate fashion. In contrast, another complex includes the [RuII(terpy)] unit at the bidentate quinone moiety of the PQQTME ligand. The difference in the coordination modes of the complexes exhibits a characteristic difference in the stability of metal coordination and also in the reversibility of the reduction processes of the PQQTME ligand. It should be noted that an additional metal-ion-binding to the PQQTME ligand largely raises the 1e−-reduction potential of the ligand. In addition, we succeeded in the characterization of the 1e−-reduced species of the complexes, where the unpaired electron was delocalized in the π-conjugated system of the PQQTME˙− ligand, using UV-Vis absorption and ESR spectroscopies.
Co-reporter:Yuta Saegusa;Dr. Tomoya Ishizuka;Dr. Tatsuhiro Kojima;Dr. Shigeki Mori;Dr. Masaki Kawano;Dr. Takahiko Kojima
Chemistry - A European Journal 2015 Volume 21( Issue 14) pp:5302-5306
Publication Date(Web):
DOI:10.1002/chem.201500389
Abstract
Molecular binding of fullerenes, C60 and C70, with the ZnII complex of a monomeric ring-fused porphyrin derivative (2-py) as a host molecule, which has a concave π-conjugated surface, has been confirmed spectroscopically. The structures of associated complexes composed of fullerenes and 2-py were explicitly established by X-ray diffraction analysis. The fullerenes in the 2:1 complexes, which consist of two 2-py molecules and one fullerene molecule, are fully covered by the concave surfaces of the two 2-py molecules in the crystal structure. In contrast, in the crystal structure of the 1:1 complex consisting of one 2-py molecule and one C60 molecule, the C60 molecule formed a π–π stacked pair with a C60 molecule in the neighboring complex using a partial surface, which was uncovered by the 2-py molecule. Additionally, the molecular size of fullerene adopted significantly affects the 1H NMR spectral changes and the redox properties of 2-py upon the molecular binding.
Co-reporter:Yuta Saegusa;Dr. Tomoya Ishizuka;Dr. Tatsuhiro Kojima;Dr. Shigeki Mori;Dr. Masaki Kawano;Dr. Takahiko Kojima
Chemistry - A European Journal 2015 Volume 21( Issue 14) pp:
Publication Date(Web):
DOI:10.1002/chem.201581461
Co-reporter:Tomoya Ishizuka, Shingo Ohzu, Hiroaki Kotani, Yoshihito Shiota, Kazunari Yoshizawa and Takahiko Kojima
Chemical Science 2014 vol. 5(Issue 4) pp:1429-1436
Publication Date(Web):06 Dec 2013
DOI:10.1039/C3SC53002G
Detailed kinetic studies on the oxidation reactions of organic substrates such as methanol with RuIVO complexes as oxidants, formed electrochemically in water, have been conducted to elucidate the reaction mechanism. The rate constants of the oxidation reactions exhibited saturation behaviours relative to the substrate concentration, regardless of the oxidants and the substrates employed. This indicates the existence of a pre-equilibrium process based on the adduct formation between the RuIVO oxidant and the substrate. Herein, we have experimentally confirmed that the driving force of the adduct formation is the hydrogen bonding between the oxidants and alcohols even in water. In addition, we have investigated the kinetic isotope effects (KIE) on the oxidation reaction using methanol and its deuterated derivatives and as a result observed moderate KIE values for the C–H bond of methanol. We have also revealed the independency of the reaction rates from the bond dissociation enthalpies of the C–H bonds of the substrates. This independency is probably derived from the tightly condensed transition state, whose energy level is strongly controlled by the activation entropy but less sensitive to the activation enthalpy.
Co-reporter:Shingo Ohzu, Tomoya Ishizuka, Hiroaki Kotani and Takahiko Kojima
Chemical Communications 2014 vol. 50(Issue 95) pp:15018-15021
Publication Date(Web):14 Oct 2014
DOI:10.1039/C4CC07488B
A mononuclear RuIII–OH complex oxidizes substrates such as hydroquinones in water through a pre-equilibrium process based on adduct formation by hydrogen bonding between the RuIII–OH complex and the substrates. The reaction mechanism switches from hydrogen atom transfer to electron transfer depending on the oxidation potential of substrates.
Co-reporter:Shingo Ohzu, Tomoya Ishizuka, Hiroaki Kotani, Yoshihito Shiota, Kazunari Yoshizawa, and Takahiko Kojima
Inorganic Chemistry 2014 Volume 53(Issue 24) pp:12677-12679
Publication Date(Web):November 20, 2014
DOI:10.1021/ic502422u
A square-shaped tetranuclear ruthenium(II) complex, [RuII4Cl5(bpmpm)2]3+ [1; bpmpm = 4,6-bis[[N,N-bis(2′-pyridylmethyl)amino]methyl]pyrimidine], exhibited four reversible and stepwise one-electron-oxidation processes: chemical oxidation of 1 formed three different mixed-valence states, in one of which the charge is partially delocalized on the two Ru centers, to be evidenced by observation of an intervalence charge-transfer absorption band, categorized into the Robin–Day class II.
Co-reporter:Dr. Takahiko Kojima;Ryosuke Kobayashi;Dr. Tomoya Ishizuka;Shinya Yamakawa;Dr. Hiroaki Kotani;Dr. Tatsuaki Nakanishi;Dr. Kei Ohkubo;Dr. Yoshihito Shiota;Dr. Kazunari Yoshizawa;Dr. Shunichi Fukuzumi
Chemistry - A European Journal 2014 Volume 20( Issue 47) pp:15518-15532
Publication Date(Web):
DOI:10.1002/chem.201403960
Abstract
A porphyrin–flavin-linked dyad and its zinc and palladium complexes (MPorFl: 2M, M=2 H, Zn, and Pd) were newly synthesized and the X-ray crystal structure of 2Pd was determined. The photodynamics of 2M were examined by femto- and nanosecond laser flash photolysis measurements. Photoinduced electron transfer (ET) in 2H2 occurred from the singlet excited state of the porphyrin moiety (H2Por) to the flavin (Fl) moiety to produce the singlet charge-separated (CS) state 1(H2Por.+Fl.−), which decayed through back ET (BET) to form 3[H2Por]*Fl with rate constants of 1.2×1010 and 1.2×109 s−1, respectively. Similarly, photoinduced ET in 2Pd afforded the singlet CS state, which decayed through BET to form 3[PdPor]*Fl with rate constants of 2.1×1011 and 6.0×1010 s−1, respectively. The rate constant of photoinduced ET and BET of 2M were related to the ET and BET driving forces by using the Marcus theory of ET. One and two Sc3+ ions bind to the flavin moiety to form the FlSc3+ and Fl(Sc3+)2 complexes with binding constants of K1=2.2×105 M−1 and K2=1.8×103 M−1, respectively. Other metal ions, such as Y3+, Zn2+, and Mg2+, form only 1:1 complexes with flavin. In contrast to 2M and the 1:1 complexes with metal ions, which afforded the short-lived singlet CS state, photoinduced ET in 2Pd⋅⋅⋅Sc3+ complexes afforded the triplet CS state (3[PdPor.+Fl.−(Sc3+)2]), which exhibited a remarkably long lifetime of τ=110 ms (kBET=9.1 s−1).
Co-reporter:Tomoya Ishizuka, Yuta Saegusa, Yoshihito Shiota, Kazuhisa Ohtake, Kazunari Yoshizawa and Takahiko Kojima
Chemical Communications 2013 vol. 49(Issue 53) pp:5939-5941
Publication Date(Web):14 May 2013
DOI:10.1039/C3CC42831A
A novel quadruply-fused porphyrin has been synthesized with a facilely prepared precursor in a high yield. A detailed comparison of the physical properties of a series of fused porphyrins revealed remarkable effects of the ring fusion on lowering LUMO levels rather than HOMO levels.
Co-reporter:Hiroumi Mitome, Tomoya Ishizuka, Yoshihiko Shiota, Kazunari Yoshizawa, and Takahiko Kojima
Inorganic Chemistry 2013 Volume 52(Issue 5) pp:2274-2276
Publication Date(Web):February 19, 2013
DOI:10.1021/ic302617b
Herein, we report the synthesis of a novel heterohexanuclear complex (1) of a heteroaromatic cofactor, pyrroloquinolinequinone (PQQ). The crystal structure of 1 was determined to reveal that two PQQ-bridged RuIIAgI units were linked by two [AgI(OTf)2]− units (OTf = CF3SO3–). A solvent-bound RuIIAgI heterodinuclear complex (2) was formed from 1 in a coordinating solvent such as acetone to show an intense metal-to-ligand charge-transfer band at 709 nm.
Co-reporter:Misaki Makino, Tomoya Ishizuka, Shingo Ohzu, Jiang Hua, Hiroaki Kotani, and Takahiko Kojima
Inorganic Chemistry 2013 Volume 52(Issue 9) pp:5507-5514
Publication Date(Web):April 22, 2013
DOI:10.1021/ic400412f
We have synthesized a mononuclear ruthenium(II) azido complex (1) and a dinuclear ruthenium(II) μ-azido complex (2) having N,N-bis(2-pyridylmethyl)-N-bis(2-pyridyl)methylamine (N4Py) as a pentadentate ancillary ligand. In the crystal structure of 2, intramolecular π–π stacking was found between the pyridine rings of the two different N4Py ligands, contributing to stabilize the dinuclear μ-azido structure. π donation from the HOMO π* orbital of the μ-azido ligand to the Ru–N(pyr) bond increases the bond order between the terminal and central N atoms in the μ-azido ligand to strengthen the N–N bonds of the μ-azido ligand. The μ-azido complex 2 was revealed to exhibit a stepwise oxidation behavior in CH3CN to afford a RuII–μ-azido–RuIII mixed-valence (MV) state upon one-electron oxidation. The MV state of one-electron-oxidized 2 was categorized in the Robin–Day class II with the electronic coupling constant (Hab) of 570 cm–1.
Co-reporter:Yuji Inui, Motoo Shiro, Shunichi Fukuzumi and Takahiko Kojima
Organic & Biomolecular Chemistry 2013 vol. 11(Issue 5) pp:758-764
Publication Date(Web):28 Nov 2012
DOI:10.1039/C2OB26877A
The formation of guanine quartets with 9-isopropylguanine (iPG) is discussed in organic solvents. Crystal structures of the iPG quartets were determined by X-ray crystallography with template cations (Na+ and Ca2+) and the structure without a template cation was also obtained by virtue of the stabilization by intermolecular hydrogen bonding with water molecules of crystallization. The difference in the quartet formation of iPG in the presence and absence of a template cation was clearly demonstrated by 1H NMR measurements in CDCl3–CH3OH mixed solvents. The quartet formation is mainly governed by the enthalpy gain due to the electrostatic interaction between the O6 oxygen in iPG and the template cations in the presence of the cations rather than the intermolecular hydrogen bonding, while desolvation of iPG is the dominant factor for the formation in the absence of cations. In the presence of Na+ and Ca2+, ΔH and ΔS values in the formation of iPG-4–Na+ and iPG-4–Ca2+ complexes were determined to be ΔH = −8.4 kcal mol−1 and ΔS = +50 cal mol−1 K−1 for Na+ and ΔH = −12.9 kcal mol−1 and ΔS = +34 cal mol−1 K−1 for Ca2+ on the basis of van't Hoff plots attained from the results of temperature-dependent UV-Vis spectroscopic measurements.
Co-reporter:Tomoya Ishizuka, Muniappan Sankar and Takahiko Kojima
Dalton Transactions 2013 vol. 42(Issue 45) pp:16073-16079
Publication Date(Web):01 Aug 2013
DOI:10.1039/C3DT51467F
Supramolecular integration of a saddle-distorted zinc(II) porphyrin complex, which has hydroxyl groups at the para-position of the four meso-aryl groups, has been demonstrated on the basis of hydrogen bonding among the peripheral hydroxyl groups. The hydrogen-bonding patterns were controlled by the recrystallization solvents and additives, and particularly, addition of a bifunctional ligand such as 4,4′-bipyridine (bpy). The coordination of bpy to form dinuclear ZnII-porphyrin complexes causes a conformational difference: the dimeric complex with four hydroxyl groups is in an eclipsed form, however, a derivative without hydroxyl groups is in a staggered form due to the presence or absence of the intermolecular hydrogen bonding. In addition, the dimerization by the bpy coordination resulted in the expansion of the intermolecular space formed in the porphyrin networks, suggesting the potential to be applied for inclusion of guest molecules.
Co-reporter:Yuji Inui, Shunichi Fukuzumi and Takahiko Kojima
Dalton Transactions 2013 vol. 42(Issue 11) pp:3779-3782
Publication Date(Web):14 Jan 2013
DOI:10.1039/C3DT33034F
Formation of a π–π stacked assembly between a Na+-templated G-quartet and octaethylporphyrinatonickel(II) was observed by spectroscopic methods in methanol/chloroform and the formation dynamics of the assembly was elucidated.
Co-reporter:Yuji Inui, Motoo Shiro, Takahiro Kusukawa, Shunichi Fukuzumi and Takahiko Kojima
Dalton Transactions 2013 vol. 42(Issue 8) pp:2773-2778
Publication Date(Web):28 Nov 2012
DOI:10.1039/C2DT32535G
[Ir6(μ-alloCl22−)3(Cp*)6(OH)3](PF6)3 (1) having 7,8-dichloroalloxazine dianion (alloCl22−) as bridging ligands was synthesized and characterized by X-ray crystallography, spectroscopic and electrochemical measurements. The alloxazine ligands showed unprecedented coordination modes to link the six Ir(III) centres. The complex exhibited remarkable stability and reversible six-electron redox processes at the bridging alloxazine ligands in organic solvents. The first reversible reduction process occurred on each of three alloxazine ligands in 1 to produce a three-electron-reduced species, [IrIII6Cp*6(μ-alloCl2˙3−)3(OH)3], and was observed as an apparent one-step reduction process at −0.65 V (vs. Fc0/+). The second reversible reduction process on each of the three alloxazine ligands in 1 was recorded at almost the same potential, −0.78 V (vs. Fc0/+), to afford the six-electron-reduced form, [IrIII6Cp*6(μ-alloCl24−)3(OH)3]3−. The radical anion of the alloxazine derivative was detected by EPR measurements at room temperature. After the six-electron reduction of 1 with cobaltocene, the backward oxidation processes of reduced forms with p-chloranil were traced by UV-Vis spectroscopy to confirm the recovery of the original spectrum of 1.
Co-reporter:Shingo Ohzu;Dr. Tomoya Ishizuka;Yuichirou Hirai;Dr. Shunichi Fukuzumi;Dr. Takahiko Kojima
Chemistry - A European Journal 2013 Volume 19( Issue 5) pp:1563-1567
Publication Date(Web):
DOI:10.1002/chem.201203430
Co-reporter:Takuya Sawaki;Dr. Tomoya Ishizuka;Dr. Masaki Kawano;Dr. Yoshihito Shiota;Dr. Kazunari Yoshizawa;Dr. Takahiko Kojima
Chemistry - A European Journal 2013 Volume 19( Issue 27) pp:8978-8990
Publication Date(Web):
DOI:10.1002/chem.201300437
Abstract
The thermal and photochemical reactions of a newly synthesized complex, [RuII(TPA)(tpphz)]2+ (1; TPA=tris(2-pyridylmethyl)amine, tpphz=tetrapyrido[3,2-a:2′,3′-c:3′′,2′′-h: 2′′′,3′′′-j]phenazine), and its derivatives have been investigated. Heating a solution of complex 1 (closed form) and its derivatives in MeCN caused the partial dissociation of one pyridylmethyl moiety of the TPA ligand and the resulting vacant site on the RuII center was occupied by a molecule of MeCN from the solvent to give a dissociated complex, [RuII(η3-TPA)(tpphz)(MeCN)]2+ (1′, open form), and its derivatives, respectively, in quantitative yields. The thermal dissociation reactions were investigated on the basis of kinetics analysis, which indicated that the reactions proceeded through a seven-coordinate transition state. Although the backwards reaction was induced by photoirradiation of the MLCT absorption bands, the photoreaction of complex 1′ reached a photostationary state between complexes 1 and 1′ and, hence, the recovery of complex 1 from complex 1′ was 67 %. Upon protonation of complex 1 at the vacant site of the tpphz ligand, the efficiency of the photoinduced recovery of complex 1+H+ from complex 1′+H+ improved to 83 %. In contrast, dinuclear μ-tpphz complexes 2 and 3, which contained the RuII(TPA)(tpphz) unit and either a RuII(bpy)2 or PdIICl2 moiety on the other coordination edge of the tpphz ligand, exhibited 100 % photoconversion from their open forms into their closed forms (2′2 and 3′3). These results are the first examples of the complete photochromic structural change of a transition-metal complex, as represented by complete interconversion between its open and closed forms. Scrutinization by performing optical and electrochemical measurements allowed us to propose a rationale for how metal coordination at the vacant site of the tpphz ligand improves the efficiency of photoconversion from the open form into the closed form. It is essential to lower the energy level of the triplet metal-to-ligand charge-transfer excited state (3MLCT*) of the closed form relative to that of the triplet metal-centered excited state (3MC*) by metal coordination. This energy-level manipulation hinders the transition from the 3MLCT* state into the 3MC* state in the closed form to block the partial photodissociation of the TPA ligand.
Co-reporter:Shunichi Fukuzumi, Tatsuhiko Honda, Takahiko Kojima
Coordination Chemistry Reviews 2012 Volume 256(21–22) pp:2488-2502
Publication Date(Web):November 2012
DOI:10.1016/j.ccr.2012.01.011
Porphyrins and phthalocyanines are planar two-dimensional π-compounds, which are normally difficult to protonate because of the low basicity. When many bulky substituents are introduced to porphyrins and phthalocyanines, however, the macrocyclic π-plane is distorted due to the steric repulsion of the bulky substituents. The π-plane distortion facilitates protonation to afford stable protonated porphyrins and phthalocyanines. Crystal structures of protonated porphyrins and phthalocyanines were determined to clarify the role of hydrogen bonding in the supramolecular assemblies. Protonated porphyrinoids can act as an electron acceptor rather than an electron donor in photoinduced electron-transfer reactions. The rate constants of photoinduced electron-transfer reactions of diprotonated porphyrin with different degrees of distortion were determined and they are evaluated in light of the Marcus theory of electron transfer to determine the reorganization energies of electron transfer, which are affected by the distortion of the π-plane. A distortion of the macrocyclic ligands also affords higher Lewis acidity at a metal center to allow facile axial coordination of ligands, due to poor overlap of the lone pair orbitals with dx2–y2 or px and py orbitals of the metal center. Thus, the distortion of the macrocyclic ligands enables one to construct various molecular and supramolecular complexes composed of porphyrins and phthalocyanines. The photodynamics of photoinduced electron-transfer reactions of various supramolecular complexes of distorted porphyrin and phthalocyanines are discussed in relation to structure and photofunction.Graphical abstractHighlights► Saddle-distorted porphyrins and phthalocyanines show high Bønsted basicity. ► Protonated porphyrins and phthalocyanines act as an electron-acceptor in electron-transfer reactions. ► Protonated porphyrins form hydrogen-bonded supramolecules. ► Saddle-distorted metal complexes of porphyrins and phthalocyanine form strong axial coordination.
Co-reporter:Tatsuhiko Honda ; Takahiko Kojima ;Shunichi Fukuzumi
Journal of the American Chemical Society 2012 Volume 134(Issue 9) pp:4196-4206
Publication Date(Web):February 2, 2012
DOI:10.1021/ja209978q
Proton-coupled electron-transfer reduction of dioxygen (O2) to afford hydrogen peroxide (H2O2) was investigated by using ferrocene derivatives as reductants and saddle-distorted (α-octaphenylphthalocyaninato)cobalt(II) (CoII(Ph8Pc)) as a catalyst under acidic conditions. The selective two-electron reduction of O2 by dimethylferrocene (Me2Fc) and decamethylferrocene (Me10Fc) occurs to yield H2O2 and the corresponding ferrocenium ions (Me2Fc+ and Me10Fc+, respectively). Mechanisms of the catalytic reduction of O2 are discussed on the basis of detailed kinetics studies on the overall catalytic reactions as well as on each redox reaction in the catalytic cycle. The active species to react with O2 in the catalytic reaction is switched from CoII(Ph8Pc) to protonated CoI(Ph8PcH), depending on the reducing ability of ferrocene derivatives employed. The protonation of CoII(Ph8Pc) inhibits the direct reduction of O2; however, the proton-coupled electron transfer from Me10Fc to CoII(Ph8Pc) and the protonated [CoII(Ph8PcH)]+ occurs to produce CoI(Ph8PcH) and [CoI(Ph8PcH2)]+, respectively, which react immediately with O2. The rate-determining step is a proton-coupled electron-transfer reduction of O2 by CoII(Ph8Pc) in the CoII(Ph8Pc)-catalyzed cycle with Me2Fc, whereas it is changed to the electron-transfer reduction of [CoII(Ph8PcH)]+ by Me10Fc in the CoI(Ph8PcH)-catalyzed cycle with Me10Fc. A single crystal of monoprotonated [CoIII(Ph8Pc)]+, [CoIIICl2(Ph8PcH)], produced by the proton-coupled electron-transfer reduction of O2 by CoII(Ph8Pc) with HCl, was obtained, and the crystal structure was determined in comparison with that of CoII(Ph8Pc).
Co-reporter:Shingo Ohzu, Tomoya Ishizuka, Yuichirou Hirai, Hua Jiang, Miyuki Sakaguchi, Takashi Ogura, Shunichi Fukuzumi and Takahiko Kojima
Chemical Science 2012 vol. 3(Issue 12) pp:3421-3431
Publication Date(Web):31 Aug 2012
DOI:10.1039/C2SC21195E
A series of Ru(IV)-oxo complexes (4–6) were synthesized from the corresponding Ru(II)-aqua complexes (1–3) and fully characterized by 1H NMR and resonance Raman spectroscopies, and ESI-MS spectrometry. Based on the diamagnetic character confirmed by the 1H NMR spectroscopy in D2O, the spin states of 5 and 6 were determined to be S = 0 in the d4 configuration, in sharp contrast to that of 4 being in the S = 1 spin state. The aqua-complexes 1–3 catalyzed oxidation of alcohols and olefins using (NH4)2[CeIV(NO3)6] (CAN) as an electron-transfer oxidant in acidic aqueous solutions. Comparison of the reactivity of electrochemically generated oxo-complexes (4–6) was made in the light of kinetic analyses for oxidation of 1-propanol and a water-soluble ethylbenzene derivative. The oxo complexes (4–6) exhibited no significant difference in the reactivity for the oxidation reactions, judging from the similar catalytic rates and the activation parameters. The slight difference observed in the reaction rates can be accounted for by the difference in the reduction potentials of the oxo-complexes, but the spin states of the oxo-complexes have hardly affected the reactivity. The activation parameters and the kinetic isotope effects (KIE) observed for the oxidation reactions of methanol indicate that the oxidation reactions of alcohols with the RuIVO complexes proceed via a concerted proton-coupled electron transfer mechanism.
Co-reporter:Tomoya Ishizuka, Muniappan Sankar, Yusuke Yamada, Shunichi Fukuzumi and Takahiko Kojima
Chemical Communications 2012 vol. 48(Issue 52) pp:6481-6483
Publication Date(Web):17 Apr 2012
DOI:10.1039/C2CC31142A
Carboxyl groups were introduced at the peripheral positions of dodecaphenylporphyrin to link nanochannel structures with intermolecular hydrogen bonds to make the supramolecular structures robust.
Co-reporter:Atsutoshi Yokoyama, Kei Ohkubo, Tomoya Ishizuka, Takahiko Kojima and Shunichi Fukuzumi
Dalton Transactions 2012 vol. 41(Issue 33) pp:10006-10013
Publication Date(Web):03 Apr 2012
DOI:10.1039/C2DT30424D
A 2:1 complex composed between a non-planar Mo(V)–porphyrin complex ([Mo(DPP)(O)]+, DPP2− = dodecaphenylporphyrin) and a ruthenium-substituted Keggin-type heteropolyoxometalate (Ru-POM), [SiW11O39RuIII(DMSO)]5−, acts as an efficient catalyst for oxidation of benzyl alcohols with iodosobenzene as an oxidant in CDCl3 at room temperature. The catalytic oxidation afforded the corresponding benzaldehydes, whereas neither the ammonium salt of Ru-POM nor [Mo(DPP)(O)]+ alone exhibited catalytic reactivity under the same experimental conditions. This enhancement can be attributed to a large anodic shift of the redox potential of the ruthenium centre due to the complexation of the Ru-POM with two cationic {Mo(DPP)(O)}+ units. The kinetic analysis demonstrated that the catalytic oxidation proceeded via formation of a catalyst-substrate complex, and electron-withdrawing substituents at the para position of benzyl alcohol accelerated the reaction. The rate constants of the oxidation reactions correlate to the bond dissociation energies of the C–H bonds of the substrate. A linear correlation was observed for logarithm of the rate constants of oxidation reactions of benzyl alcohols with that of hydrogen abstraction by cumyl peroxyl radical, indicating the reaction proceeds via hydrogen abstraction. The observed kinetic isotope effect (KIE) indicates that the hydrogen abstraction occurs from the benzyl group rather than the hydroxy group.
Co-reporter:Yuji Inui;Dr. Soushi Miyazaki;Dr. Kei Ohkubo;Dr. Shunichi Fukuzumi;Dr. Takahiko Kojima
Angewandte Chemie International Edition 2012 Volume 51( Issue 19) pp:4623-4627
Publication Date(Web):
DOI:10.1002/anie.201108827
Co-reporter:Takahiko Kojima ; Kazuya Nakayama ; Kenichiro Ikemura ; Takashi Ogura ;Shunichi Fukuzumi
Journal of the American Chemical Society 2011 Volume 133(Issue 30) pp:11692-11700
Publication Date(Web):June 22, 2011
DOI:10.1021/ja2037645
A coordinatively saturated ruthenium(II) complex having tetradentate tris(2-pyridylmethyl)amine (TPA) and bidentate 2,2′-bipyridine (bpy), [Ru(TPA)(bpy)]2+ (1), was oxidized by a Ce(IV) ion in H2O to afford a Ru(IV)-oxo complex, [Ru(O)(H+TPA)(bpy)]3+ (2). The crystal structure of the Ru(IV)-oxo complex 2 was determined by X-ray crystallography. In 2, the TPA ligand partially dissociates to be in a facial tridentate fashion and the uncoordinated pyridine moiety is protonated. The spin state of 2, which showed paramagnetically shifted NMR signals in the range of 60 to −20 ppm, was determined to be an intermediate spin (S = 1) by the Evans’ method with 1H NMR spectroscopy in acetone-d6. The reaction of 2 with various oraganic substrates in acetonitrile at room temperature afforded oxidized and oxygenated products and a solvent-bound complex, [Ru(H+TPA)(bpy)(CH3CN)], which is intact in the presence of alcohols. The oxygenation reaction of saturated C–H bonds with 2 proceeds by two-step processes: the hydrogen abstraction with 2, followed by the dissociation of the alcohol products from the oxygen-rebound complexes, Ru(III)-alkoxo complexes, which were successfully detected by ESI-MS spectrometry. The kinetic isotope effects in the first step for the reaction of dihydroanthrathene (DHA) and cumene with 2 were determined to be 49 and 12, respectively. The second-order rate constants of C–H oxygenation in the first step exhibited a linear correlation with bond dissociation energies of the C–H bond cleavage.
Co-reporter:Takahiko Kojima ; Kazuya Nakayama ; Miyuki Sakaguchi ; Takashi Ogura ; Kei Ohkubo ;Shunichi Fukuzumi
Journal of the American Chemical Society 2011 Volume 133(Issue 44) pp:17901-17911
Publication Date(Web):September 26, 2011
DOI:10.1021/ja207572z
Ruthenium(II)–acetonitrile complexes having η3-tris(2-pyridylmethyl)amine (TPA) with an uncoordinated pyridine ring and diimine such as 2,2′-bipyridine (bpy) and 2,2′-bipyrimidine (bpm), [RuII(η3-TPA)(diimine)(CH3CN)]2+, reacted with m-chloroperbenzoic acid to afford corresponding Ru(II)–acetonitrile complexes having an uncoordinated pyridine-N-oxide arm, [RuII(η3-TPA-O)(diimine)(CH3CN)]2+, with retention of the coordination environment. Photoirradiation of the acetonitrile complexes having diimine and the η3-TPA with the uncoordinated pyridine-N-oxide arm afforded a mixture of [RuII(TPA)(diimine)]2+, intermediate-spin (S = 1) Ru(IV)–oxo complex with uncoordinated pyridine arm, and intermediate-spin Ru(IV)–oxo complex with uncoordinated pyridine-N-oxide arm. A Ru(II) complex bearing an oxygen-bound pyridine-N-oxide as a ligand and bpm as a diimine ligand was also obtained, and its crystal structure was determined by X-ray crystallography. Femtosecond laser flash photolysis of the isolated O-coordinated Ru(II)–pyridine-N-oxide complex has been investigated to reveal the photodynamics. The Ru(IV)–oxo complex with an uncoordinated pyridine moiety was alternatively prepared by reaction of the corresponding acetonitrile complex with 2,6-dichloropyridine-N-oxide (Cl2py-O) to identify the Ru(IV)–oxo species. The formation of Ru(IV)–oxo complexes was concluded to proceed via intermolecular oxygen atom transfer from the uncoordinated pyridine-N-oxide to a Ru(II) center on the basis of the results of the reaction with Cl2py-O and the concentration dependence of the consumption of the starting Ru(II) complexes having the uncoordinated pyridine-N-oxide moiety. Oxygenation reactions of organic substrates by [RuII(η3-TPA-O)(diimine)(CH3CN)]2+ were examined under irradiation (at 420 ± 5 nm) and showed selective allylic oxygenation of cyclohexene to give cyclohexen-1-ol and cyclohexen-1-one and cumene oxygenation to afford cumyl alcohol and acetophenone.
Co-reporter:Tomoya Ishizuka ; Kengo Tobita ; Yuichi Yano ; Yoshihito Shiota ; Kazunari Yoshizawa ; Shunichi Fukuzumi
Journal of the American Chemical Society 2011 Volume 133(Issue 46) pp:18570-18573
Publication Date(Web):October 24, 2011
DOI:10.1021/ja208141b
A heterodinuclear complex based on a RuII–TPA [TPA = tris(2-pyridylmethyl)amine] complex having a peripheral CuII(bpy)2 (bpy = 2,2′-bipyridine) group bonded through an amide linkage displayed reversible intramolecular electron transfer between the Ru and Cu complex units that can be controlled by protonation and deprotonation of the bridging amide moiety.
Co-reporter:Tatsuhiko Honda, Takahiko Kojima and Shunichi Fukuzumi
Chemical Communications 2011 vol. 47(Issue 28) pp:7986-7988
Publication Date(Web):17 Jun 2011
DOI:10.1039/C1CC12710A
Facile protonation of α-octabutoxyphthalocyaninato zinc(II) (Zn(OBu)8Pc) occurs to afford up to tetra-protonated species stabilized by intramolecular hydrogen bonding, resulting in positive shifts of the reduction potentials of Zn(OBu)8PcHnn+ (n = 1–4) with increasing the number of protons attached to facilitate electron-transfer reduction.
Co-reporter:Atsutoshi Yokoyama, Takahiko Kojima and Shunichi Fukuzumi
Dalton Transactions 2011 vol. 40(Issue 24) pp:6445-6450
Publication Date(Web):13 May 2011
DOI:10.1039/C0DT01708F
A 2:1 supramolecular assembly composed of a non-planar Mo(V)-porphyrin, [Mo(DPP)(O)(H2O)]+ (1) (DPP2+; dodecaphenylporphyrin), and a Keggin-type heteropolyoxometalate (POM), α-[(n-butyl)4N]2[SW12O40] (2), was formed via hydrogen bonds. The crystal structure was determined by X-ray crystallography to clarify that the POM was enclosed into a π-space of a supramolecular porphyrin nanotube by virtue of a hydrogen-bond network. In contrast to the formation of the 2:1 assembly ([{Mo(DPP)(O)(H2O)}2(SW12O40)] (3)) between 1 and [SW12O40]2− in the crystal, it was revealed that those two components form a 1:1 assembly in solution, in light of the results of MALDI-TOF-MS measurements in PhCN. Variable-temperature UV-vis spectroscopic titration allowed us to determine the thermodynamic parameters for the formation of the 1:1 supramolecular assembly in solution, the heat of formation (ΔH) and the entropy change (ΔS). These results provide the first thermodynamic data set to elucidate the formation process of supramolecuar structures emerged by hydrogen bonding between metalloporphyrin complexes and POMs, indicating that the formation of the assembly is an entropy-controlled process rather than an enthalpy-controlled one. Comparisons of the thermodynamic parameters with those of a planar Mo(V)-porphyrin complex also highlighted high Lewis acidity of the Mo(V) centre in the distorted porphyrin.
Co-reporter:Yuichi Yano, Takahiko Kojima, Shunichi Fukuzumi
Inorganica Chimica Acta 2011 Volume 374(Issue 1) pp:104-111
Publication Date(Web):1 August 2011
DOI:10.1016/j.ica.2011.01.100
A novel TPA derivative (TPA = tris(2-pyridylmethyl)amine) having two 1,10-phenanthroline (phen) moieties via amide linkage was synthesized and this ligand reacted with [Ru(hmb)Cl2]2 (hmb: hexamethylbenzene) to give a trinuclear Ru(II) complex, [RuCl(TPA-{phenRuCl(hmb)}2-H+)](PF6)2 (1-Cl), in a moderate yield. The complex involves a deprotonated and oxygen-coordinated amide linkage, which exhibits reversible protonation–deprotonation equilibrium. The chlorido complex was converted to be an aqua complex, [Ru(H2O)(TPA-{phenRu(H2O)2(hmb)}2-H+)](SO4)5/2 (1-H2O), by the reaction of 1-Cl with Ag2SO4 in H2O. Transfer hydrogenation of ketones was examined by using 1-Cl as a catalyst and HCOONa as a hydride source in H2O/CH3OH (1:1 v/v) at 50 °C under Ar. The time-course of the transfer hydrogenation of cyclohexanone to give cyclohexanol revealed that 1-Cl showed a cooperative effect on the catalytic reactivity as compared with that of mononuclear [RuCl(hmb)(phen)] (3-Cl) and [RuCl((1-Naph)2-TPA)]PF6 in H2O/CH3OH (1:2 v/v) under the same conditions. The detailed kinetic study has revealed that the catalytic transfer hydrogenation proceeds via the formato complex, which interacts with a substrate rather than via the hydrido complex. The two Ru centers placed at close proximity in 1-H2O enhanced the interaction of the formato complex with a substrate, resulting in an increase in the catalytic reactivity as compared with the mononuclear complex.Graphical abstractA trinuclear Ru(II) complex, [RuCl(TPA-{phenRuCl(C6Me6)}2-H+)](PF6)2, acts as an efficient catalyst for transfer hydrogenation of ketones with formic acid as compared with the corresponding mononuclear Ru(II) complex, because the two Ru centers placed at close proximity enhances the interaction of the formato complex with a substrate.Research highlights► A trinuclear Ru(II) complex was synthesized with an amide linkage. ► The two Ru centers are converged to a vicinity by the coordination of amide oxygen. ► The two Ru centers facilitated the catalytic activity for transfer hydrogenation .
Co-reporter:Dr. Takahiko Kojima;Dr. Shunichi Fukuzumi
Angewandte Chemie 2011 Volume 123( Issue 17) pp:3936-3937
Publication Date(Web):
DOI:10.1002/ange.201100719
Co-reporter:Tatsuhiko Honda;Dr. Takahiko Kojima;Dr. Nagao Kobayashi;Dr. Shunichi Fukuzumi
Angewandte Chemie International Edition 2011 Volume 50( Issue 12) pp:2725-2728
Publication Date(Web):
DOI:10.1002/anie.201006607
Co-reporter:Dr. Takahiko Kojima;Dr. Shunichi Fukuzumi
Angewandte Chemie International Edition 2011 Volume 50( Issue 17) pp:3852-3853
Publication Date(Web):
DOI:10.1002/anie.201100719
Co-reporter:Atsutoshi Yokoyama, Takahiko Kojima, Kei Ohkubo, Motoo Shiro, and Shunichi Fukuzumi
The Journal of Physical Chemistry A 2011 Volume 115(Issue 6) pp:986-997
Publication Date(Web):January 26, 2011
DOI:10.1021/jp109863d
Nonplanar Sn(IV)−porphyrin complexes, [Sn(TMPP(Ph)8)-Cl2] (1) and [Sn(TMPP(Ph)8)(OMe)2] (2) (TMPP(Ph)8: 5,10,15,20-tetrakis(4-methoxyphenyl)-2,3,7,8,12,13,17,18-octaphenylporphyrinato), were prepared and characterized by spectroscopic and electrochemical methods together with X-ray crystallography. Variable-temperature 1H NMR study revealed that the coordination of the methoxo ligand of 2 is weak enough in solution to enhance the axial ligand exchange with a Keggin-type phosphotungstate (α-[PW12O40]3−) due to the steric stress between the axial methoxo ligand and the peripheral phenyl groups of the porphyrin ligand. The formation of a novel 1:1 donor−acceptor complex, [Sn(TMPP(Ph)8)(OMe)(α-[PW12O40])]2− (4) was confirmed by 1H NMR and UV−vis spectral titrations, and also by MALDI-TOF-MS measurements. Electrochemical measurements for the donor−acceptor complex in PhCN revealed that the Sn(IV)−TMPP(Ph)8 moiety acts as an electron donor and the α-[PW12O40]3− moiety acts as an electron acceptor and that the energy level of the electron-transfer (ET) state of the 1:1 complex (1.17 eV) is lower than that of the triplet excited states of the SnTMPP(Ph)8 complex (1.31 eV). Femtosecond and nanosecond laser flash photolysis measurements indicate that intersystem crossing from the singlet excited sate to the triplet excited state occurs followed by intramolecular photoinduced electron transfer from the triplet excited state of the Sn(IV)−TMPP(Ph)8 moiety to the α-[PW12O40]3− moiety in the 1:1 complex in benzonitrile.
Co-reporter:Dr. Tomoya Ishizuka;Takuya Sawaki;Dr. Soushi Miyazaki;Dr. Masaki Kawano;Dr. Yoshihito Shiota;Dr. Kazunari Yoshizawa;Dr. Shunichi Fukuzumi;Dr. Takahiko Kojima
Chemistry - A European Journal 2011 Volume 17( Issue 24) pp:6652-6662
Publication Date(Web):
DOI:10.1002/chem.201003522
Abstract
The pterin-coordinated ruthenium complex, [RuII(dmdmp)(tpa)]+ (1) (Hdmdmp=N,N-dimethyl-6,7-dimethylpterin, tpa=tris(2-pyridylmethyl)amine), undergoes photochromic isomerization efficiently. The isomeric complex (2) was fully characterized to reveal an apparent 180° pseudorotation of the pterin ligand. Photoirradiation to the solution of 1 in acetone with incident light at 460 nm resulted in dissociation of one pyridylmethyl arm of the tpa ligand from the RuII center to give an intermediate complex, [Ru(dmdmp)(tpa)(acetone)]2+ (I), accompanied by structural change and the coordination of a solvent molecule to occupy the vacant site. The quantum yield (ϕ) of this photoreaction was determined to be 0.87 %. The subsequent thermal process from intermediate I affords an isomeric complex 2, as a result of the rotation of the dmdmp2− ligand and the recoordination of the pyridyl group through structural change. The thermal process obeyed first-order kinetics, and the rate constant at 298 K was determined to be 5.83×10−5 s−1. The activation parameters were determined to be ΔH≠=81.8 kJ mol−1 and ΔS≠=−49.8 J mol−1 K−1. The negative ΔS≠ value indicates that this reaction involves a seven-coordinate complex in the transition state (i.e., an interchange associative mechanism). The most unique point of this reaction is that the recoordination of the photodissociated pyridylmethyl group occurs only from the direction to give isomer 2, without going back to starting complex 1, and thus the reaction proceeds with 100 % conversion efficiency. Upon heating a solution of 2 in acetonitrile, isomer 2 turned back into starting complex 1. The backward reaction is highly dependent on the solvent: isomer 2 is quite stable and hard to return to 1 in acetone; however, 2 was converted to 1 smoothly by heating in acetonitrile. The activation parameters for the first-order process in acetonitrile were determined to be ΔH≠=59.2 kJ mol−1 and ΔS≠=−147.4 kJ mol−1 K−1. The largely negative ΔS≠ value suggests the involvement of a seven-coordinate species with the strongly coordinated acetonitrile molecule in the transition state. Thus, the strength of the coordination of the solvent molecule to the RuII center is a determinant factor in the photoisomerization of the RuII–pterin complex.
Co-reporter:Masanori Kanematsu;Dr. Pan&x10d;e Naumov;Dr. Takahiko Kojima;Dr. Shunichi Fukuzumi
Chemistry - A European Journal 2011 Volume 17( Issue 44) pp:12372-12384
Publication Date(Web):
DOI:10.1002/chem.201100493
Abstract
The rate constants of intermolecular photoinduced electron transfer from triplet excited states of metalloporphyrins to a series of p-benzoquinone derivatives in benzonitrile were determined to examine the effects of the driving force, the metal, and the conformational distortion of the porphyrin ring on the reorganization energies (λ) of electron transfer by laser flash photolysis. The λ values were evaluated from the determined rate constants on the basis of the Marcus theory of electron transfer. The λ values of planar metalloporphyrins, [Al(TPP)(PhCOO)] and [Zn(TPP)] (TPP2−=tetraphenylporphyrin dianion), are approximately the same, but they are 0.27 eV smaller than those of the corresponding nonplanar (saddle-distorted) metalloporphyrins [Al(DPP)(PhCOO)] and [Zn(DPP)] (DPP2−=dodecaphenylporphyrin dianion) when they are compared for the same driving force of photoinduced electron transfer. The axial ligand PhCOO− of [Al(TPP)]+ and [Al(DPP)]+ was replaced by anthraquinone-2-carboxylate (AqCOO−) to afford the electron donor–acceptor complexes [Al(TPP)(AqCOO)] and [Al(DPP)(AqCOO)], respectively. The X-ray crystal structure of [Al(TPP)(AqCOO)] revealed strong coordination of AqCOO− to the Al3+ ion of [Al(TPP)]+ and the existence of π–π interactions between AqCOO− and the porphyrin ring. In the case of the saddle-distorted [Al(DPP)(AqCOO)], however, the AqCOO− moiety is nearly perpendicular to the porphyrin ring. The photodynamics of intracomplex photoinduced electron transfer from the singlet excited state of [Al(TPP)]+ and [Al(DPP)]+ to the AqCOO− moiety were also examined in comparison with the intermolecular photoinduced electron-transfer reactions, and the determined rate constants were evaluated in light of the Marcus theory of electron transfer to reveal that the electron transfer is adiabatic in each case.
Co-reporter:Tatsuhiko Honda;Dr. Takahiko Kojima;Dr. Nagao Kobayashi;Dr. Shunichi Fukuzumi
Angewandte Chemie 2011 Volume 123( Issue 12) pp:2777-2780
Publication Date(Web):
DOI:10.1002/ange.201006607
Co-reporter:Tatsuhiko Honda ; Tatsuaki Nakanishi ; Kei Ohkubo ; Takahiko Kojima ;Shunichi Fukuzumi
Journal of the American Chemical Society 2010 Volume 132(Issue 29) pp:10155-10163
Publication Date(Web):July 2, 2010
DOI:10.1021/ja103889f
The excited-state photodynamics of intrasupramolecular photoinduced electron transfer was investigated in a series of hydrogen-bonded supramolecular complexes composed of diprotonated 2,3,5,7,8,10,12,13,15,17,18,20-dodecaphenylporphyrin (H4DPP2+) and electron donors bearing a carboxylate group. The formation of supramolecular complexes was examined by spectroscopic measurements. The binding constants obtained by spectroscopic titration indicate the strong binding (108−1010 M−2) even in a polar and coordinating solvent, benzonitrile (PhCN). The crystal structure of the supramolecular assembly using ferrocenecarboxylate (FcCOO−) was determined to reveal a new structural motif involving two-point and single-point hydrogen bonding among saddle-distorted H4DPP2+ dication and two FcCOO− anions. Femtosecond laser flash photolysis was applied to investigate the photodynamics in the hydrogen-bonded supramolecular complexes. Rate constants obtained were evaluated in light of the Marcus theory of electron transfer, allowing us to determine the reorganization energy and the electronic coupling matrix constant of photoinduced electron transfer and back electron transfer to be 0.68 eV and 43 cm−1, respectively. The distance dependence of electron transfer was also examined by using a series of ferrocenecarboxylate derivatives connected by linear phenylene linkers, and the distance dependence of the rate constant of electron transfer (kET) was determined to be kET = k0 exp(−βr), in which β = 0.64 Å−1.
Co-reporter:Takahiko Kojima ; Norihisa Hirasa ; Daisuke Noguchi ; Tomoya Ishizuka ; Soushi Miyazaki ; Yoshihito Shiota ; Kazunari Yoshizawa ;Shunichi Fukuzumi
Inorganic Chemistry 2010 Volume 49(Issue 8) pp:3737-3745
Publication Date(Web):March 23, 2010
DOI:10.1021/ic902070q
A novel tris(2-pyridylmethyl)amine (TPA) derivate having two catechol moieties linked by amide linkages at the 6-positions of two pyridyl groups was synthesized. The ligand, N,N-bis[6-{3,4-(dihydroxy)benzamide}-2-pyridyl-methyl]-N-(2-pyridylmethyl)amine (Cat2-TPA; L2), and its precursor, N,N-bis[6-{3,4-bis(benzyloxy)-benzamide}-2-pyridyl-methyl]-N-(2-pyridylmethyl)-amine ((Bn2Cat)2-TPA; L1), formed stable ruthenium(II) complexes, [RuCl(L2)]PF6 (2) and [RuCl(L1)]PF6 (1), respectively. The crystal structure of [RuCl(L2)]Cl (2′) was determined by X-ray crystallography to show two isomers in terms of the orientation of one catechol moiety. In complex 2, the ligand bearing catechols acts as a pentadentate ligand involving coordination of one of the amide oxygen atoms in addition to that of the tetradentate TPA moiety and two metal-free catechol moieties as metal-binding sites. The coordination of L2 results in the preorganization of the two catechols to converge them to undergo intramolecular π−π interactions. The 1H NMR spectrum of 2 in DMSO-d6 revealed that only one isomer was present in the solution. This selective formation could be ascribed to the formation of an intramolecular hydrogen-bonding network among the hydroxyl groups of the catechol moieties, as suggested by X-ray analysis. This intramolecular hydrogen bonding could differentiate the pKa values of the hydroxy groups of the catechol moieties into three kinds, as indicated by spectroscopic titration with tetramethylammonium hydroxide (TMAOH) in DMF. The complexation of 2 with other metal ions was also examined. The reaction of 2 with [Cu(NO3)2(TMEDA)] (TMEDA = N,N,N′,N′-tetramethylethylenediamine) in methanol allowed us to observe the selective formation of a binuclear complex, [RuCl(L22−){Cu(TMEDA)}]PF6 (3), which was characterized by ESI-MS, UV−vis, and ESR spectroscopies. Its ESR spectrum in methanol suggested that the coordination of the Cu(II)-TMEDA unit to the converged catechol moieties would be different from conventional κ2-O,O′:η2-coordination: it exhibits a novel bridging coordination mode, bis-κ1-O:η1-coordination, to form the binuclear Ru(II)−Cu(II) complex.
Co-reporter:Atsutoshi Yokoyama, Takahiko Kojima, Kei Ohkubo, and Shunichi Fukuzumi
Inorganic Chemistry 2010 Volume 49(Issue 23) pp:11190-11198
Publication Date(Web):November 10, 2010
DOI:10.1021/ic1019586
Reactions of a saddle-distorted Mo(V)-porphyrin complex, [Mo(DPP)(O)(H2O)]ClO4 (1·ClO4; DPP2− = dodecaphenylporphyrin dianion), with tetra-n-butylammonium (TBA) salts of Keggin-type heteropolyoxomatalates (POMs), α-[XW12O40]n− (X = P, n = 3, 2; X = Si, n = 4, 3; X = B, n = 5; 4), in ethyl acetate/acetonitrile gave 2:1 complexes formulated as [{Mo(DPP)(O)}2(HPW12O40)] (5), [{Mo(DPP)(O)}2(H2SiW12O40)] (6), and [(n-butyl)4N]2[{Mo(DPP)(O)}2(HBW12O40)] (7) under mild reaction conditions. The crystal structures of the complexes were determined by X-ray crystallography. In these three complexes, named Porphyrin Hamburgers, the POM binds to two Mo(V) centers of porphyrin units directly via coordination of two terminal oxo groups. In spite of the similarity of those POM’s structures, those Porphyrin Hamburgers exhibit different coordination bond angles between POM and the Mo(V) center in the porphyrin: 5 and 7 show two different coordination bond angles in one molecule in contrast to 6, which exhibits only one coordination bond angle. The Porphyrin Hamburgers involve protonation of the POM moieties to adjust the charge balance, as confirmed by spectroscopic titration with bases. In the crystals, the Porphyrin Hamburgers form two-dimensional (2D) sheets in the ac plane based on π−π interactions among peripheral phenyl substituents. Stacking of the 2D sheets toward the b axis constructs a 3D layered structure involving channels running into the crystallographic [1 0 0] and [0 0 1] directions in the crystal to include solvent molecules of crystallization for 5−7, and also counter cations for 7. Three complexes were revealed to be stable enough to maintain their structures even in solutions to show molecular ion peaks in the MALDI-TOF-MS measurements. They also exhibited different electron paramagnetic resonance (EPR) signals because of the Mo(V) (S = 1/2, I = 0) centers, reflecting the difference in the crystal structures. In addition, these complexes showed reversible multistep redox processes as observed in their cyclic voltammograms in benzonitrile to demonstrate high stability throughout the redox reactions in solution.
Co-reporter:Dr. Takahiko Kojima;Yuichirou Hirai;Dr. Tomoya Ishizuka;Dr. Yoshihito Shiota;Dr. Kazunari Yoshizawa;Dr. Kenichiro Ikemura;Dr. Takashi Ogura;Dr. Shunichi Fukuzumi
Angewandte Chemie 2010 Volume 122( Issue 45) pp:8627-8631
Publication Date(Web):
DOI:10.1002/ange.201002733
Co-reporter:Takahiko Kojima Dr.;Kakeru Hanabusa;Kei Ohkubo Dr.;Motoo Shiro Dr.;Shunichi Fukuzumi Dr.
Chemistry - A European Journal 2010 Volume 16( Issue 12) pp:3646-3655
Publication Date(Web):
DOI:10.1002/chem.200902939
Abstract
Novel conglomerates consisting of saddle-distorted SnIV(DPP) (H2DPP=dodecaphenylporphyrin) complexes and μ3-O-centered and carboxylato-bridged trinuclear RuIII clusters linked by pyridine carboxylates were synthesized and characterized. SnIV–DPP complexes with Cl−, OH−, and 3- and 4-pyridine carboxylates ligands were characterized by spectroscopic methods and X-ray crystallography. Reactions of [Sn(DPP)(pyridinecarboxylato)2] with trinuclear RuIII clusters gave novel conglomerates in moderate yields. The conglomerates are stable in solution as demonstrated by 1H NMR and electrospray ionization mass spectrometry (ESI-MS) measurements, which show consistent spectra with those expected from their structures, and also by electrochemical measurements, which exhibit reversible multistep redox processes. This stability stems from the saddle distortion of the DPP2− ligand to enhance the Lewis acidity of the SnIV center that strengthens the axial coordination of the linker. The fast intramolecular photoinduced electron transfer from the SnIV(DPP) unit to trinuclear RuIII clusters, affording the electron-transfer (ET) state {Sn(DPP.+)–RuIIRuIII2}, was observed by femtosecond laser flash photolysis. The lifetimes of ET states of the conglomerates were determined to be in the range 98–446 ps, depending on the clusters and energies of the ET states. The reorganization energy of the electron transfer was determined to be 0.58±0.08 eV in light of the Marcus theory of electron transfer. The rate constants of both the photoinduced electron transfer and the back electron transfer in the conglomerates fall in the Marcus inverted region due to the small reorganization energy of electron transfer.
Co-reporter:Takahiko Kojima Dr.;Kakeru Hanabusa;Kei Ohkubo Dr.;Motoo Shiro Dr.;Shunichi Fukuzumi Dr.
Chemistry - A European Journal 2010 Volume 16( Issue 12) pp:
Publication Date(Web):
DOI:10.1002/chem.201090053
Co-reporter:Dr. Takahiko Kojima;Yuichirou Hirai;Dr. Tomoya Ishizuka;Dr. Yoshihito Shiota;Dr. Kazunari Yoshizawa;Dr. Kenichiro Ikemura;Dr. Takashi Ogura;Dr. Shunichi Fukuzumi
Angewandte Chemie International Edition 2010 Volume 49( Issue 45) pp:8449-8453
Publication Date(Web):
DOI:10.1002/anie.201002733
Co-reporter:Takahiko Kojima, Yuji Inui, Soushi Miyazaki, Motoo Shiro and Shunichi Fukuzumi
Chemical Communications 2009 (Issue 43) pp:6643-6645
Publication Date(Web):25 Sep 2009
DOI:10.1039/B911033J
A novel tetranuclear Ir(III) complex involving unprecedented coordination modes of alloxazine formed a closed π-space by intermolecular hydrogen bonding and the counter anions encapsulated in the space could be exchanged viaself-assembly.
Co-reporter:Tatsuhiko Honda, Takahiko Kojima and Shunichi Fukuzumi
Chemical Communications 2009 (Issue 33) pp:4994-4996
Publication Date(Web):13 Jul 2009
DOI:10.1039/B910077F
A stable monoprotonated porphyrin (porphyrin monoacid) was obtained by reaction of saddle-distorted dodecaphenylporphyrin with anthracene sulfonic acids and the crystal structures of the supramolecular assemblies were determined.
Co-reporter:Shunichi Fukuzumi, Takahiko Kojima, Yong-Min Lee, Wonwoo Nam
Coordination Chemistry Reviews (15 February 2017) Volume 333() pp:
Publication Date(Web):15 February 2017
DOI:10.1016/j.ccr.2016.09.018
•Generation of metal-oxo complexes by proton-coupled electron transfer.•Catalytic oxygenation of substrates by metal complexes using water as an oxygen source.•Catalytic water oxidation by one-electron oxidants with metal complexes.•Photocatalytic oxidation of water and substrates with metal complexes.High-valent metal-oxo complexes are produced by successive electron-transfer oxidation of metal complexes with one-electron oxidants in the presence of water, which is an oxygen source in the generation of the metal-oxo complexes. Then, metal-oxo complexes oxidize substrates to yield oxygenated substrates, accompanied by the regeneration of reduced metal complexes. Thus, the oxidation of substrates using one-electron oxidants can be catalyzed by metal complexes via formation of high-valent metal-oxo complexes by the electron-transfer oxidation of metal complexes in the presence of water as an oxygen source. When water is used as a substrate, water is oxidized by one-electron oxidants to evolve dioxygen via an OO bond formation process. The one-electron oxidants used for the formation of high-valent metal-oxo complexes can be replaced by much weaker oxidants, when a photosensitizing metal complex, such as [Ru(bpy)3]2+ (bpy = 2,2′-bipyridine), is employed as a photocatalyst, an oxidized form of the photocatalyst, which is generated via photoinduced electron transfer from the excited state to a weaker oxidant, can oxidize metal complexes in the presence of water to afford the high-valent metal-oxo complexes. Thus, the oxidation of substrates, including water oxidation, by weak oxidants can be catalyzed by metal complexes under photoirradiation of the photocatalyst using water as an oxygen source.
Co-reporter:Yuji Inui, Motoo Shiro, Shunichi Fukuzumi and Takahiko Kojima
Organic & Biomolecular Chemistry 2013 - vol. 11(Issue 5) pp:NaN764-764
Publication Date(Web):2012/11/28
DOI:10.1039/C2OB26877A
The formation of guanine quartets with 9-isopropylguanine (iPG) is discussed in organic solvents. Crystal structures of the iPG quartets were determined by X-ray crystallography with template cations (Na+ and Ca2+) and the structure without a template cation was also obtained by virtue of the stabilization by intermolecular hydrogen bonding with water molecules of crystallization. The difference in the quartet formation of iPG in the presence and absence of a template cation was clearly demonstrated by 1H NMR measurements in CDCl3–CH3OH mixed solvents. The quartet formation is mainly governed by the enthalpy gain due to the electrostatic interaction between the O6 oxygen in iPG and the template cations in the presence of the cations rather than the intermolecular hydrogen bonding, while desolvation of iPG is the dominant factor for the formation in the absence of cations. In the presence of Na+ and Ca2+, ΔH and ΔS values in the formation of iPG-4–Na+ and iPG-4–Ca2+ complexes were determined to be ΔH = −8.4 kcal mol−1 and ΔS = +50 cal mol−1 K−1 for Na+ and ΔH = −12.9 kcal mol−1 and ΔS = +34 cal mol−1 K−1 for Ca2+ on the basis of van't Hoff plots attained from the results of temperature-dependent UV-Vis spectroscopic measurements.
Co-reporter:Yuji Inui, Motoo Shiro, Takahiro Kusukawa, Shunichi Fukuzumi and Takahiko Kojima
Dalton Transactions 2013 - vol. 42(Issue 8) pp:NaN2778-2778
Publication Date(Web):2012/11/28
DOI:10.1039/C2DT32535G
[Ir6(μ-alloCl22−)3(Cp*)6(OH)3](PF6)3 (1) having 7,8-dichloroalloxazine dianion (alloCl22−) as bridging ligands was synthesized and characterized by X-ray crystallography, spectroscopic and electrochemical measurements. The alloxazine ligands showed unprecedented coordination modes to link the six Ir(III) centres. The complex exhibited remarkable stability and reversible six-electron redox processes at the bridging alloxazine ligands in organic solvents. The first reversible reduction process occurred on each of three alloxazine ligands in 1 to produce a three-electron-reduced species, [IrIII6Cp*6(μ-alloCl2˙3−)3(OH)3], and was observed as an apparent one-step reduction process at −0.65 V (vs. Fc0/+). The second reversible reduction process on each of the three alloxazine ligands in 1 was recorded at almost the same potential, −0.78 V (vs. Fc0/+), to afford the six-electron-reduced form, [IrIII6Cp*6(μ-alloCl24−)3(OH)3]3−. The radical anion of the alloxazine derivative was detected by EPR measurements at room temperature. After the six-electron reduction of 1 with cobaltocene, the backward oxidation processes of reduced forms with p-chloranil were traced by UV-Vis spectroscopy to confirm the recovery of the original spectrum of 1.
Co-reporter:Tomoya Ishizuka, Hiroaki Kotani and Takahiko Kojima
Dalton Transactions 2016 - vol. 45(Issue 42) pp:NaN16750-16750
Publication Date(Web):2016/09/19
DOI:10.1039/C6DT03024F
In this perspective, we have surveyed the synthetic procedure, characteristics, and reactivity of high-valent ruthenium–oxo complexes. The ruthenium–oxo complexes have served as ideal species to elucidate the characteristics of metal–oxo complexes in terms of not only geometrical and electronic structures but also oxidation reactivity and mechanisms of oxidation reactions. Due to the high stability and excellent reversibility of redox processes, ruthenium–oxo complexes have provided significant mechanistic insights into the oxidation of organic compounds including alcohols, alkenes, and alkanes and also water on the basis of detailed kinetic analysis.
Co-reporter:Wataru Suzuki, Hiroaki Kotani, Tomoya Ishizuka, Yoshihito Shiota, Kazunari Yoshizawa and Takahiko Kojima
Chemical Communications 2017 - vol. 53(Issue 47) pp:NaN6362-6362
Publication Date(Web):2017/05/19
DOI:10.1039/C7CC03635C
The thermodynamic stability of diprotonated saddle-distorted dodecaphenylporphyrin (H4DPP2+(X−)2) was controlled by the hydrogen-bonding strength of conjugate bases (X−) of strong acids (HX) or acids (R+-COOH) having positively charged moieties. The thermodynamic control of H4DPP2+(X−)2 made it possible to achieve selective formation of supramolecular hetero-triads, H4DPP2+(X−)(Cl−).
Co-reporter:Tomoya Ishizuka, Yuta Saegusa, Yoshihito Shiota, Kazuhisa Ohtake, Kazunari Yoshizawa and Takahiko Kojima
Chemical Communications 2013 - vol. 49(Issue 53) pp:NaN5941-5941
Publication Date(Web):2013/05/14
DOI:10.1039/C3CC42831A
A novel quadruply-fused porphyrin has been synthesized with a facilely prepared precursor in a high yield. A detailed comparison of the physical properties of a series of fused porphyrins revealed remarkable effects of the ring fusion on lowering LUMO levels rather than HOMO levels.
Co-reporter:Tatsuhiko Honda, Takahiko Kojima and Shunichi Fukuzumi
Chemical Communications 2009(Issue 33) pp:
Publication Date(Web):
DOI:10.1039/B910077F
Co-reporter:Takahiko Kojima, Yuji Inui, Soushi Miyazaki, Motoo Shiro and Shunichi Fukuzumi
Chemical Communications 2009(Issue 43) pp:NaN6645-6645
Publication Date(Web):2009/09/25
DOI:10.1039/B911033J
A novel tetranuclear Ir(III) complex involving unprecedented coordination modes of alloxazine formed a closed π-space by intermolecular hydrogen bonding and the counter anions encapsulated in the space could be exchanged viaself-assembly.
Co-reporter:Shingo Ohzu, Tomoya Ishizuka, Yuichirou Hirai, Hua Jiang, Miyuki Sakaguchi, Takashi Ogura, Shunichi Fukuzumi and Takahiko Kojima
Chemical Science (2010-Present) 2012 - vol. 3(Issue 12) pp:NaN3431-3431
Publication Date(Web):2012/08/31
DOI:10.1039/C2SC21195E
A series of Ru(IV)-oxo complexes (4–6) were synthesized from the corresponding Ru(II)-aqua complexes (1–3) and fully characterized by 1H NMR and resonance Raman spectroscopies, and ESI-MS spectrometry. Based on the diamagnetic character confirmed by the 1H NMR spectroscopy in D2O, the spin states of 5 and 6 were determined to be S = 0 in the d4 configuration, in sharp contrast to that of 4 being in the S = 1 spin state. The aqua-complexes 1–3 catalyzed oxidation of alcohols and olefins using (NH4)2[CeIV(NO3)6] (CAN) as an electron-transfer oxidant in acidic aqueous solutions. Comparison of the reactivity of electrochemically generated oxo-complexes (4–6) was made in the light of kinetic analyses for oxidation of 1-propanol and a water-soluble ethylbenzene derivative. The oxo complexes (4–6) exhibited no significant difference in the reactivity for the oxidation reactions, judging from the similar catalytic rates and the activation parameters. The slight difference observed in the reaction rates can be accounted for by the difference in the reduction potentials of the oxo-complexes, but the spin states of the oxo-complexes have hardly affected the reactivity. The activation parameters and the kinetic isotope effects (KIE) observed for the oxidation reactions of methanol indicate that the oxidation reactions of alcohols with the RuIVO complexes proceed via a concerted proton-coupled electron transfer mechanism.
Co-reporter:Atsutoshi Yokoyama, Takahiko Kojima and Shunichi Fukuzumi
Dalton Transactions 2011 - vol. 40(Issue 24) pp:NaN6450-6450
Publication Date(Web):2011/05/13
DOI:10.1039/C0DT01708F
A 2:1 supramolecular assembly composed of a non-planar Mo(V)-porphyrin, [Mo(DPP)(O)(H2O)]+ (1) (DPP2+; dodecaphenylporphyrin), and a Keggin-type heteropolyoxometalate (POM), α-[(n-butyl)4N]2[SW12O40] (2), was formed via hydrogen bonds. The crystal structure was determined by X-ray crystallography to clarify that the POM was enclosed into a π-space of a supramolecular porphyrin nanotube by virtue of a hydrogen-bond network. In contrast to the formation of the 2:1 assembly ([{Mo(DPP)(O)(H2O)}2(SW12O40)] (3)) between 1 and [SW12O40]2− in the crystal, it was revealed that those two components form a 1:1 assembly in solution, in light of the results of MALDI-TOF-MS measurements in PhCN. Variable-temperature UV-vis spectroscopic titration allowed us to determine the thermodynamic parameters for the formation of the 1:1 supramolecular assembly in solution, the heat of formation (ΔH) and the entropy change (ΔS). These results provide the first thermodynamic data set to elucidate the formation process of supramolecuar structures emerged by hydrogen bonding between metalloporphyrin complexes and POMs, indicating that the formation of the assembly is an entropy-controlled process rather than an enthalpy-controlled one. Comparisons of the thermodynamic parameters with those of a planar Mo(V)-porphyrin complex also highlighted high Lewis acidity of the Mo(V) centre in the distorted porphyrin.
Co-reporter:Atsutoshi Yokoyama, Kei Ohkubo, Tomoya Ishizuka, Takahiko Kojima and Shunichi Fukuzumi
Dalton Transactions 2012 - vol. 41(Issue 33) pp:NaN10013-10013
Publication Date(Web):2012/04/03
DOI:10.1039/C2DT30424D
A 2:1 complex composed between a non-planar Mo(V)–porphyrin complex ([Mo(DPP)(O)]+, DPP2− = dodecaphenylporphyrin) and a ruthenium-substituted Keggin-type heteropolyoxometalate (Ru-POM), [SiW11O39RuIII(DMSO)]5−, acts as an efficient catalyst for oxidation of benzyl alcohols with iodosobenzene as an oxidant in CDCl3 at room temperature. The catalytic oxidation afforded the corresponding benzaldehydes, whereas neither the ammonium salt of Ru-POM nor [Mo(DPP)(O)]+ alone exhibited catalytic reactivity under the same experimental conditions. This enhancement can be attributed to a large anodic shift of the redox potential of the ruthenium centre due to the complexation of the Ru-POM with two cationic {Mo(DPP)(O)}+ units. The kinetic analysis demonstrated that the catalytic oxidation proceeded via formation of a catalyst-substrate complex, and electron-withdrawing substituents at the para position of benzyl alcohol accelerated the reaction. The rate constants of the oxidation reactions correlate to the bond dissociation energies of the C–H bonds of the substrate. A linear correlation was observed for logarithm of the rate constants of oxidation reactions of benzyl alcohols with that of hydrogen abstraction by cumyl peroxyl radical, indicating the reaction proceeds via hydrogen abstraction. The observed kinetic isotope effect (KIE) indicates that the hydrogen abstraction occurs from the benzyl group rather than the hydroxy group.
Co-reporter:Yuta Saegusa, Tomoya Ishizuka, Keiyu Komamura, Soji Shimizu, Hiroaki Kotani, Nagao Kobayashi and Takahiko Kojima
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 22) pp:NaN15011-15011
Publication Date(Web):2015/05/13
DOI:10.1039/C5CP01420D
Here, we report the effects of ring fusion, which causes expansion of the π-conjugation circuits of the porphyrin derivatives to the fused meso-aryl groups, on the aromaticity and the magnetic properties of porphyrin derivatives. These studies revealed the facts that the ring fusion with five-membered rings causes not only the remarkable red shifts of the absorption bands and narrowed HOMO–LUMO gaps, but also the contribution of anti-aromatic resonance forms to the magnetic properties as observed in the 1H NMR spectra. The optical absorption and magnetic circular dichroism (MCD) spectroscopies indicate that the increase in the number of the fused rings causes stabilization of the LUMO level of the porphyrin derivatives and as a result induces the loosening of the LUMO degeneracy that is generally observed for porphyrins. The electronic structure of a quadruply fused porphyrin derivative was experimentally clarified by the ESR studies on the 1e−-oxidized and 1e−-reduced species in THF. Furthermore, we revealed the substituent effects of the fused meso-aryl groups of quadruply fused porphyrins (QFPs) on the crystal structures, absorption spectra and redox potentials; the sensitiveness of the substituent effects shows that the π-conjugation circuits extended to the fused meso-aryl groups. Additionally, the elongation of the bond lengths between the pyrrolic nitrogen and the central metal ions in QFP–metal complexes causes a remarkable increase of the Lewis acidity of the central metal ions.
Co-reporter:Yuji Inui, Shunichi Fukuzumi and Takahiko Kojima
Dalton Transactions 2013 - vol. 42(Issue 11) pp:NaN3782-3782
Publication Date(Web):2013/01/14
DOI:10.1039/C3DT33034F
Formation of a π–π stacked assembly between a Na+-templated G-quartet and octaethylporphyrinatonickel(II) was observed by spectroscopic methods in methanol/chloroform and the formation dynamics of the assembly was elucidated.
Co-reporter:Tomoya Ishizuka, Muniappan Sankar and Takahiko Kojima
Dalton Transactions 2013 - vol. 42(Issue 45) pp:NaN16079-16079
Publication Date(Web):2013/08/01
DOI:10.1039/C3DT51467F
Supramolecular integration of a saddle-distorted zinc(II) porphyrin complex, which has hydroxyl groups at the para-position of the four meso-aryl groups, has been demonstrated on the basis of hydrogen bonding among the peripheral hydroxyl groups. The hydrogen-bonding patterns were controlled by the recrystallization solvents and additives, and particularly, addition of a bifunctional ligand such as 4,4′-bipyridine (bpy). The coordination of bpy to form dinuclear ZnII-porphyrin complexes causes a conformational difference: the dimeric complex with four hydroxyl groups is in an eclipsed form, however, a derivative without hydroxyl groups is in a staggered form due to the presence or absence of the intermolecular hydrogen bonding. In addition, the dimerization by the bpy coordination resulted in the expansion of the intermolecular space formed in the porphyrin networks, suggesting the potential to be applied for inclusion of guest molecules.
Co-reporter:Hiroumi Mitome, Tomoya Ishizuka, Yoshihito Shiota, Kazunari Yoshizawa and Takahiko Kojima
Dalton Transactions 2015 - vol. 44(Issue 7) pp:NaN3158-3158
Publication Date(Web):2014/12/22
DOI:10.1039/C4DT03358B
Ruthenium(II) complexes of PQQTME, a trimethyl ester derivative of redox-active PQQ (pyrroloquinolinequinone), were prepared using a tridentate ligand, 2,2′:6′,2′′-terpyridine (terpy) as an auxiliary ligand. The characterization of the complexes was performed by spectroscopic methods, X-ray crystallography, and electrochemical measurements. In one complex, the pyridine site of PQQTME binds to the [RuII(terpy)] unit as a tridentate ligand, and a silver(I) ion is coordinated by the quinone moiety in a bidentate fashion. In contrast, another complex includes the [RuII(terpy)] unit at the bidentate quinone moiety of the PQQTME ligand. The difference in the coordination modes of the complexes exhibits a characteristic difference in the stability of metal coordination and also in the reversibility of the reduction processes of the PQQTME ligand. It should be noted that an additional metal-ion-binding to the PQQTME ligand largely raises the 1e−-reduction potential of the ligand. In addition, we succeeded in the characterization of the 1e−-reduced species of the complexes, where the unpaired electron was delocalized in the π-conjugated system of the PQQTME˙− ligand, using UV-Vis absorption and ESR spectroscopies.
Co-reporter:Hiroaki Kotani, Suzue Kaida, Tomoya Ishizuka, Miyuki Sakaguchi, Takashi Ogura, Yoshihito Shiota, Kazunari Yoshizawa and Takahiko Kojima
Chemical Science (2010-Present) 2015 - vol. 6(Issue 2) pp:NaN955-955
Publication Date(Web):2014/10/17
DOI:10.1039/C4SC02285H
A mononuclear Cr(V)–oxo complex, [CrV(O)(6-COO−-tpa)](BF4)2 (1; 6-COO−-tpa = N,N-bis(2-pyridylmethyl)-N-(6-carboxylato-2-pyridylmethyl)amine) was prepared through the reaction of a Cr(III) precursor complex with iodosylbenzene as an oxidant. Characterization of 1 was achieved using ESI-MS spectrometry, electron paramagnetic resonance, UV-vis, and resonance Raman spectroscopies. The reduction potential (Ered) of 1 was determined to be 1.23 V vs. SCE in acetonitrile based on analysis of the electron-transfer (ET) equilibrium between 1 and a one-electron donor, [RuII(bpy)3]2+ (bpy = 2,2′-bipyridine). The reorganization energy (λ) of 1 was also determined to be 1.03 eV in ET reactions from phenol derivatives to 1 on the basis of the Marcus theory of ET. The smaller λ value in comparison with that of an Fe(IV)–oxo complex (2.37 eV) is caused by the small structural change during ET due to the dπ character of the electron-accepting LUMO of 1. When benzyl alcohol derivatives (R-BA) with different oxidation potentials were employed as substrates, corresponding aldehydes were obtained as the 2e−-oxidized products in moderate yields as determined from 1H NMR and GC-MS measurements. One-step UV-vis spectral changes were observed in the course of the oxidation reactions of BA derivatives by 1 and a kinetic isotope effect (KIE) was observed in the oxidation reactions for deuterated BA derivatives at the benzylic position as substrates. These results indicate that the rate-limiting step is a concerted proton-coupled electron transfer (PCET) from substrate to 1. In sharp contrast, in the oxidation of trimethoxy-BA (Eox = 1.22 V) by 1, trimethoxy-BA radical cation was observed by UV-vis spectroscopy. Thus, it was revealed that the mechanism of the oxidation reaction changed from one-step PCET to stepwise ET–proton transfer (ET/PT), depending on the redox potentials of R-BA.
Co-reporter:Tomoya Ishizuka, Shingo Ohzu, Hiroaki Kotani, Yoshihito Shiota, Kazunari Yoshizawa and Takahiko Kojima
Chemical Science (2010-Present) 2014 - vol. 5(Issue 4) pp:NaN1436-1436
Publication Date(Web):2013/12/06
DOI:10.1039/C3SC53002G
Detailed kinetic studies on the oxidation reactions of organic substrates such as methanol with RuIVO complexes as oxidants, formed electrochemically in water, have been conducted to elucidate the reaction mechanism. The rate constants of the oxidation reactions exhibited saturation behaviours relative to the substrate concentration, regardless of the oxidants and the substrates employed. This indicates the existence of a pre-equilibrium process based on the adduct formation between the RuIVO oxidant and the substrate. Herein, we have experimentally confirmed that the driving force of the adduct formation is the hydrogen bonding between the oxidants and alcohols even in water. In addition, we have investigated the kinetic isotope effects (KIE) on the oxidation reaction using methanol and its deuterated derivatives and as a result observed moderate KIE values for the C–H bond of methanol. We have also revealed the independency of the reaction rates from the bond dissociation enthalpies of the C–H bonds of the substrates. This independency is probably derived from the tightly condensed transition state, whose energy level is strongly controlled by the activation entropy but less sensitive to the activation enthalpy.
Co-reporter:Tomoya Ishizuka, Muniappan Sankar, Yusuke Yamada, Shunichi Fukuzumi and Takahiko Kojima
Chemical Communications 2012 - vol. 48(Issue 52) pp:NaN6483-6483
Publication Date(Web):2012/04/17
DOI:10.1039/C2CC31142A
Carboxyl groups were introduced at the peripheral positions of dodecaphenylporphyrin to link nanochannel structures with intermolecular hydrogen bonds to make the supramolecular structures robust.
Co-reporter:Tatsuhiko Honda, Takahiko Kojima and Shunichi Fukuzumi
Chemical Communications 2011 - vol. 47(Issue 28) pp:NaN7988-7988
Publication Date(Web):2011/06/17
DOI:10.1039/C1CC12710A
Facile protonation of α-octabutoxyphthalocyaninato zinc(II) (Zn(OBu)8Pc) occurs to afford up to tetra-protonated species stabilized by intramolecular hydrogen bonding, resulting in positive shifts of the reduction potentials of Zn(OBu)8PcHnn+ (n = 1–4) with increasing the number of protons attached to facilitate electron-transfer reduction.
Co-reporter:Shingo Ohzu, Tomoya Ishizuka, Hiroaki Kotani and Takahiko Kojima
Chemical Communications 2014 - vol. 50(Issue 95) pp:NaN15021-15021
Publication Date(Web):2014/10/14
DOI:10.1039/C4CC07488B
A mononuclear RuIII–OH complex oxidizes substrates such as hydroquinones in water through a pre-equilibrium process based on adduct formation by hydrogen bonding between the RuIII–OH complex and the substrates. The reaction mechanism switches from hydrogen atom transfer to electron transfer depending on the oxidation potential of substrates.
Co-reporter:Hiroaki Kotani, Tomomi Yagi, Tomoya Ishizuka and Takahiko Kojima
Chemical Communications 2015 - vol. 51(Issue 69) pp:NaN13388-13388
Publication Date(Web):2015/07/13
DOI:10.1039/C5CC03012A
We have synthesised a novel copper(II) complex with a pyridine pendant as a proton relay port for electrocatalytic 4e− reduction of O2 in water. The enhancement of the electrocatalytic O2 reduction via protonation of the pyridine pendant is demonstrated in comparison with a copper(II) complex without the pyridine pendant.
Co-reporter:Tomoya Ishizuka, Hiroaki Kotani and Takahiko Kojima
Dalton Transactions 2016 - vol. 45(Issue 48) pp:NaN19529-19529
Publication Date(Web):2016/11/22
DOI:10.1039/C6DT90217K
Correction for ‘Characteristics and reactivity of ruthenium–oxo complexes’ by Tomoya Ishizuka et al., Dalton Trans., 2016, 45, 16727–16750.

![Benzenamine, 4-[10,15,20-tris[3,5-bis(1,1-dimethylethyl)phenyl]-21H,23H-porphin-5-yl]-](/data/chemimg/3724200/163811-80-1.png)
![Benzenamine, 4-[10,15,20-tris[3,5-bis(1,1-dimethylethyl)phenyl]-21H,23H-porphin-5-yl]-](/data/chemimg/3724200/163811-80-1_b.png)
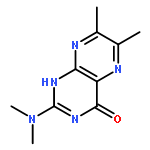
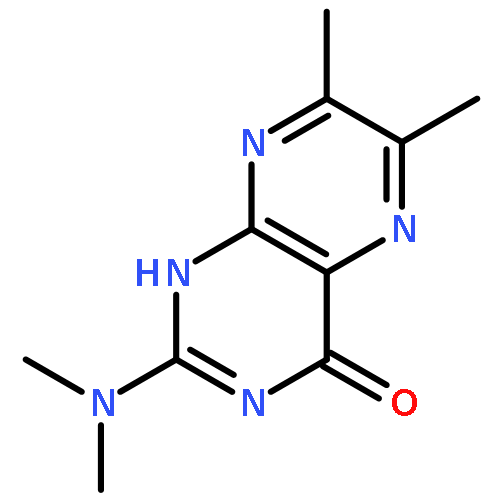
![BENZO[G]PTERIDINE-2,4(3H,10H)-DIONE, 10-DECYL-](http://img.cochemist.com/ccimg/81600/81569-78-0.png)
![BENZO[G]PTERIDINE-2,4(3H,10H)-DIONE, 10-DECYL-](http://img.cochemist.com/ccimg/81600/81569-78-0_b.png)
![1H-Pyrrole, 2,2'-[[3,5-bis(1,1-dimethylethyl)phenyl]methylene]bis-](http://img.cochemist.com/ccimg/181800/181762-72-1.png)
![1H-Pyrrole, 2,2'-[[3,5-bis(1,1-dimethylethyl)phenyl]methylene]bis-](http://img.cochemist.com/ccimg/181800/181762-72-1_b.png)
![21H,23H-PORPHINE, 5,10,15,20-TETRAKIS[4-(TRIFLUOROMETHYL)PHENYL]-](http://img.cochemist.com/ccimg/56500/56420-24-7.png)
![21H,23H-PORPHINE, 5,10,15,20-TETRAKIS[4-(TRIFLUOROMETHYL)PHENYL]-](http://img.cochemist.com/ccimg/56500/56420-24-7_b.png)









![2-Propanamine,2-methyl-N-[(1-oxido-4-pyridinyl)methylene]-, N-oxide](http://img.cochemist.com/ccimg/66900/66893-81-0.png)
![2-Propanamine,2-methyl-N-[(1-oxido-4-pyridinyl)methylene]-, N-oxide](http://img.cochemist.com/ccimg/66900/66893-81-0_b.png)
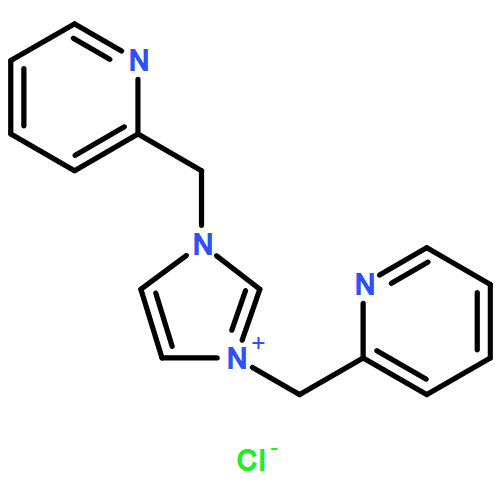







![TRIMETHYL 4,5-DIOXO-1H-PYRROLO[2,3-F]QUINOLINE-2,7,9-TRICARBOXYLATE](http://img.cochemist.com/ccimg/74500/74447-88-4.png)
![TRIMETHYL 4,5-DIOXO-1H-PYRROLO[2,3-F]QUINOLINE-2,7,9-TRICARBOXYLATE](http://img.cochemist.com/ccimg/74500/74447-88-4_b.png)
![4,5-Dioxo-4,5-dihydro-1H-pyrrolo[2,3-f]quinoline-2,7,9-tricarboxylic acid](/data/chemimg/654800/72909-34-3.png)
![4,5-Dioxo-4,5-dihydro-1H-pyrrolo[2,3-f]quinoline-2,7,9-tricarboxylic acid](/data/chemimg/654800/72909-34-3_b.png)


![21H,23H-Porphine, 5,10,15,20-tetrakis[4-(1,1-dimethylethyl)phenyl]-](/data/chemimg/133300/110452-48-7.png)
![21H,23H-Porphine, 5,10,15,20-tetrakis[4-(1,1-dimethylethyl)phenyl]-](/data/chemimg/133300/110452-48-7_b.png)